

Abstract
Autism Spectrum Disorders (ASD) currently affects approximately 1% of the population causing grave disability and necessitating a better understanding of the currently enigmatic etiology of these disorders. Recent data suggest that some patients with ASD may have a dysfunction in brain plasticity (specifically data from animal models and human studies suggest a propensity toward excessive amount of plasticity). Plasticity is essential to the establishment and maintenance of brain circuitry; however, too much plasticity may lead to instability of structural connections and compromise of functional systems necessary for cognition and behavior. Multiple lines of evidence suggest that plasticity declines throughout the age-span and may underlie age-related cognitive decline. We hypothesize that individuals whose cortex begins as relatively “hyperplastic” (such as may be seen in ASD) should then be relatively protected from age-related cognitive decline (which we suggest is related to a reduction in plasticity). In the current study, we conducted a multiple linear regression using age and diagnosis as predictor variables in order to evaluate strength of the relationship between age, diagnosis or an interaction of the two factors and the degree of modulation in cortical excitability by transcranial magnetic stimulation as an index of cortical plasticity. Results indicate that across the age-span individuals with ASD show a consistently increased modulation of cortical excitability as compared to typically developing individuals, such that the general slope of decline across the age span is matched across both groups. We have argued that an individual’s risk of age-related cognitive decline (and risk for manifesting symptoms of dementia) depends on the individual’s starting point and slopes of change in plasticity efficiency over the lifespan. Therefore, our results suggest that individuals with ASD might be relatively protected from age-related cognitive decline and the risk of dementia.
Keywords: Autism Spectrum Disorder, Plasticity, Alzheimer’s Disease, Transcranial Magnetic Stimulation, Protection
Introduction
According to the latest reports from the centers for disease control and prevention (CDC), Autism Spectrum Disorders (ASD) currently affect approximately 1.5% of the population [1] necessitating an urgent and coordinated response by scientists and clinicians working together to better understand the currently enigmatic etiology of these disorders in order to prevent and treat them more effectively. Even though the genetic cause is not identified, autism is a highly heritable neurodevelopmental disorder (with a heritability of approximately 90% [2, 3]. Clinical phenotype is quite variable with symptoms including qualitative impairments in communication and social skills as well as the presence of restricted, repetitive, and stereotyped patterns of behavior, interests, and activities [4]. Underlying genetics are complex, with over 700 genes having been identified being associated with ASD (http://genotator.hms.harvard.edu/geno/disorder/autism). The underlying etiology of ASD is thus likely to be found in a complex interaction of multiple genes with epigenetic and environmental factors.
Why would so many genes be associated with such a devastating condition? Certainly ASD are phenotypically diverse and it might be argued that the different genes involved lead to slightly different clinical manifestations and thus ASD is an amalgamation of diverse pathologies. On the other hand it is plausible that the core symptoms of ASD reflect a fundamental malfunction default of the human nervous system. Relevant to this perspective, it is noteworthy that the vast majority of the large number of candidate genes that have been identified code for proteins critical to synaptic development and plasticity [5]. This has led multiple researchers to suggest that ASD is a result of a dysfunction in the mechanisms of brain plasticity [6–9]. If so, an intriguing notion emerges: aberrant mechanisms of plasticity due to the genes associated with ASD may simultaneously cause pathology while conferring traits that are beneficial to the individual, and hence lead to evolutionary advantages that may tend to select for the preservation of the underlying gene pool.
Brain plasticity is an intrinsic property of the nervous system that allows an individual to adapt to a rapidly changing environment through strengthening, weakening, pruning, or adding of synaptic connections and by promoting neurogenesis [10]. Plasticity is essential to the establishment and maintenance of brain circuitry, but when plasticity is aberrant, it can account for the symptoms of disease. In older age a deficit in plasticity may render the brain unable to adjust to changing demands. On the other hand, if the brain is too plastic during development (as we suggest may be the case in ASD), structural connections may become unstable and functional systems necessary for cognitive and behavioral development may be compromised. Thus, plasticity must be kept at a homeostatic level, making moderators of such processes essential to healthy functioning across the lifespan. Plasticity is a complex phenomenon, mediated by a number of molecular substrates, and serving a variety of functions across different stages of development and different brain systems. We have argued [11] and provided direct empirical evidence [12, 13] that, for any given individual, the efficiency of neuronal plasticity declines throughout the age-span. Such age-related changes in plasticity are linked to an individual’s cognitive ability and age-related cognitive decline may be associated to them. An individual’s risk of age-related cognitive decline (and ultimately the manifestation of symptoms of dementia) might then depend on the individual’s starting point and slopes of change in plasticity efficiency over the lifespan. Indeed, studies in patients with early Alzheimer’s Disease, the most common dementing illness, reveal an abnormally suppressed efficacy of plasticity mechanisms [13, 14]. As schematically summarized in Figure 1, we argue that individuals whose cortex begins as relatively “hyperplastic” (easily adapting to the environment and quick to modulate synaptic connections) may be relatively protected from age-related cognitive decline.
Figure 1.

Schematic of Plasticity Hypothesis. We hypothesize that a number of environmental and genetic factors influence both the starting point and slopes of change in plasticity across the lifespan. Furthermore, assuming a similar slope of change, those individuals whose cortex begins as relatively “hyperplastic” (easily adapting to the environment and quick to modulate synaptic connections) are relatively protected from age-related cognitive decline (which we suggest is related to a reduced capacity for plastic change).
We have provided direct empirical evidence that adults with ASD show a greater duration of modulation to a repetitive transcranial magnetic stimulation (rTMS) paradigm called theta burst stimulation (TBS), designed to induce a modulation in cortical excitability and proposed to be a putative measure of plasticity [15]. Our original study focused on group effects finding the modulation of cortical excitability through the TBS protocol to be significantly longer lasting in the ASD group as compared to age matched controls. In the present study, we combined two data sets collected using the same protocol, and conducted a multiple linear regression on both the duration (“time to baseline”) as well as degree (“area under the curve”) of modulation of corticospinal excitability following cTBS the using age and diagnosis as predictor variables
Material and Methods
Participants
The data analyzed in this study came from the same cohort of patients recruited for a previous study (Oberman et al., 2012). Two cohorts of participants with Asperger’s Syndrome (AS) and matching neurotypical controls were studied. Data from cohort one were collected in Boston, Massachusetts, and were recorded from 20 individuals with AS [16 male (M), four female (F); age 18–64 (mean ± SD, 34.3 ± 16.4) years; mean ± SD IQ, 118.2 ± 17.3)] and 20 age-, gender- and full-scale IQ-matched typically developing (TD) individuals (16 M, four F; mean age, 34.9 ± 16.2 years; mean IQ, 112.0 ± 13.0). Data from the second cohort were collected in Barcelona, Spain, and were recorded from 15 individuals with AS [(14 M, one F; mean age, 42.4 ± 7.36 years; mean IQ, 110.4 ± 18.75) and 15 age-, gender- and IQ-matched TD individuals (14 M, one F; mean age, 42.4.1 ± 7.36 years; mean IQ, 115.3 (SD = 16.41)]. Thus a total of 70 participants were entered into this analysis. All participants gave informed consent to the study, which was reviewed and approved by the institutional review boards at each participating institution. Participants were recruited through local community advertisement and local Asperger’s Associations and clinics.
All AS participants in both cohorts had an IQ > 80 based on the Weschler Abbreviated Scale of Intelligence (WASI) and a formal clinical diagnosis from an independent clinician prior to participation in the study. All met DSM-IV-TR criteria for Asperger’s Syndrome and met criteria for ASD on the Autism Diagnostic Observation Schedule, Module 4 (ADOS) (mean ± SD Social and Communication score, 10.2 ± 4.6). Additionally, the Autism Diagnostic Interview Revised was completed on 11 participants whose parents were available for interview. For these individuals the mean Social score was 18.2 ± 5.1, Communication score was 20.0 ± 2.6 and Repetitive Behavior score was 6.0 ± 2.3. Cognitive and clinical evaluation was identical for the two cohorts, with Spanish-translated versions of the ADOS and WASI used for the participants in cohort two.
Participants in the neurotypical group were healthy controls with no neurological or psychiatric disorders. This group was matched with respect to chronological age, gender and full-scale IQ with the AS group. All participants were given a comprehensive neurological exam by a board-certified neurologist to confirm normal gross motor and fine motor functioning. Lastly, all participants were screened following published recommendations [16] to ensure that they did not have any condition that would put them at greater risk of an adverse event related to TMS (e.g. a personal or family history of epilepsy).
Stimulation and recording
Study procedures were identical in the two study locations. The experimenters who collected the data at each location used the same equipment and procedures described herein. continuous Theta Burst Stimulation (cTBS) was applied as described in Huang et al., 2005. The cTBS paradigm consisted of three pulses of 50 Hz stimulation repeated at 200-ms intervals for 40 s (for a total of 600 pulses) at an intensity of 80% of active motor threshold (AMT)(Fig. 2). Corticospinal excitability was assessed prior to and following cTBS by measuring peak-to-peak amplitude of MEPs induced in the contralateral first dorsal interosseus (FDI) muscle in response to single-pulse TMS at a rate of approximately 0.1 Hz (a random jitter of ± 1 s was introduced to avoid any train effects). Three batches of 10 MEPs were recorded prior to cTBS and used as a baseline. Following cTBS, batches of 10 MEPs were measured at periodic intervals for a total of 120 min to track changes in MEP amplitude over time.
Figure 2.

Schematic summary of Theta Burst Stimulation. Corticospinal excitability can be evaluated by comparing the peak-to-peak amplitude of motor evoked potentials (MEPs) recorded from a given target muscle in response to a single pulse of TMS to the primary motor cortex. Comparison of the TMS-induced MEPs at baseline and following a train of continuous theta burst stimulation (TBS) offers a measure of cortical plasticity (darker blue shaded area under the curve is a measure of LTD-like plasticity).
In order to measure TMS induced MEPs, Ag-AgCl EMG surface electrodes were placed in a belly-tendon montage over the FDI muscle of participants’ right hands. Raw signals were amplified and bandpass-filtered between 20 and 2000 Hz. EMG signals were sampled at a rate of 5000 Hz. All stimulation (single-pulse TMS and TBS) was delivered using a hand-held figure-of-eight coil attached to a Magstim Super Rapid stimulator. The coil was placed tangentially to the scalp with the handle pointing posteriorly and an orientation of approximately 45 degree from the midsagital plane. All stimulation was applied over the hand area of the left motor cortex and individually localized for each participant based on the optimal position for eliciting MEPs of maximal peak-to-peak amplitude in the right FDI. The stimulation intensity for baseline and post-TBS single pulses was set at 120% of each individual’s resting motor threshold (RMT) while the TBS itself was delivered at 80% of AMT. RMT and AMT were defined following recommendation from the International Federation of Clinical Neurophysiology. RMT was defined as the minimum single-pulse TMS intensity required to induce an MEP in the contralateral FDI of > 50 lV peak-to-peak amplitude on more than five out of ten consecutive trials while the target muscle was at rest. AMT was defined as the minimum single-pulse TMS intensity required to induce an MEP in the contralateral FDI of 200 lV peak-to-peak amplitude on more than five out of ten consecutive trials while the target muscle was held at approximately 20% of the maximal contraction. In order to precisely target the stimulation site (primary motor cortex) and keep the brain target constant throughout the stimulation session, we used a frameless stereotactic neuronavigation system (Brainsight, Rogue Inc.).
Data analysis
For all experiments across both cohorts data were analyzed using SPSS version 19 by an experimenter blind to the identities of the participants. MEP amplitude at a given time-point was defined as the mean amplitude of the 10 MEPs to single TMS pulses recorded in a given 2-min time window. As an index of the duration of the TBS induced modulation of corticospinal excitability, we defined, for each participant, the time-point at which the average MEP amplitude at a given time following TBS returned to within the 95% confidence interval of the baseline amplitude and did not return to outside that interval on subsequent time-point measures. MEP amplitudes were standardized, forming a ratio of MEP amplitudes following TBS relative to average baseline MEP amplitude for each individual.
Smooth curves through the data points were calculated using spline interpolation. Spline interpolation is a method of interpolation where the interpolant is a cubic spline, a piecewise continuous function defined by third-degree polynomials in the intervals of a limited range of known data points (in this case, the time-points at which MEP data were collected with batches of 10 single TMS pulses). Spline interpolation has advantages over both linear and polynomial interpolation: It is more precise than linear interpolation and minimizes oscillation inaccuracies often found with high-degree polynomials. Spline interpolation has previously been described and successfully applied spatially to TMS data [17]. As an index of the duration of the TBS-induced modulation of cortico-spinal excitability, we defined, for each participant, the time-point (“time-to-baseline”) at which post-cTBS MEP amplitude returned to the average MEP amplitude at baseline, i.e., the time-point at which the spline crossed the MEP threshold. In addition to time-to-baseline values, for each individual we calculated the area representing the amount of inhibition (“area of inhibition”) following cTBS until return to baseline, i.e., the positive-valued area bounded below by the spline and above by each subject’s MEP threshold.
Tests of normality indicated that the data were significantly different than normal, thus a natural log transformation was applied to the data and the log-transformed data were used for analysis. A multiple regression analysis was conducted with age, diagnosis and an interaction term as predictor variables for both “time-to-baseline” and “area of inhibition”. Statistical significance was defined at p < 0.05.
Results
A multiple regression analysis was used to test if a model including age, diagnosis and an interaction of the two factors significantly predicted participants’ response to the cTBS protocol. As described above, response to the cTBS protocol was defined as duration of effect as defined by the number of minutes following cTBS before the participant returned to baseline excitability levels (“time-to-baseline”) and degree of effect as defined by the total area under the curve defined by a ratio of average MEP amplitude at each time point following cTBS divided by the average baseline MEP amplitude (“area of inhibition”). The results of the regression indicated that this model explained 56% of the variance in “time-to-baseline” (R2=.56, F(3,66)=28.09, p<.001) and 45% of the variance in “area of inhibition” (R2=.45, F(3,66)=17.72, p<.001). It was found that diagnosis significantly predicted both “time-to-baseline” and “area of inhibition” (β = .66, p<.01 and β = .81, p<.01 respectively) neither age nor the interaction term contributed significantly to the model (Figure 3A,B).
Figure 3.

Results of logistic regression. Diagnosis significantly predicted both the duration (A) and degree (B) of modulation of excitability following cTBS.
Discussion
The current study aimed to explore the predictive value of diagnosis and age on the response to a novel index of cortical plasticity (cTBS) in healthy control individuals and individuals with ASD age 18–64. Results indicate that diagnosis, but not age significantly predicts the response to the cTBS protocol. Understanding the pattern of response to this novel index of plasticity across the lifespan in health and disease is critically important. We have previously suggested that an individual’s cognitive functioning or “Brain Health” is dependent on maintaining an appropriate balance of local and global (network) cortical plasticity. Furthermore, that there are age-related changes in cortical plasticity with a progressive decline in local cortical plasticity and a counterbalancing increase in global (network) cortical plasticity [18]. We propose that an individual’s risk of age-related cognitive decline and in a pathological state, dementia, is dependent on the individual’s starting point and slopes of change in plasticity efficiency over the lifespan. A wealth of in vitro and in vivo evidence demonstrates alterations in neuroplasticity and impaired synaptic function in AD. In fact, alteration in synaptic plasticity is thought to be a very early process in the cascade of pathophysiologic insults that lead up to dementia in AD. Oligomeric amyloid, which antecedes amyloid deposition in the brain, is thought to alter synaptic plasticity years before tau-mediated neuronal dysfunction and cell death. In this context, the hyperplasticity state associated with ASD may offer protection from the initial insult that triggers the neuropathological cascade that leads to symptomatic AD.
Recent advances in informatics open opportunities to collect preliminary empirical data exploring the validity of the hypothesis that having the neuropathology characteristic of ASD may carry with it an evolutionary benefit of relative protection from developing dementia in older adulthood. The Harvard Clinical and Translational Science Center (Harvard Catalyst) has created a secure, anonymous web-based query tool, called Shrine, to allow investigators to determine the aggregate total number of patients at participating hospitals who meet a given set of inclusion and exclusion criteria. After obtaining approval to use Shrine, we did a query of the number of patients over the age of 55 with a diagnosis on the ASD. This resulted in 265 patients. We then did a query of the number of patients over the age of 55 with a diagnosis of AD and related dementia. This resulted in 9045 patients. If patients with ASD had a greater risk of developing AD, one would expect to find over 33 individuals (13% of 265) who would carry both an ASD and AD diagnosis, representing a proportion greater than the general prevalence number (Herbert et al., 2003). We then conducted an additional query, including patients over the age of 55 with both ASD and AD or age-related dementia. The results support our hypothesis indicating that out of the 265 patients over 55 with ASD that there were less than 10 individuals who had a comorbid diagnosis of AD or related dementia (the default minimal amount reported by Shrine). To further confirm that this result was not simply a matter of reduced prevalence due to dual diagnoses, a final query was conducted including patients over the age of 55 with Schizophrenia and AD or age-related dementia. The results indicated that out of the 22,494 patients with Schizophrenia over 55 in the database, 3932 or 17% have a comorbid diagnosis of AD (Figure 4).
Figure 4.
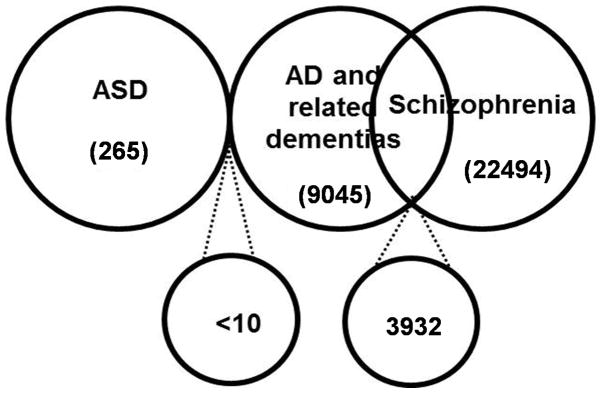
Venn diagram showing the results of the Shrine query indicating relatively less individuals with ASD and AD and relatively more individuals with Schizophrenia and AD as compared to general population prevalence numbers.
The availability of a database, such as Shrine, for researchers and clinicians to query is the first step in obtaining the necessary empirical data in support of hypotheses such as that presented here, however the ability to verify diagnoses is limited by what information is present in the patient’s medical record. This is greatly influenced by a number of factors including primary purpose of medical visit, selective deposit of information on the part of the patient or their caregiver, ability of the clinician to evaluate cognitive state, and validity of diagnoses. Short of bringing these patients in for evaluation, it is difficult to ascertain the concurrent validity of the prevalence numbers obtained through Shrine or a medical record review to the actual prevalence of AD in the ASD population. Ideally, in order to obtain empirical data on the relative risk of a patient with ASD developing AD or an age-related dementia, one would need to establish a large cohort of individuals with ASD and follow them prospectively throughout their lifespan. Studies such as this are underway.
We have previously argued that hyperplasticity in ASD leads to cognitive and behavioral symptoms [9, 19] that are evident in early childhood. However, there is no evidence to suggest that patients with ASD have a shorter life expectancy than the general population. This study indicates that individual’s with ASD maintain a higher capacity for modulation, a putative index of local cortical plasticity, across the lifespan. Therefore, it follows that individuals with ASD should be relatively protected from age-related cognitive decline. Thus, what may be a pathological state during development may be evolutionarily advantageous in older adulthood as it may protect the individual from developing age-related cognitive decline and dementia. At this time, this hypothesis is purely speculative. There is indeed evidence for hyperplasticity in ASD and hypoplasticity in AD using the same indices (cTBS) [11]. However, as ASD is highly heterogeneous both from a behavioral and physiological standpoint, and as there are multiple substrates that contribute to the multiple forms of plasticity at various times across the lifespan, it is unclear whether the mechanisms that lead to the observed hyperplasticity in ASD are the same that are deficient in AD or the extent to which these abnormalities in plasticity are directly related to the behavioral phenotype of these disorders. That being said, at a time when the world’s population is aging and the incidence of age-related cognitive decline is growing, we provide preliminary data to suggest that individuals with ASD may represent a group of individuals who may be protected.
In conclusion, we present herein a novel hypothesis as well as supporting experimental and epidemiologic data that patients with ASD show hyperplasticity across adulthood and that this may provide protection for this population against development of AD and age-related cognitive decline. Further studies are currently underway to confirm the relationship between hyperplasticity and the observed reduced prevalence of AD and age-related cognitive decline in these individuals.
Acknowledgments
Work on the project is supported by grants from the National Institutes of Health and National Institute of Mental Health (1R01MH100186), and Harvard Catalyst | The Harvard Clinical and Translational Science Center (NCRR and the NCATS NIH 8KL2TR000168-05) Dr. Oberman is further supported by grants from the Harvard Clinical and Translational Science Center (8UL1TR000170-05), the Epilepsy Research Foundation, the Simons Foundation and the Nancy Lurie Marks Family Foundation. Dr. Pascual-Leone is further supported by grants from the National Institutes of Health (R01HD069776, R01NS073601, R21 MH099196, R21 NS082870, R21 NS085491, R21 HD07616, UL1 RR025758), Michael J. Fox Foundation and Sidney R. Baer Foundation.
Work on this study was supported by grants from the National Center for Research Resources: Harvard-Thorndike Clinical Research Center at BIDMC (NCRR MO1 RR01032) and Harvard Clinical and Translational Science Center (UL1 RR025758); NIH grant K24 RR018875 and grants from Autism Speaks, the Sidney Baer Foundation, and the Nancy Lurie Marks Family Foundation to A.P.-L. L. Oberman was supported by NIH fellowship F32MH080493 and 1KL2RR025757-01. We would like to thank Elissa Wilker for her help with the statistical analysis. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the Sidney Baer Foundation, Nancy Lurie Marks Family Foundation, National Center for Research Resources or the National Institutes of Health.
Footnotes
Conflict of Interest Statement:
Dr. Pascual-Leone serves on the scientific advisory boards for Nexstim, Neuronix, Starlab Neuroscience, Neuroelectrics, and Neosync; and is listed as an inventor on several issued and pending patents on the real-time integration of transcranial magnetic stimulation (TMS) with electroencephalography (EEG) and magnetic resonance imaging (MRI).
The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centers, the National Institutes of Health, the Epilepsy Research Foundation, the Simons Foundation, the Nancy Lurie Marks Family Foundation, the Michael J. Fox Foundation or the Sidney R. Baer Jr. Foundation.
References
- 1.Baio J. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years — Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2010. MMWR Surveillance Summary. 2014;63(SS02):1–21. [PubMed] [Google Scholar]
- 2.Folstein SE, Rosen-Sheidley B. Genetics of autism: complex aetiology for a heterogeneous disorder. Nat Rev Genet. 2001;2(12):943–55. doi: 10.1038/35103559. [DOI] [PubMed] [Google Scholar]
- 3.Veenstra-VanderWeele J, Cook EH., Jr Molecular genetics of autism spectrum disorder. Mol Psychiatry. 2004;9(9):819–32. doi: 10.1038/sj.mp.4001505. [DOI] [PubMed] [Google Scholar]
- 4.APA. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington DC: American Psychiatric Association; 1994. (DSM-IV) [Google Scholar]
- 5.Walsh CA, Morrow EM, Rubenstein JL. Autism and brain development. Cell. 2008;135(3):396–400. doi: 10.1016/j.cell.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Markram H, Rinaldi T, Markram K. The intense world syndrome--an alternative hypothesis for autism. Front Neurosci. 2007;1(1):77–96. doi: 10.3389/neuro.01.1.1.006.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dolen G, Bear MF. Fragile x syndrome and autism: from disease model to therapeutic targets. J Neurodev Disord. 2009;1(2):133–40. doi: 10.1007/s11689-009-9015-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2(5):255–67. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oberman LM, Rotenberg A, Pascual-Leone A, Tracy BHJ, Sathian K, editors. Plasticity of Cognition in Neurologic Disorders. Oxford University Press; New York: Aberrant brain plasticity in autism spectrum disorders. in press. [Google Scholar]
- 10.Pascual-Leone A, et al. The plastic human brain cortex. Annu Rev Neurosci. 2005;28:377–401. doi: 10.1146/annurev.neuro.27.070203.144216. [DOI] [PubMed] [Google Scholar]
- 11.Pascual-Leone A, et al. Characterizing brain cortical plasticity and network dynamics across the age-span in health and disease with TMS-EEG and TMS-fMRI. Brain Topogr. 2011;24(3–4):302–15. doi: 10.1007/s10548-011-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freitas C, et al. Changes in cortical plasticity across the lifespan. Front Aging Neurosci. 2011;3:5. doi: 10.3389/fnagi.2011.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freitas C, Mondragon-Llorca H, Pascual-Leone A. Noninvasive brain stimulation in Alzheimer’s disease: systematic review and perspectives for the future. Exp Gerontol. 2011;46(8):611–27. doi: 10.1016/j.exger.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koch G, et al. Impaired LTP- but not LTD-like cortical plasticity in Alzheimer’s disease patients. J Alzheimers Dis. 2012;31(3):593–9. doi: 10.3233/JAD-2012-120532. [DOI] [PubMed] [Google Scholar]
- 15.Oberman L, et al. Abnormal modulation of corticospinal excitability in adults with Asperger’s syndrome. Eur J Neurosci. 2012;36(6):2782–8. doi: 10.1111/j.1460-9568.2012.08172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rossi S, et al. Safety, ethical considerations, and application guidelines for the use of transcranial magnetic stimulation in clinical practice and research. Clin Neurophysiol. 2009;120(12):2008–39. doi: 10.1016/j.clinph.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borghetti D, et al. Transcranial magnetic stimulation mapping: a model based on spline interpolation. Brain Res Bull. 2008;77(2–3):143–8. doi: 10.1016/j.brainresbull.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Freitas C, Farzan F, Pascual-Leone A. Assessing brain plasticity across the lifespan with transcranial magnetic stimulation: why, how, and what is the ultimate goal? Front Neurosci. 2013;7:42. doi: 10.3389/fnins.2013.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oberman LaP-LA. Cortical plasticity: A proposed mechanism by which genomic factors lead to the behavioral and neurological phenotype of autism spectrum and psychotic spectrum disorders. Behavioral and Brain Sciences. 2008;31:241–320. [Google Scholar]