

Abstract
The mechanisms that predispose to hypertension, coronary artery disease (CAD), and type 2 diabetes (T2D) in individuals of normal weight are poorly understood. In contrast, in monogenic primary lipodystrophy—a reduction in subcutaneous adipose tissue—it is clear that it is adipose dysfunction that causes severe insulin resistance (IR), hypertension, CAD, and T2D. We aimed to test the hypothesis that common alleles associated with IR also influence the wider clinical and biochemical profile of monogenic IR. We selected 19 common genetic variants associated with fasting insulin–based measures of IR. We used hierarchical clustering and results from genome-wide association studies of eight nondisease outcomes of monogenic IR to group these variants. We analyzed genetic risk scores against disease outcomes, including 12,171 T2D cases, 40,365 CAD cases, and 69,828 individuals with blood pressure measurements. Hierarchical clustering identified 11 variants associated with a metabolic profile consistent with a common, subtle form of lipodystrophy. A genetic risk score consisting of these 11 IR risk alleles was associated with higher triglycerides (β = 0.018; P = 4 × 10−29), lower HDL cholesterol (β = −0.020; P = 7 × 10−37), greater hepatic steatosis (β = 0.021; P = 3 × 10−4), higher alanine transaminase (β = 0.002; P = 3 × 10−5), lower sex-hormone-binding globulin (β = −0.010; P = 9 × 10−13), and lower adiponectin (β = −0.015; P = 2 × 10−26). The same risk alleles were associated with lower BMI (per-allele β = −0.008; P = 7 × 10−8) and increased visceral-to-subcutaneous adipose tissue ratio (β = −0.015; P = 6 × 10−7). Individuals carrying ≥17 fasting insulin–raising alleles (5.5% population) were slimmer (0.30 kg/m2) but at increased risk of T2D (odds ratio [OR] 1.46; per-allele P = 5 × 10−13), CAD (OR 1.12; per-allele P = 1 × 10−5), and increased blood pressure (systolic and diastolic blood pressure of 1.21 mmHg [per-allele P = 2 × 10−5] and 0.67 mmHg [per-allele P = 2 × 10−4], respectively) compared with individuals carrying ≤9 risk alleles (5.5% population). Our results provide genetic evidence for a link between the three diseases of the “metabolic syndrome” and point to reduced subcutaneous adiposity as a central mechanism.
Introduction
Some individuals are at increased risk of metabolic diseases, including hypertension, coronary artery disease (CAD), and type 2 diabetes (T2D) despite a normal BMI. These individuals are often referred to as “metabolically obese, normal weight” (1,2). The mechanisms that cause an adverse metabolic phenotype in individuals of normal weight are poorly understood. One possible mediator is insulin resistance (IR), which was proposed 25 years ago as a potential link among hypertension, CAD, and T2D and which led to the idea of a “metabolic syndrome” (3).
Three broad categories of unusually severe IR exist caused by mutations in single genes. The mechanisms underlying these monogenic disorders are better understood (4) compared with the “common” IR for which genome-wide association studies (GWAS) have identified numerous associated alleles. The first category is lipodystrophy, a partial or complete lack of subcutaneous fat, most often caused by mutations in genes involved in fat cell differentiation or function (e.g., PPARG [5]) or, rarely, by more distal insulin signaling defects (AKT2 [6]). Lipodystrophy is characterized by varying degrees of adipose tissue deficiency, severe dyslipidemia (high triglycerides, low HDL cholesterol [HDL-C]), severe fatty liver, low adiponectin, low sex-hormone-binding globulin (SHBG), and an increased risk of hypertension, CAD, and T2D (7–9). The uncoupling of indices of adiposity from severe metabolic disease in lipodystrophy is one of the key pieces of evidence for the notion that expandable, metabolically flexible adipose tissue is essential for health (9). The second category includes syndromes (such as those caused by leptin deficiency) in which IR is driven by a primary effect on energy homeostasis, leading to severe obesity and a biochemical profile resembling the “common” metabolic syndrome. These disorders are usually characterized by hyperphagia from an early age and extreme childhood obesity and are most commonly attributable to mutations affecting key components of hypothalamic neurocircuitry (10). The third category includes syndromes caused by loss-of-function mutations in the insulin receptor called “receptoropathies.” These disorders are characterized by extremely high insulin levels but lack dyslipidemia and fatty liver disease and often exhibit normal or raised levels of plasma adiponectin and SHBG (11), quite unlike common IR.
In this study, we tested the hypothesis that common alleles associated with IR influence the metabolic outcomes of monogenic forms of IR, including increased risk of hypertension, CAD, and T2D. We aimed to use the subclinical phenotypes that characterize different forms of monogenic IR to determine whether mechanistically informative subphenotypes of common IR may be identified. Recent GWAS have identified 19 common genetic variants associated with indices of IR based on fasting plasma insulin concentration (12,13). One of these variants is in the FTO gene, and the allele associated with greater risk of IR is associated with higher BMI and an adverse metabolic profile (12). This pattern is closely similar to that of severe obesity. A second common variant, near the IRS1 gene, is associated with IR, lower total body fat content, lower subcutaneous fat amount, dyslipidemia, lower adiponectin, and increased risk of diabetes and CAD (14). This pattern is closely similar to that of “lipodystrophic” IR, but quite unlike the severe obesity or “receptoropathy” patterns.
We show that a cluster of 11 variants among 19 common genetic variants associated with indices of IR are collectively associated with metabolic features similar to those of lipodystrophy, including a lower BMI, higher visceral-to-subcutaneous adipose tissue ratio, and a predisposition to T2D, hypertension, and CAD. This finding provides genetic evidence for an association between reduced subcutaneous adiposity and BMI and increased risk of “obesity-associated” diseases.
Research Design and Methods
Selection of Genetic Variants
We selected the 19 independent single nucleotide polymorphisms associated with fasting insulin as reported in the most recent Meta-Analysis of Glucose and Insulin Consortium (MAGIC) GWAS (12) (Supplementary Table 1). Of these 19 variants, 14 were detected at genome-wide significance before correction for BMI, and 5 only reached significance after correcting for BMI. All were at least nominally associated with uncorrected fasting insulin at P < 1.19 × 10−5. Although not the optimum measure of IR, collectively these variants are associated with gold standard measures of IR (15). For comparison, we selected the 32 variants associated with BMI from the Genetic Investigation of ANthropometric Traits (GIANT) consortium (16) (Supplementary Table 2).
Metabolic Traits
We used summary statistics data from publicly available GWAS (Table 1).
Table 1.
Source of summary statistic data from GWAS of metabolic traits and their changes in monogenic forms of IR
| Trait | Consortia | Maximum N (all or cases vs. controls) | Reference | Monogenic obesity* | Monogenic lipodystrophy* | Monogenic receptoropathy* |
|---|---|---|---|---|---|---|
| Nondisease metabolic traits | ||||||
| SHBG | CHARGE | 21,000 | (22) | – | – | 0/+ |
| HDL-C | GLGC | 99,900 | (17) | – | – | 0 |
| Adiponectin | ADIPOGEN | 29,346 | (21) | – | – | 0/+ |
| BMI | GIANT | 123,865 | (16) | + | – | 0 |
| VATSAT ratio | VATGen | 10,557 | (18) | + | + | 0 |
| CT-measured hepatic steatosis | GOLD | 7,176 | (19) | + | + | 0 |
| ALT | — | 55,474 | (20) | + | + | 0 |
| Triglyceride | GLGC | 96,598 | (17) | + | + | 0 |
| Metabolic disease and disease-related outcomes | ||||||
| T2D | DIAGRAM | 12,171 vs. 56,862 | (26) | + | + | + |
| CAD | CARDIoGRAM | 40,365 vs. 63,714 | (25) | + | + | 0 |
| Systolic blood pressure | ICBP | 69,828 | (24) | + | + | 0 |
| Diastolic blood pressure | ICBP | 69,816 | (24) | + | + | 0 |
| cIMT | CHARGE | 31,210 | (23) | + | + | 0 |
| Carotid plaque | CHARGE | 25,179 | (23) | + | + | 0 |
VATSAT, visceral-to-subcutaneous adipose tissue.
*+ refers to high in condition, – refers to low in condition, and 0 refers to not changed.
Study Design
Selection of Nondisease Metabolic Traits
We selected eight traits known to be nondisease markers of the three different subtypes of monogenic IR (Table 1). These traits were HDL-C (17) and triglyceride levels (17) to represent dyslipidemia; BMI (16) and visceral-to-subcutaneous adipose tissue ratio (18) to represent adiposity; computed tomography (CT)-measured hepatic steatosis (19) and plasma levels of the liver enzyme alanine transaminase (ALT) (20) to represent the spectrum of nonalcoholic fatty liver disease; and circulating levels of the fat cell–derived protein adiponectin (21) and circulating levels of the liver-derived protein SHBG (22).
Selection of Metabolic Disease Outcome
We selected six metabolic diseases or disease-related outcomes known to be increased in some forms of monogenic IR. These outcome traits represent the diseases that the metabolic syndrome tries to predict: carotid intima-media thickness (cIMT) and carotid plaque as surrogates of atherosclerosis (23); diastolic and systolic blood pressure to represent hypertension (24); and CAD (25) and T2D (26). Further details of disease outcomes are available in Supplementary Table 3.
Cluster Analysis of Fasting Insulin–Associated Genetic Variants Using Nondisease Metabolic Traits
We used a hierarchical clustering analysis and the eight nondisease traits to group fasting insulin variants into those likely to predispose to IR through similar mechanisms. This method is used for clustering gene expression profiles to discover coregulated and functionally related genes or to identify subtypes of related samples (27) and is similar to principal components analyses. This approach was recently used to group 37 genetic variants associated with T2D into those with primary effects on glucose sensing, β-cell dysfunction, or IR (28). We used the summary statistics of the 19 fasting insulin variants (12) from the GWAS of the 8 nondisease metabolic traits (Table 1) and aligned all effects to the fasting insulin increasing alleles. We used the “pvclust” package in R, which performs agglomerative hierarchical clustering and bootstrapping, to estimate the stability and significance of the resulting clusters (29). In this method, variants are assigned to clusters based on associations with a similar set of phenotypes. We used Euclidean metrics in the algorithm to calculate pairwise distance between the effect sizes on biomarker levels of the variants as input data. We used “ward” as a cluster method. The P values in this method represent the relative frequency of how often the variants are in the same cluster.
Meta-analysis of Fasting Insulin– and BMI-Associated Genetic Variants Against Nondisease Metabolic Traits and Metabolic Disease Outcomes
We calculated genetic risk scores of variants that clustered together by meta-analyzing summary statistics of genotype-phenotype associations across the variants for a given trait. This method has been described and validated previously (30). We repeated this analysis for the 32 variants known to be associated with BMI. We compared per-allele effects of fasting insulin clusters to each other and to per-allele effects of the BMI variants using Z test. We considered a conservative nominal P ≤ 0.001 as significant, corresponding to a Bonferroni correction of 42 tests (14 traits tested [8 nondisease metabolic traits and 6 metabolic disease outcomes] against 3 clusters).
Sensitivity Analyses
We performed three sets of sensitivity analyses to test whether weighting changes the association of each genetic risk score with nondisease marker and disease outcome traits. We calculated the genetic risk score using summary statistics of genotype-phenotype associations weighted in three different ways: 1) by each variant’s corresponding effect size with the primary trait (fasting insulin) as weight; 2) by each variant’s corresponding effect size with the primary trait (fasting insulin) using the formula used before (21),
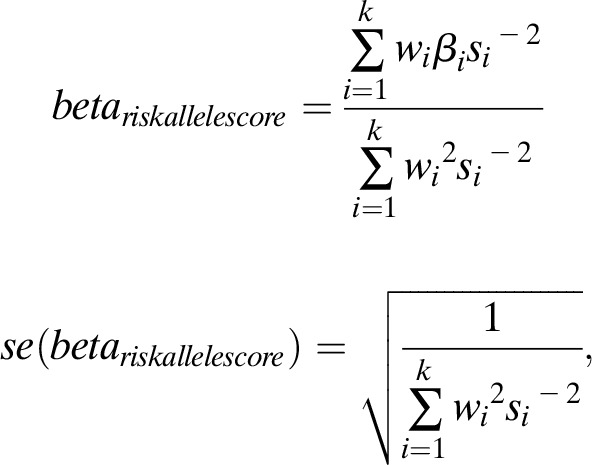 |
where 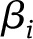 refers to the effect of varianti on the outcome data,
refers to the effect of varianti on the outcome data, 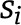 is the associated SE estimate, and
is the associated SE estimate, and 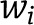 is the effect of varianti on fasting insulin; and 3) by weighting variants and taking into account allele frequencies,
is the effect of varianti on fasting insulin; and 3) by weighting variants and taking into account allele frequencies,
where 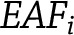 is the effect allele frequency of the varianti and
is the effect allele frequency of the varianti and 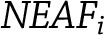 is the frequency of the other allele for varianti.
is the frequency of the other allele for varianti.
Results
A Cluster of 11 Fasting Insulin–Associated Genetic Variants Resembles the Metabolic Profile of Monogenic Lipodystrophic IR
Using the 8 nondisease metabolic traits of monogenic IR and all 19 common fasting insulin associated genetic variants (12), we identified 11 genetic variants that together could be distinguished statistically from the remaining 8 variants (P of the robustness of the branching event = 96%) (Fig. 1). This cluster of 11 common variants included those in or near the genes IRS1, GRB14, ARL15, FAM13A, LYPLAL1, PEPD, PDGFC, RSPO3, PPARG, TET2, and ANKRD55. The 11-variant genetic risk score was associated with all 8 nondisease markers of monogenic IR. As expected, given the traits used to define the cluster, fasting insulin–raising alleles were associated with higher triglycerides (β = 0.018; P = 4 × 10−29), lower HDL-C (β = −0.020; P = 7 × 10−37), greater hepatic steatosis (β = 0.021; P = 3 × 10−4) higher ALT (β = 0.002; P = 3 × 10−5), lower SHBG (β = −0.010; P = 9 × 10−13), and lower adiponectin (β = −0.015; P = 2 × 10−26). In contrast to this adverse metabolic profile, the fasting insulin–raising alleles were associated with lower BMI (per-allele β = −0.008; P = 7 × 10−8) (Table 2). The fasting insulin–raising alleles were also associated with increased visceral-to-subcutaneous adipose tissue ratio (β = −0.015; P = 6 × 10−7) (Table 2). The association with visceral-to-subcutaneous adipose tissue ratio was primarily driven by reduced subcutaneous adipose tissue (−0.014; 2 × 10−6). As an example, these effects meant that the 5.5% of individuals carrying ≥17 fasting insulin–raising alleles were 0.30 kg/m2 of BMI slimmer but had triglyceride levels 0.16 SD units higher and HDL levels 0.18 SD units lower, compared with the 5.5% of individuals carrying ≤9 fasting insulin–raising alleles. Note that the units for each biomarker may be different depending on the source of GWAS data (Table 2).
Figure 1.

Cluster analysis of fasting insulin variants using eight traits known to be nondisease metabolic traits of monogenic IR, including those representing dyslipidemia (HDL and triglyceride), adiposity (BMI and visceral-to-subcutaneous adipose tissue ratio), fatty liver (CT-measured hepatic steatosis and the liver enzyme ALT), and adiponectin and SHBG levels. The dendrogram (A) shows that 11 variants and 5 variants are grouped in two significant clusters (the approximate unbiased values = 96% [P = 0.04] and 98% [P = 0.02], respectively). The heatmap (B) shows this cluster is consistent with monogenic lipodystrophic IR; the stronger the green color, the stronger the effect of the insulin-raising allele with reduced trait levels; the stronger the pink color, the stronger the effect of the insulin-raising allele with higher trait levels. VATSAT, visceral-to-subcutaneous adipose tissue ratio; NAFLD, nonalcoholic fatty liver disease.
Table 2.
Results of genotype risk score analysis of the 11 “lipodystrophy-like” variants
| Category/trait | Unit | Per-allele β | 95% CI | P | N |
|---|---|---|---|---|---|
| Nondisease metabolic traits of monogenic IR | |||||
| SHBG (BMI adjusted) | Natural log | −0.010 | −0.012, −0.008 | 9 × 10−13 | 21,000 |
| HDL-C | SD | −0.020 | −0.024, −0.016 | 7 × 10−37 | 99,900 |
| Adiponectin (BMI adjusted) | log | −0.015 | −0.017, −0.013 | 2 × 10−26 | 29,346 |
| BMI | SD | −0.008 | −0.012, −0.004 | 7 × 10−8 | 123,865 |
| VATSAT ratio | z-score | 0.015 | 0.009, 0.021 | 6 × 10−7 | 10,557 |
| CT-measured hepatic steatosis | SD | 0.021 | 0.009, 0.033 | 3 × 10−4 | 7,176 |
| ALT | log10 | 0.002 | 0.001, 0.003 | 3 × 10−5 | 55,474 |
| Triglyceride | SD | 0.018 | 0.014, 0.022 | 4 × 10−29 | 96,598 |
| Metabolic disease and disease-related outcomes | |||||
| T2D | OR | 1.043 | 1.031, 1.055 | 5 × 10−13 | 12,171 vs. 56,862 |
| CAD | OR | 1.013 | 1.007, 1.019 | 1 × 10−5 | 40,365 vs. 63,714 |
| Systolic blood pressure (BMI adjusted) | mmHg | 0.135 | 0.072, 0.198 | 2 × 10−5 | 69,828 |
| Diastolic blood pressure (BMI adjusted) | mmHg | 0.075 | 0.036, 0.114 | 2 × 10−4 | 69,816 |
| cIMT | log | 0.000 | −0.002, 0.002 | 0.70 | 31,210 |
| Carotid plaques | OR | 1.005 | 0.991, 1.019 | 0.50 | 25,179 |
The 8 nondisease metabolic traits were used to select the 11 variants, so associations are not independent of the clustering process. The metabolic disease outcomes were not used in the clustering process and so represent independent tests. VATSAT, visceral-to-subcutaneous adipose tissue. P values ≤ 0.001 are in bold.
A second cluster of five genetic variants (P of the robustness of the branching event = 98%) included those in or near the genes YSK4, UKRF1BP1, TCF7L2, IGF1, and HIP1 (Fig. 1) but was not associated with any of the nondisease markers of monogenic IR (all P values > 0.001) except higher BMI (β = 0.012; P = 3 × 10−6) (Supplementary Table 4) and so was not studied further. Variants in or near FTO, GCKR, and PPP1R3B did not cluster with other variants, most likely because of their primary effects on BMI, triglycerides, and HDL-C, respectively (Fig. 1; Supplementary Table 4). The variant in FTO provides a proof-of-principle example of how a BMI-associated variant can be separated from variants associated with fasting insulin through mechanisms other than higher BMI. The GCKR variant shows how the use of multiple metabolic phenotypes can be used to separate out different mechanisms—we know this variant is likely to operate through a particular mechanism linking glucose to lipid metabolism (31) in the liver and it was individually associated with multiple liver-based phenotypes (Supplementary Table 4).
A Cluster of 11 Fasting Insulin Associated Variants Resembling Lipodystrophy Is Associated With Metabolic Disease Outcomes
The genetic risk score consisting of the 11 fasting insulin variants was associated with 4 of the 6 metabolic disease outcomes, in directions consistent with the lipodystrophy-like phenotype. The 11-variant genetic risk score was associated with a higher risk of T2D (per-allele odds ratio [OR] = 1.043; 95% CIs 1.031, 1.055; P = 5 × 10−13) and CAD (per-allele OR = 1.013; 95% CIs 1.007, 1.019; P = 1 × 10−5), and higher systolic (β = 0.135; 95% CIs 0.072, 0.198; P = 2 × 10−5) and diastolic blood pressure (β = 0.075; 95% CIs 0.036, 0.114; P = 2 × 10−4) but was not associated with cIMT or risk of carotid plaques (P values > 0.001) (Fig. 2; Table 2). As an example, these effects meant that individuals carrying ≥17 fasting insulin–raising alleles (5.5% of the population) were 0.30 kg/m2 slimmer but had an estimated OR of T2D and coronary heart disease of 1.46 and 1.12, respectively, and an estimated increase in systolic and diastolic blood pressure of 1.21 and 0.67 mmHg, respectively, compared with individuals carrying ≤9 fasting insulin–raising alleles (5.5% of the population) (Supplementary Fig. 1A and C). Sensitivity analyses using three different methods to weight individual variants provided very similar results (Supplementary Table 5), and exclusion of the previously reported variant near IRS1 did not appreciably change the results (Supplementary Table 6).
Figure 2.

Forest plots of the effect of the 11 “lipodystrophy-like” variants on six metabolic disease outcomes. The x-axis is the effect size per fasting insulin–increasing alleles on each trait. The dashed line is the null effect. For cIMT, actual effects ranged from −0.003 to 0.003 but here are shown rounded to two decimal places. FE, fixed effect.
Comparing the Cluster of 11 Fasting Insulin Variants Resembling Lipodystrophy to the 32 Known BMI Variants
We next compared the per-allele effects of the cluster of fasting insulin–associated variants with the per-allele effects of the known 32 BMI variants. They were both associated with nondisease metabolic traits, with two notable differences. 1) The cluster of 11 fasting insulin variants was associated with higher visceral-to-subcutaneous adipose tissue ratio, whereas the 32-variant BMI group was associated with lower visceral-to-subcutaneous adipose tissue ratio (Supplementary Table 4). 2) The 32-variant BMI cluster was not associated with CT-measured hepatic steatosis (Supplementary Table 4). Compared with the 32-BMI-variant risk score, the 11 variant fasting insulin cluster was more strongly associated with risk of T2D (BMI per-allele OR = 1.021 [1.016–1.028], P = 1 × 10−12; the 11-variant cluster per-allele OR = 1.043 [1.031–1.055], P = 5 × 10−13; P of difference = 0.002) but did not have detectably different per-allele associations with CAD (Supplementary Fig. 1B and D; Supplementary Table 4). As with the fasting insulin cluster, the 32-BMI-variant score was not associated with cIMT or carotid plaque.
Discussion
We have shown that common alleles associated with IR (as measured by fasting insulin) are associated with metabolic outcomes of monogenic forms of IR. Most notably, a cluster of 11 common genetic variants show a pattern of metabolic trait associations consistent with a common, subtle “lipodystrophy-like” phenotype, conferring an increased risk of hypertension, T2D, and CAD despite a lower BMI. Our findings are consistent with “adipose expandability” hypotheses (32), which propose that the expandability of adipose tissue is limited and that it is the exceeding of this intrinsic storage capacity, which is partly genetically determined, which contributes to common forms of IR. More broadly, our results suggest that there is a partly genetic basis to the widely reported “metabolically obese, normal-weight” phenotype (1,2).
Our study has a number of implications. First, our results provide evidence that the 11 variants associated with a lipodystrophy-like metabolic phenotype exert their effect through a primary role in adipose tissue. Consistent with this notion, the genetic loci involved include PPARG, which is the most important transcription factor in the control of adipogenesis (33,34) and which harbors dominant negative mutations in a Mendelian subtype of partial lipodystrophy featuring reduced body fat and severely dyslipidemic IR (35). Thus, although our findings do not prove causality in the observed associations, they indicate that future in vitro and in vivo functional studies could start with adipose tissue–based hypotheses and the genes located near the 11 variants highlighted by our cluster analysis.
Second, we have shown that using phenotypic features of monogenic diseases may be an informative method to help understand likely mechanisms of more common, less penetrant alleles. We show that common fasting insulin–associated variants identified by GWAS may be clustered into discrete subgroups using a panel of nondisease markers that discriminate different forms of monogenic severe IR (9). These markers included circulating factors such as lipids, adiponectin, and SHBG as well as adiposity. The approach is analogous to that recently used to cluster T2D variants into different groups according to subclinical phenotypes (28).
Third, our results raise the possibility that individuals carrying large numbers of fasting insulin–raising alleles at these 11 loci represent human “in vivo” models of adipose tissue dysfunction. Although individually the common alleles have very subtle effects, ∼5.5% of people in the general population will have an ∼1.46 increased odds of T2D and 1.12 increased odds of CAD compared with people from the other end of the distribution of these alleles, but these people will have lower BMIs. More in depth studies of the fat distribution and adipose tissue morphology and function of these individuals may lead to further understanding of the BMI-independent mechanisms of metabolic disease.
Finally, if more lipodystrophy-like variants are identified through ongoing genetic studies, testing such variants in combination may have clinical utility in identifying individuals at high genetic risk of “adipose failure.” These individuals have previously been termed the metabolically obese, normal weight (1,2). In such people, it may be appropriate to use lower BMI-based thresholds for access to adipose offloading therapies, including oral weight loss medications and bariatric surgery, as they have the potential to develop metabolic disease at a lower BMI.
Our study has some notable strengths and some limitations. The main strength is that we used a genetic approach to dissect the associations between multiple metabolic traits. While we cannot be certain of the primary trait involved, genetic variants are far less susceptible to confounding and bias than most nongenetic measures, and so our results provide strong support for a causal link between the traits tested. The second main strength is that we used a wider panel of phenotypic features than IR alone as a basis for subgrouping fasting insulin–associated variants while, critically, constraining this panel to those markers that have shown utility in discriminating different forms of monogenic IR.
There are some important limitations to our results. Firstly and most importantly, the associations we observed, while statistically very robust, are very subtle in terms of effects on metabolic traits. More than 50,000 individuals were required to identify the variants associated with fasting insulin, and so they explain only a small proportion of the variation in IR and the other metabolic traits we have examined. Hence our results do not provide evidence that the “polygenic lipodystrophy-like” phenotype is the primary mechanism of IR and its link to metabolic disease but instead establish the principle that reduced adipose expandability is one mechanism that links these traits in the general population. Further genetic discoveries will help us understand the relative role of this mechanism. A second limitation was that our hierarchical clustering approach was based on eight nondisease markers that are correlated with each other, and many of the same studies and individuals contributed to multiple phenotypes. Further studies with individual level data will be needed to able to account for these correlations. However, using two correlated, but not perfectly correlated measures, such as adiponectin and SHBG, will increase the resolution of the clustering approach, as recently demonstrated in a similar approach to cluster T2D variants by correlated glycemic traits. A third limitation is that the fasting insulin–associated variants were ascertained from truncated distributions of glycemic traits because the MAGIC study excluded individuals with fasting glucose above 7.0 mmol/L to limit the possibility of confounding effects from diabetes disease processes and treatments. This ascertainment issue may explain why the diabetes risk allele at TCF7L2 and possibly other alleles may be associated with apparently greater insulin sensitivity—carriers of T2D risk alleles will need to be subtly more insulin sensitive to remain nondiabetic, for example. It is therefore reassuring that TCF7L2 does not cluster with the 11 “lipodystrophy-like” variants. A fourth main limitation is that some of the phenotypes used were not gold standard measures. We used genetic variants associated with fasting insulin, which is not the best measure of IR, but we note that the genetic risk score we used is strongly associated with more direct measures of IR in the accompanying article (15). We also used cIMT and carotid plaque as surrogates for atherosclerosis, and it is well-known that these traits are suboptimal measures of atherosclerosis (23). The lack of association between the cluster of 11 fasting insulin variants and cIMT or carotid plaque could be due to reduced statistical power due to sample size or the intraindividual variation and measurement error involved in these phenotypes. Alternatively, the lack of association could be because the fasting insulin–associated variants predispose to CAD through pathways other than those captured by cIMT and carotid plaque measures.
In summary, the group of genetic variants associated with a “lipodystrophy-like” phenotype provides evidence that subtle genetically influenced higher visceral-to-subcutaneous adipose tissue ratio, fasting insulin, and dyslipidemia in combination can increase the risk of hypertension, CAD, and T2D in the absence of increased BMI. Our results provide genetic evidence for a link between the three diseases of the “metabolic syndrome.” Our results may help elucidate the mechanistic pathways of how common genetic loci are linked to IR and the mechanisms behind how some individuals can remain relatively healthy despite obesity while others are susceptible to heart disease and diabetes despite relative leanness.
Our results highlight the potential role of adipose tissue dysfunction as one of the underlying mechanisms for IR, hypertension, T2D, and CAD.
Supplementary Material
Article Information
Acknowledgments. The authors are grateful to all GWAS consortia who provided the lookups, including CHARGE, GLGC, ADIPOGEN, GIANT, VATGen, GOLD, GLGC, DIAGRAM, CARDIoGRAM, and ICBP.
Funding. The authors thank the European Research Council (ERS: 323195), Wellcome Trust, and University of Exeter Medical School. P.B.M. acknowledges the National Institute for Health Research Barts Cardiovascular Biomedical Research Unit for supporting this work.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. H.Y. conceived and designed the study, designed the statistical analysis, analyzed the data, and wrote the first draft of the manuscript. R.A.S. provided data from published GWAS, commented on the manuscript, and agreed with manuscript results and conclusions. C.C.W., W.Z., G.B.E., J.C.B., C.O., N.F., and Z.D. provided data from published GWAS and agreed with manuscript results and conclusions. E.S. provided data from published GWAS, commented on the manuscript, and agreed with manuscript results and conclusions. P.B.M. provided data from published GWAS. P.B.M., C.S.F., M.-F.H., and J.B.R. commented on the manuscript and agreed with manuscript results and conclusions. M.W., I.B.B., J.W.K., L.Y.-A., J.R.B.P, J.C.C., J.S.K., and C.L. agreed with manuscript results and conclusions. R.K.S. wrote the first draft of the manuscript and commented on the manuscript. T.M.F. conceived and designed the study and wrote the first draft of the manuscript. T.M.F. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented as a poster at the 74th Scientific Sessions of the American Diabetes Association, San Francisco, CA, 13–17 June 2014.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-0318/-/DC1.
See accompanying article, p. 4004.
References
- 1.Ruderman N, Chisholm D, Pi-Sunyer X, Schneider S. The metabolically obese, normal-weight individual revisited. Diabetes 1998;47:699–713 [DOI] [PubMed] [Google Scholar]
- 2.Ruderman NB, Schneider SH, Berchtold P. The “metabolically-obese,” normal-weight individual. Am J Clin Nutr 1981;34:1617–1621 [DOI] [PubMed] [Google Scholar]
- 3.Reaven GM. Banting Lecture 1988. Role of insulin resistance in human disease. 1988. Nutrition 1997;13:65; discussion 64, 66 [DOI] [PubMed]
- 4.Semple RK, Savage DB, Cochran EK, Gorden P, O’Rahilly S. Genetic syndromes of severe insulin resistance. Endocr Rev 2011;32:498–514 [DOI] [PubMed] [Google Scholar]
- 5.Semple RK, Chatterjee VK, O’Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest 2006;116:581–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.George S, Rochford JJ, Wolfrum C, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science 2004;304:1325–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semple RK, Cochran EK, Soos MA, et al. Plasma adiponectin as a marker of insulin receptor dysfunction: clinical utility in severe insulin resistance. Diabetes Care 2008;31:977–979 [DOI] [PubMed] [Google Scholar]
- 8.Semple RK, Halberg NH, Burling K, et al. Paradoxical elevation of high-molecular weight adiponectin in acquired extreme insulin resistance due to insulin receptor antibodies. Diabetes 2007;56:1712–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stears A, O’Rahilly S, Semple RK, Savage DB. Metabolic insights from extreme human insulin resistance phenotypes. Best Pract Res Clin Endocrinol Metab 2012;26:145–157 [DOI] [PubMed] [Google Scholar]
- 10.Bochukova EG, Huang N, Keogh J, et al. Large, rare chromosomal deletions associated with severe early-onset obesity. Nature 2010;463:666–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Semple RK, Soos MA, Luan J, et al. Elevated plasma adiponectin in humans with genetically defective insulin receptors. J Clin Endocrinol Metab 2006;91:3219–3223 [DOI] [PubMed] [Google Scholar]
- 12.Scott RA, Lagou V, Welch RP, et al. DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium . Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet 2012;44:991–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manning AK, Hivert MF, Scott RA, et al. DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium. Multiple Tissue Human Expression Resource (MUTHER) Consortium . A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 2012;44:659–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kilpeläinen TO, Zillikens MC, Stančákova A, et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat Genet 2011;43:753–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scott RA, Fall T, Pasko D, et al. Common genetic variants highlight the role of insulin resistance and body fat distribution in type 2 diabetes, independent of obesity. Diabetes 2014;63:4378–4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Speliotes EK, Willer CJ, Berndt SI, et al. MAGIC. Procardis Consortium . Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010;42:937–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010;466:707–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fox CS, Liu Y, White CC, et al. GIANT Consortium. MAGIC Consortium. GLGC Consortium . Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet 2012;8:e1002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Speliotes EK, Yerges-Armstrong LM, Wu J, et al. NASH CRN. GIANT Consortium. MAGIC Investigators. GOLD Consortium . Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 2011;7:e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chambers JC, Zhang W, Sehmi J, et al. Alcohol Genome-wide Association (AlcGen) Consortium. Diabetes Genetics Replication and Meta-analyses (DIAGRAM+) Study. Genetic Investigation of Anthropometric Traits (GIANT) Consortium. Global Lipids Genetics Consortium. Genetics of Liver Disease (GOLD) Consortium. International Consortium for Blood Pressure (ICBP-GWAS) Meta-analyses of Glucose and Insulin-Related Traits Consortium (MAGIC) . Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet 2011;43:1131–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dastani Z, Hivert MF, Timpson N, et al. DIAGRAM+ Consortium. MAGIC Consortium. GLGC Investigators. MuTHER Consortium. DIAGRAM Consortium. GIANT Consortium. Global B Pgen Consortium. Procardis Consortium. MAGIC investigators. GLGC Consortium . Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet 2012;8:e1002607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coviello AD, Haring R, Wellons M, et al. A genome-wide association meta-analysis of circulating sex hormone-binding globulin reveals multiple Loci implicated in sex steroid hormone regulation. PLoS Genet 2012;8:e1002805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bis JC, Kavousi M, Franceschini N, et al. CARDIoGRAM Consortium . Meta-analysis of genome-wide association studies from the CHARGE consortium identifies common variants associated with carotid intima media thickness and plaque. Nat Genet 2011;43:940–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehret GB, Munroe PB, Rice KM, et al. International Consortium for Blood Pressure Genome-Wide Association Studies. CARDIoGRAM consortium. CKDGen Consortium. KidneyGen Consortium. EchoGen consortium. CHARGE-HF consortium . Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011;478:103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deloukas P, Kanoni S, Willenborg C, et al. CARDIoGRAMplusC4D Consortium. DIAGRAM Consortium. CARDIOGENICS Consortium. MuTHER Consortium. Wellcome Trust Case Control Consortium . Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 2013;45:25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris AP, Voight BF, Teslovich TM, et al. Wellcome Trust Case Control Consortium. Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) Investigators. Genetic Investigation of ANthropometric Traits (GIANT) Consortium. Asian Genetic Epidemiology Network–Type 2 Diabetes (AGEN-T2D) Consortium. South Asian Type 2 Diabetes (SAT2D) Consortium. DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium . Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012;44:981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 1998;95:14863–14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dimas AS, Lagou V, Barker A, et al.; MAGIC Investigators. Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes 2014;63:2158–2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006;22:1540–1542 [DOI] [PubMed] [Google Scholar]
- 30.Yaghootkar H, Lamina C, Scott RA, et al. Mendelian randomization studies do not support a causal role for reduced circulating adiponectin levels in insulin resistance and type 2 diabetes. Diabetes 2013;62:3589–3598 [DOI] [PMC free article] [PubMed]
- 31.Rees MG, Raimondo A, Wang J, et al. Inheritance of rare functional GCKR variants and their contribution to triglyceride levels in families. Hum Mol Genet. 30 May 2014 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome--an allostatic perspective. Biochim Biophys Acta 2010;1801:338–349 [DOI] [PubMed]
- 33.Barak Y, Nelson MC, Ong ES, et al. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell 1999;4:585–595 [DOI] [PubMed] [Google Scholar]
- 34.Medina-Gomez G, Gray SL, Yetukuri L, et al. PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet 2007;3:e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barroso I, Gurnell M, Crowley VE, et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999;402:880–883 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.