

Abstract
Methylmercury is a potent neurotoxin that is produced by anaerobic microorganisms from inorganic mercury by a recently discovered pathway. A two-gene cluster, consisting of hgcA and hgcB, encodes two of the proteins essential for this activity. hgcA encodes a corrinoid protein with a strictly conserved cysteine proposed to be the ligand for cobalt in the corrinoid cofactor, whereas hgcB encodes a ferredoxin-like protein thought to be an electron donor to HgcA. Deletion of either gene eliminates mercury methylation by the methylator Desulfovibrio desulfuricans ND132. Here, site-directed mutants of HgcA and HgcB were constructed to determine amino acid residues essential for mercury methylation. Mutations of the strictly conserved residue Cys93 in HgcA, the proposed ligand for the corrinoid cobalt, to Ala or Thr completely abolished the methylation capacity, but a His substitution produced measurable methylmercury. Mutations of conserved amino acids near Cys93 had various impacts on the methylation capacity but showed that the structure of the putative “cap helix” region harboring Cys93 is crucial for methylation function. In the ferredoxin-like protein HgcB, only one of two conserved cysteines found at the C terminus was necessary for methylation, but either cysteine sufficed. An additional, strictly conserved cysteine, Cys73, was also determined to be essential for methylation. This study supports the previously predicted importance of Cys93 in HgcA for methylation of mercury and reveals additional residues in HgcA and HgcB that facilitate the production of this neurotoxin.
INTRODUCTION
Methylmercury (MeHg), a neurotoxin present in the environment, is a significant risk to human health in many regions of the world (1). Sources of the mercury (Hg) substrate from which MeHg is derived are numerous and include both natural and anthropogenic sources (2, 3). Currently, only anaerobic microbes are known to produce MeHg (4), predominantly in sediments of aquatic environments. Once MeHg is produced, it accumulates in the aquatic food chain, becoming concentrated in top predators (5). Human exposure to MeHg results from eating those predators, for example, in marine food webs, shark, swordfish, albacore tuna, and eel. Once in the body, MeHg passes through the intestinal epithelium, usually complexed with thiol groups in proteins (6). Eventually, MeHg buildup in humans can lead to neuropathies in adults and developmental disorders in children exposed in utero (2).
Anaerobic Deltaproteobacteria are thought to be the major contributors of MeHg found in the environment (7, 8), although potentially significant contributions by methanogens and fermenting bacteria have recently been discovered (9–11). In 2013, we showed that the proteins encoded by two genes in two bacteria of the Deltaproteobacteria phylum, Desulfovibrio desulfuricans ND132 and Geobacter sulfurreducens PCA, are essential for the conversion of Hg(II) to MeHg (12). That study was stimulated by the work of Choi and colleagues (13–15) implicating a 40-kDa corrinoid protein in the methylation of Hg in cell extracts of Desulfovibrio desulfuricans LS, a strain that was subsequently lost. Hg methylation was thought to involve the reductive acetyl coenzyme A (acetyl-CoA) pathway and perhaps the corrinoid protein therein (14, 16). With this information as a starting point, the amino acid sequence of the corrinoid iron-sulfur protein (CFeSP) from the reductive acetyl-CoA pathway of Carboxydothermus hydrogenoformans Z-2901 (17, 18) was used to search for related proteins encoded in the genomes of known Hg methylators. By comparing sequences of Hg-methylating and nonmethylating bacteria, a gene that is conserved in all methylators and absent in nonmethylators was identified and appears to encode a corrinoid protein of the approximate size of that reported by Choi et al. (14). This gene was subsequently confirmed by mutational studies of ND132 and PCA to be essential for Hg methylation and was designated hgcA (12).
Immediately downstream of hgcA, 14 bp in the ND132 genome and 12 bp in the PCA genome, is the start codon of hgcB, a predicted gene encoding a ferredoxin-like protein that was also shown to be essential for Hg methylation (11). It was hypothesized that the two genes, essential for methylation and occurring in close proximity in the genomes of Hg methylators, produce proteins that function together in the methylation reaction. Furthermore, any organism whose genome contains homologs of hgcA and hgcB was predicted to be capable of Hg methylation. Microorganisms from diverse origins and environments containing homologs of both hgcA and hgcB have been identified (11, 19–21). All cultured microbes assayed thus far that possess hgcA and hgcB methylate Hg, whereas all microbes assayed that lack homologs of hgcA and hgcB are unable to methylate Hg (11).
Although the genes essential for Hg methylation have been identified, confirmation of the predictions of critical amino acid residues from bioinformatics analysis and structural modeling has not been obtained. Currently, there is no atomic-resolution structure of HgcA; however, a model of HgcA was generated from the X-ray crystal structure of the CFeSP from C. hydrogenoformans. CFeSP functions as a complex comprising two subunits, CfsA and CfsB (18, 22). The large subunit, CfsA, binds a corrinoid cofactor but has several features not shared with HgcA. CfsA has three domains, an N-terminal domain binding a single [4Fe-4S] center, a central (βα)8 barrel domain, and the domain with homology to HgcA, its C terminus that adopts a Rossmann fold binding the corrinoid in a “base-off” configuration (23). The C-terminal domain of CfsA also contains an alpha helix, the so called “cap helix,” that interacts with the lower face of the cofactor (17). CfsB, having no ortholog in ND132, folds as a (βα)8 barrel and interacts with the upper axial face (the face that receives and donates the methyl group) of the corrinoid (24). The interaction of the CfsA and CfsB subunits of CFeSP on either side of the corrinoid has been proposed to stabilize the Co(I) state of the cofactor (17).
Homology between CfsA and HgcA is limited to the C-terminal corrinoid-binding domain of CfsA and the N-terminal region of HgcA (∼166 residues). Like CfsA, HgcA is predicted to bind its cofactor in a base-off state where the cap helix is oriented to interact with the corrinoid lower face. The HgcA C-terminal domain contains four or five predicted transmembrane helices with no homology to CFeSP or any other protein. For the catalytic function of CFeSP, the cob(I)amide cofactor receives a methyl group as a carbocation from methyltetrahydrofolate via the methyltetrahydrofolate:CFeSP methyltransferase (MeTr) to produce methylcob(III)amide (25, 26). This route has been suggested for HgcA (12); however, subsequent methyl group transfer as a carbanion to Hg(II) would result in a Co(III) intermediate (25, 27). Regeneration of the Co(I) state would be required for continued catalytic function. For many corrinoid proteins, accidental oxidation of the low-potential Co(I) to Co(II) causes inactivation of the enzyme, requiring a dedicated activase capable of reducing the Co(II) to Co(I) (28, 29). We hypothesize that the two-electron reduction that we predict to be needed to reactivate HgcA involves the ferredoxin-like protein HgcB.
Of possibly critical importance for Hg methylation is the strictly conserved cysteine (Cys93 in ND132) (Fig. 1A) in the soluble portion of HgcA, predicted to be located in the cap helix and to be the lower axial ligand to Co (Fig. 1B). The proposed “Cys-on” Co coordination in HgcA is unique among corrinoid proteins, as most CFeSPs have threonine at the analogous position (Thr374 in CFeSP from C. hydrogenoformans Z-2901). By using density functional theory calculations on model complexes, it has been shown that the proposed transfer of a methyl carbanion from HgcA to Hg(II) substrates is facilitated by Cys-on coordination to Co(III) (30).
FIG 1.
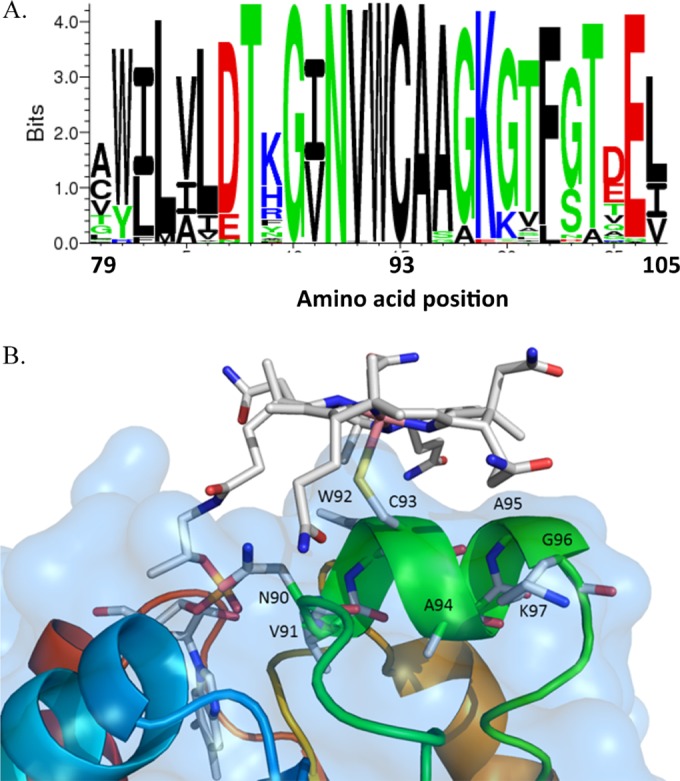
(A) Protein sequence conservation profile for residues in the region surrounding Cys93 (ND132 numbering) in 77 known HgcA homologs found by an NCBI BLAST search as of November 2014. The larger the size of the letter for the amino acid correlates with a higher level of conservation (created by Seq2Logo [61]). (B) Homology model of HgcA. Only residues in the putative cap helix (ND132 numbering) and cobalamin in a “base-off, Cys-on” configuration are shown. The methylcobalamin planar ring is at the top. Important features of the corrinoid ring are shown in the following colors: pink for cobalt, red for oxygen, blue for nitrogen, and gray for carbon. The cap helix region of HgcA is shown in green, and the sulfur of Cys93 is shown in yellow.
HgcB has two [4Fe-4S] cluster-binding motifs, suggesting a ferredoxin-like function but with an architecture distinct from that of other [4Fe-4S] ferredoxins (31–33). Apart from the cysteines needed for the [4Fe-4S] clusters, HgcB has three additional highly conserved cysteines, two at the C terminus and a third at residue 73. These cysteines could play a role in electron or Hg(II) delivery to HgcA or serve to maintain the proper folding of HgcB.
We chose the known Hg methylator ND132 (34) to examine the conserved residues and HgcA/HgcB structural predictions, as this strain is genetically accessible and a deletion mutant lacking hgcAB has already been generated (12). Results of mutations in key regions of HgcA and HgcB that affect Hg methylation are reported and discussed. Although we can state definitively that many of the mutations have altered HgcA and HgcB Hg methylation, future studies are needed to determine whether these mutations change the catalytic capacity only or also alter the structure of the protein.
MATERIALS AND METHODS
Reagents and chemicals.
All chemicals used were of analytical grade and obtained from either Sigma (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA), unless otherwise stated.
Bacteria, culture media, growth conditions, and antibiotics.
All bacterial strains and plasmids used in this work are listed in Tables 1 and 2. Medium compositions, cell growth, Hg exposure for methylation assays, and DNA manipulations for mutagenesis were previously described (12). For standard culturing or for methylation assays, our basal salts medium with 0.1% (wt/vol) yeast extract (MOY salts) (35) contained 60 mM lactate and 40 mM sulfate (MOYLS4) or 40 mM pyruvate and 40 mM fumarate (MOYPF). D. desulfuricans ND132 and Desulfovibrio vulgaris Hildenborough (ATCC 29579) were always cultured anaerobically. Escherichia coli was grown aerobically for cloning purposes (35) or anaerobically for Hg(II) exposure experiments. Antibiotic resistance genes employed for selection included kanamycin resistance (Kmr; npt), spectinomycin resistance (Spr; aadA), and ampicillin resistance (Apr; bla) genes. For selection in E. coli, all antibiotics were used at a concentration of 100 μg/ml, whereas ND132 was sensitive to Km, Sp, and Ap at 400 μg/ml, 200 μg/ml, and 100 μg/ml, respectively. Kmr and Spr D. vulgaris Hildenborough cells were selected with 400 μg G418 (a kanamycin analog) and 200 μg spectinomycin/ml.
TABLE 1.
Bacterial strains used in this work
| Strain | Genotype and/or description | Source or reference |
|---|---|---|
| Escherichia coli Silver | F− deoR endA1 recA1 relA1 gyrA96 hsdR17(rK− mK+) supE44 thi-1 phoA Δ(lacZYA-argF)U169 ϕ80lacZΔM15 λ− | Bioline |
| Desulfovibrio desulfuricans | 11 | |
| ND132 | Wild type | |
| JWN1001 | ND132 (ΔhgcAB)1::(Pnpt-npt-upp); Kmr | 12 |
| JWN1005 | JWN1001 hgcA(1–498) hgcB+-aadAa; Kms Spr; truncation of HgcA after amino acid 166 | This study |
| JWN1006 | JWN1001 hgcA(C93T) hgcB+-aadA; Kms Spr | This study |
| JWN1016 | JWN1001 hgcA(C93A) hgcB+-aadA; Kms Spr | This study |
| JWN1017 | JWN1001 hgcA(W92A) hgcB+-aadA; Kms Spr | This study |
| JWN1018 | JWN1001 hgcA(V91A) hgcB+-aadA; Kms Spr | This study |
| JWN1019 | JWN1001 hgcA(N90A) hgcB+-aadA; Kms Spr | This study |
| JWN1020 | JWN1001 hgcA(I89A) hgcB+-aadA; Kms Spr | This study |
| JWN1021 | JWN1001 hgcA(G88A) hgcB+-aadA; Kms Spr | This study |
| JWN1022 | JWN1001 hgcA(R87A) hgcB+-aadA; Kms Spr | This study |
| JWN1024 | JWN1001 hgcA(K97A) hgcB+-aadA; Kms Spr | This study |
| JWN1025 | JWN1001 hgcA(G98A) hgcB+-aadA; Kms Spr | This study |
| JWN1026 | JWN1001 hgcA(L99A) hgcB+-aadA; Kms Spr | This study |
| JWN1027 | JWN1001 hgcA(F100A) hgcB+-aadA; Kms Spr | This study |
| JWN1028 | JWN1001 hgcA(T101A) hgcB+-aadA; Kms Spr | This study |
| JWN1029 | JWN1001 hgcA(A94S) hgcB+-aadA; Kms Spr | This study |
| JWN1030 | JWN1001 hgcA(A95S) hgcB+-aadA; Kms Spr | This study |
| JWN1031 | JWN1001 hgcA(E312A) hgcB+-aadA; Kms Spr | This study |
| JWN1045 | JWN1001 hgcA(C93H) hgcB+-aadA; Kms Spr | This study |
| JWN1064 | JWN1001 hgcA(N90P) hgcB+-aadA; Kms Spr | This study |
| JWN1065 | JWN1001 hgcA(V91P) hgcB+-aadA; Kms Spr | This study |
| JWN1072 | JWN1001 hgcA(A94P) hgcB+-aadA; Kms Spr | This study |
| JWN1067 | JWN1001 hgcA(A95P) hgcB+-aadA; Kms Spr | This study |
| JWN1068 | JWN1001 hgcA(W92P) hgcB+-aadA; Kms Spr | This study |
| JWN1069 | JWN1001 hgcA(W92F) hgcB+-aadA; Kms Spr | This study |
| JWN1066 | JWN1001 hgcA(Q187 stop) hgcB+-aadA; Kms Spr; HgcA with Glu187 mutated to TAA stop codon | This study |
| JWN1073 | JWN1001 hgcA(G98P) hgcB+-aadA; Kms Spr | This study |
| JWN1013 | JWN1001 hgcA+ hgcB(C95A)-aadA; Kms Spr | This study |
| JWN1014 | JWN1001 hgcA+ hgcB(C96A)-aadA; Kms Spr | This study |
| JWN1015 | JWN1001 hgcA+ hgcB(C95A/C96A)-aadA; Kms Spr | This study |
| JWN1081 | JWN1001 hgcA+ hgcB(C73A)-aadA; Kms Spr | This study |
aadA encodes aminoglycoside 3′-adenyltransferase, which confers spectinomycin resistance.
TABLE 2.
| Primer name | Sequence (5′–3′) | Purpose | Resulting plasmid |
|---|---|---|---|
| 1056 R87A F | GTG GTG GAT ACG GCC GGC ATC AAC GTC TGG | To make the point mutation of Arg87 to Ala87 in hgcA | pMO4676 |
| 1056 R87A R | CCA GAC GTT GAT GCC GGC CGT ATC CAC CAC | To make the point mutation of Arg87 to Ala87 in hgcA | pMO4676 |
| 1056 G88A F | GTG GAT ACG CGC GCC ATC AAC GTC TGG TGC | To make the point mutation of Gly88 to Ala88 in hgcA | pMO4675 |
| 1056 G88A R | GCA CCA GAC GTT GAT GGC GCG CGT ATC CAC | To make the point mutation of Gly88 to Ala88 in hgcA | pMO4675 |
| 1056 I89A F | GAT ACG CGC GGC GCC AAC GTC TGG TGC GC | To make the point mutation of Ile89 to Ala89 in hgcA | pMO4674 |
| 1056 I89A R | GCG CAC CAG ACG TTG GCG CCG CGC GTA TC | To make the point mutation of Ile89 to Ala89 in hgcA | pMO4674 |
| 1056 N90A F | GCG CGG CAT CGC CGT CTG GTG CGC G | To make the point mutation of Asn90 to Ala90 in hgcA | pMO4673 |
| 1056 N90A R | CGC GCA CCA GAC GGC GAT GCC GCG C | To make the point mutation of Asn90 to Ala90 in hgcA | pMO4673 |
| hgcA N90P F | TACGCGCGGCATCCCCGTCTGGTGCGCGGCGGGCA | To make the point mutation of Asn90 to Pro90 in hgcA | pMO4800 |
| hgcA N90P R | TGCCCGCCGCGCACCAGACGGGGATGCCGCGCGTA | To make the point mutation of Asn90 to Pro90 in hgcA | pMO4800 |
| 1056 V91A F | CGG CAT CAA CGC CTG GTG CGC GGC | To make the point mutation of Val91 to Ala91 in hgcA | pMO4672 |
| 1056 V91A R | GCC GCG CAC CAG GCG TTG ATG CCG | To make the point mutation of Val91 to Ala91 in hgcA | pMO4672 |
| hgcA V91P F | TGGATACGCGCGGCATCAACCCCTGGTGCGCGGCGGGCAAGGGGT | To make the point mutation of Val91 to Pro91 in hgcA | pMO4801 |
| hgcA V91P R | ACCCCTTGCCCGCCGCGCACCAGGGGTTGATGCCGCGCGTATCCA | To make the point mutation of Val91 to Pro91 in hgcA | pMO4801 |
| 1056 W92A F | CAT CAA CGT CGC GTG CGC GGC GGG | To make the point mutation of Trp92 to Ala92 in hgcA | pMO4671 |
| 1056 W92A R | CCC GCC GCG CAC GCG ACG TTG ATG | To make the point mutation of Trp92 to Ala92 in hgcA | pMO4671 |
| hgcA W92P F | ACGCGCGGCATCAACGTCCCGTGCGCGGCGGGCAAGGGGT | To make the point mutation of Trp92 to Pro92 in hgcA | pMO4802 |
| hgcA W92P R | ACCCCTTGCCCGCCGCGCACGGGACGTTGATGCCGCGCGT | To make the point mutation of Trp92 to Pro92 in hgcA | pMO4802 |
| hgcA W92F-F | CGCGGCATCAACGTCTTCTGCGCGGCGGGCAAGG | To make the point mutation of Trp92 to Phe92 in hgcA | pMO4806 |
| hgcA W92F-R | CCTTGCCCGCCGGCGAGAAGACGTTGATGCCGCG | To make the point mutation of Trp92 to Phe92 in hgcA | pMO4806 |
| 1056 C delta T- F | CAACGTCTGGACCGCGGCGGGCAA | To make the point mutation of Cys93 to Thr93 in hgcA | pMO4658 |
| 1056 C delta T- R | TTGCCCGCCGCGGTCCAGACGTTG | To make the point mutation of Cys93 to Thr93 in hgcA | pMO4658 |
| 1056 C93A-F | CAACGTCTGGGCCGCGGCGGGCAA | To make the point mutation of Cys93 to Ala93 in hgcA | pMO4670 |
| 1056 C93A-R | TTGCCCGCCGCGGCCCAGACGTTG | To make the point mutation of Cys93 to Ala93 in hgcA | pMO4670 |
| hgcA C93H Fwd | ATCAACGTCTGGCACGCGGCGGGCAAG | To make the point mutation of Cys93 to His93 in hgcA | pMO4699 |
| hgcA C93H Rev | CTTGCCCGCCGCGTGCCAGACGTTGAT | To make the point mutation of Cys93 to His93 in hgcA | pMO4699 |
| hgcA A94S-F | CAACGTCTGGTGCTCGGCGGGCAAGGGGT | To make the point mutation of Ala94 to Ser94 in hgcA | pMO4683 |
| hgcA A94S-R | ACCCCTTGCCCGCCGAGCACCAGACGTTG | To make the point mutation of Ala94 to Ser94 in hgcA | pMO4683 |
| hgcA A94P F | GGCATCAACGTCTGGTGCCCGGCGGGCAAGGGGTTGTTC | To make the point mutation of Ala94 to Pro94 in hgcA | pMO4803 |
| hgcA A94P R | GAACAACCCCTTGCCCGCCGGGCACCAGACGTTGATGCC | To make the point mutation of Ala94 to Pro94 in hgcA | pMO4803 |
| hcgA A95S-F | ACGTCTGGTGCGCGTCGGGCAAGGGGTTGT | To make the point mutation of Ala95 to Ser95 in hgcA | pMO4684 |
| hgcA A95S-R | ACAACCCCTTGCCCGAGCCGCACCAGACGT | To make the point mutation of Ala95 to Ser95 in hgcA | pMO4684 |
| hgcA A95P F | ATCAACGTCTGGTGCGCGCCGGGCAAGGGGTTGTTCACCGCT | To make the point mutation of Ala95 to Pro95 in hgcA | pMO4804 |
| hgcA A95P R | AGCGGTGAACAACCCCTTGCCCGGCGCGCACCAGACGTTGAT | To make the point mutation of Ala95 to Pro95 in hgcA | pMO4804 |
| 1056 G96A F | TGG TGC GCG GCG GCC AAG GGG TTG TTC A | To make the point mutation of Gly96 to Ala96 in hgcA | pMO4677 |
| 1056 G96A R | TGA ACA ACC CCT TGG CCG CCG CGC ACC A | To make the point mutation of Gly96 to Ala96 in hgcA | pMO4677 |
| 1056 K97A F | TGC GCG GCG GGC GCG GGG TTG TTC ACC G | To make the point mutation of Lys97 to Ala97 in hgcA | pMO4678 |
| 1056 K97A R | CGG TGA ACA ACC CCG CGC CCG CCG CGC A | To make the point mutation of Lys97 to Ala97 in hgcA | pMO4678 |
| 1056 G98A F | GCG GCG GGC AAG GCG TTG TTC ACC GCT T | To make the point mutation of Gly98 to Ala98 in hgcA | pMO4679 |
| 1056 G98A R | AAG CGG TGA ACA ACG CCT TGC CCG CCG C | To make the point mutation of Gly98 to Ala98 in hgcA | pMO4679 |
| hgcA G98P-F | TGCGCGGCGGGCAAGCCGTTGTTCACCGCTTCCGA | To make the point mutation of Gly98 to Pro98 in hgcA | pMO4805 |
| hgcA G98P-R | TCGGAAGCGGTGAACAACGGCTTGCCCGCCGCGCA | To make the point mutation of Gly98 to Pro98 in hgcA | pMO4805 |
| 1056 L99A F | GCG GGC AAG GGG GCG TTC ACC GCT TCC G | To make the point mutation of Leu99 to Ala99 in hgcA | pMO4680 |
| 1056 L99A R | CGG AAG CGG TGA ACG CCC CCT TGC CCG C | To make the point mutation of Leu99 to Ala99 in hgcA | pMO4680 |
| 1056 F100A F | GGC AAG GGG TTG GCC ACC GCT TCC GAG G | To make the point mutation of Phe100 to Ala100 in hgcA | pMO4681 |
| 1056 F100A R | CCT CGG AAG CGG TGG CCA ACC CCT TGC C | To make the point mutation of Phe100 to Ala100 in hgcA | pMO4681 |
| 1056 T101A F | CAA GGG GTT GTT CGC CGC TTC CGA GGT GG | To make the point mutation of Thr101 to Ala101 in hgcA | pMO4682 |
| 1056 T101A R | CCA CCT CGG AAG CGG CGA ACA ACC CCT TG | To make the point mutation of Thr101 to Ala101 in hgcA | pMO4682 |
| F into 1057 for 1056 trunc | CCTTCCTGCGCAACGGCAACAAGGCGTGACCGGGAGACTGATGATGAAGGATTTCCG | To truncate hgcA after aa 166c | pMO4695 |
| R into 1056 for 1056 trunc | CGGAAATCCTTCATCATCAGTCTCCCGGTCACGCCTTGTTGCCGTTGCGCAGGAAGG | To truncate hgcA after aa 166c | pMO4695 |
| hgcA STOP 187-F | CGCTCATCCCGGTGTAACTGTACCAGCTGCGCAA | To mutate Glu187 of hgcA to stop codon (TAA)d | pMO4807 |
| hgcA STOP 187-R | TTGCGCAGCTGGTACAGTTACACCGGGATGAGCG | To mutate Glu187 of hgcA to stop codon (TAA)d | pMO4807 |
| hgcB C73A Fwd | GTCACCCCCGGCACGGGCGCGGCCGCCTACCTGGTCTCGGT | To make the point mutation of Cys73 to Ala73 in hgB | pMO4808 |
| hgcB C73A Rev | ACCGAGACCAGGTAGGCGGCCGCGCCCGTGCCGGGGGTGAC | To make the point mutation of Cys73 to Ala73 in hgB | pMO4808 |
| 1057 C95A-F | ATCGACGCCGCCGCCTGCTAGCGTATGCGCT | To make the point mutation of Cys94 to Ala94 in hgcB | pMO4667 |
| 1057 C95A-R | AGCGCATACGCTAGCAGGCGGCGGCGTCGAT | To make the point mutation of Cys94 to Ala94 in hgcB | pMO4667 |
| 1057 C96A-F | ATCGACGCCGCCTGCGCCTAGCGTATGCGCT | To make the point mutation of Cys95 to Ala95 in hgcB | pMO4668 |
| 1057 C96A-R | AGCGCATACGCTAGGCGCAGGCGGCGTCGAT | To make the point mutation of Cys95 to Ala95 in hgcB | pMO4668 |
| 1057 C95A C96A-F | ATCGACGCCGCCGCCGCCTAGCGTATGCGCT | To make the point mutation of Cys94/95 to Ala94/95 in hgcB | pMO4669 |
| 1057 C95A C96A-R | AGCGCATACGCTAGGCGGCGGCGGCGTCGAT | To make the point mutation of Cys94/95 to Ala94/95 in hgcB | pMO4669 |
| pUC Amp Rev | ATGTGAGCAAAAGGCCAGCAAAAGGCC | For SLIC of all of the mutants listed above using pMO4661e | All |
| 1056 upstream Fwd with pUC ovhng | GGCCTTTTGCTGGCCTTTTGCTCACATGTCTACAGGGAGCCGTTCACC | For SLIC of all of the mutants listed above using pMO4661e | All |
| Amp pUC Fwd | GAAGTTTTAAATCAATCTAAAGTATAT | For SLIC of all of the mutants listed above using pMO4661e | All |
| 1057 downstream Rev w/Amp ovhng | ATTCCCTGTGCGTCGTCTGGGAAGTTTTAAATCAATCTAAAGTATAT | For SLIC of all of the mutants listed above using pMO4661e | All |
All fragments were amplified from pMO4661, the insertional complement of the hgcAB deletion, and therefore share the same traits as pMO4661 with regard to upstream and downstream homology and antibiotic resistance but differ in their respective described HgcA or HgcB mutations. The resulting plasmids were used to introduce the respective mutations and create the strains listed in Table 1.
See Fig. 3C.
See Fig. 2.
Site-directed mutant constructions.
All primers utilized and constructed for this study are listed in Table 2. For site-directed mutations in hgcA and hgcB, all plasmids were constructed to ensure that the correct reading frame was conserved when integrated into the ND132 chromosome. Plasmid pMO4661 (Fig. 2A) was used as a template for all hgcA and hgcB mutations. pMO4661, a nonreplicating plasmid in ND132, includes the following six regions, in the 5′-to-3′ direction: a 731-bp DNA region upstream of the predicted GTG start codon of hgcA, hgcA, and hgcB; a gene conferring Spr; and the region downstream of the hgcB stop codon consisting of 762 bp, followed by an Apr gene and a pUC origin of replication (Fig. 2A). Three PCR amplicons were generated from pMO4661 to amplify the plasmid in three separate pieces (blue primers and red primers in Fig. 2). Point mutations in hgcA or hgcB were introduced by PCR with primers designed to change the amino acid codons desired (red primers in Fig. 2). With the three pieces reassembled in E. coli by sequence- and ligation-independent cloning (36), the new plasmid with the desired point mutation was confirmed by sequencing of both strands of hgcA and hgcB containing the introduced mutation(s) and the homologous flanking regions. Constructed plasmids were selected in E. coli with Spr and Apr. When this nonreplicating plasmid was introduced into JWN1001 (the ND132 ΔhgcAB strain containing npt [Kmr] replacing the two genes), selection for Spr transformants and screening for Kms allowed the identification of double homologous recombination events where the Kmr cassette of JWN1001 was replaced with the mutated hgcA or hgcB gene linked to the Spr marker.
FIG 2.

Diagram of the plasmids used in the strategy for creating site-directed mutations in D. desulfuricans ND132 hgcA. (A) Plasmid pMO4661. The pMO4661 origin of replication and ampicillin resistance-encoding gene are from pCR4-TOPO (Invitrogen). Green segments represent the chromosomal DNA regions flanking hgcA (purple) and hgcB (orange) that provide the homology for recombination events. (B) Example of the specific strategy for amino acid mutagenesis in HgcA and delivery to the JWN1001 (ΔhgcA-hgcB)::(Pnpt-npt-upp) strain through transformation. The red primers were designed to mutate amino acids, while the blue primers were designed to amplify sections of the plasmid backbone. PCR amplicons were generated and assembled by using sequence- and ligation-independent cloning procedures (36). (C) Enlargement of the position of the PCR primers used for introducing changes in the Cys93 codon in the sequence of hgcA.
HgcA truncations.
For the C-terminal truncation of HgcA, pMO4661 was once again used as the template. Primers were designed to either delete the C-terminal domain of HgcA after amino acid 166 (Fig. 3A and B and Table 2) or mutate GAA of Glu187 to the nonsense mutation TAA stop codon (Fig. 3C and Table 2). Mutant constructs were verified by sequencing on both strands.
FIG 3.

Primer design for removal of the C-terminal transmembrane domain of hgcA. (A) Genetic deletion strategy. The primer color reflects the complementary strands. Red indicates that the stop codon of hgcA was retained. Yellow represents the 14-bp intergenic region between hgcA and hgcB in ND132. (B) Resulting construct showing a deletion of the DNA encoding the C-terminal region of HgcA. (C) Construct showing the mutation of Glu187 to a translational stop codon that retains the 3′ region of the hgcA gene, which harbors a putative transcriptional start site of hgcB.
ND132 electroporations.
Electroporations and selection of recombinant ND132 colonies were performed as previously described (12).
Alignment of HgcA and HgcB from ND132 and Dethiobacter alkaliphilus.
Sequence alignments of the HgcA and HgcB proteins from ND132 and Dethiobacter alkaliphilus were performed with T-Coffee (37), and colors were converted for publication with BoxShade (http://www.ch.embnet.org/software/BOX_form.html).
Determination of the Hg methylation potential by measurement of MeHg.
All exposures of bacteria to Hg(II) were performed in a chamber (Coy Laboratory Products, Grass Lake, MI) containing an N2-H2 (95:5) anaerobic atmosphere and <0.5% (vol/vol) O2. Deletion mutants of ND132 were subcultured in fresh medium as described above (1%, vol/vol) and grown until the optical density at 600 nm (OD600) reached ∼0.5. These cultures were then diluted to an OD600 of ∼0.2 in fresh medium. Hg(II) exposure was performed in triplicate in 2-ml microcentrifuge tubes containing 400 μl of culture, to which HgCl2 (prepared at 74 μmol/ml in deionized water in a Teflon-coated glass vial and kept at 4°C in the dark) was added (final concentration, 37 pmol/ml [10 ng/ml]). To allow mercury methylation, amended cultures were incubated in the dark for 2 h at 34°C. The cells were then immediately acidified with HNO3 (final concentration, 3 M) and digested at room temperature (RT) for 24 h before analysis. Wild-type strain ND132 was used as a positive Hg methylation control. Nonmethylating controls were heat-killed strain ND132, viable ΔhgcAB mutant strain JWN1001 (12), and sterile medium as an abiotic control. Mutants were also tested as heat-killed cultures. None of the nonmethylating controls, JWN1001 or abiotic medium, showed MeHg production above either that of ND132 heat-killed controls or the detection limits (see below).
Following acidification of samples, all manipulations were performed at room temperature in a fume hood. MeHg production was determined by ethylation purge and trap gas chromatography atomic fluorescence spectrometry (EPT-GC-AFS) according to EPA Method 1630 (http://nepis.epa.gov/Exe/ZyPURL.cgi?Dockey=P100IKBQ.TXT). An aliquot (80 μl) of the acidified sample was added to 40 ml sodium acetate buffer (1 M acetic acid, 1 M sodium acetate [pH 3.9]). Hg species were then ethylated with 50 μl sodium tetraethyl borate (1%, wt/vol) and extracted from solution into an automated purge-and-trap system followed by GC separation and quantification by AFS with a MerX Hg speciation GC and pyrolysis module coupled to a model III cold vapor atomic fluorescence spectrophotometer. Data were integrated with Mercury Guru 4.1 software, and peaks were quantified by comparison with a MeHg standard curve. HgCl2 and CH3HgCl were used for Hg methylation assays and external calibration, respectively, and were both >95% pure. The experimental detection limits for MeHg determined with analytical blanks (n = 15) for each experimental batch were 45 ± 20 fmol/ml. All equipment, reagents, standards, and software for MeHg detection were obtained from Brooks Rand Labs, Seattle, WA.
Antibody development and Western blot analysis.
A rabbit polyclonal antibody to amino acids 18 to 29 of ND132 HgcA (epitope YLRRDDRVGDLR) was developed and affinity purified by Anaspec (Fremont, CA). Western analysis, including SDS-PAGE, protein transfer onto membranes, and immunoblotting, was generally performed according to procedures described previously by Shapiro et al., Towbin et al., and Burnett (38–40), with the following details and modifications. ND132 cultures grown anaerobically in 200 ml MOYPF to an OD600 of 0.5 were centrifuged at 4,000 × g for 12 min at 4°C. The cell pellet was resuspended, washed, and centrifuged again in 50 ml of cold 50 mM potassium phosphate (KPi) buffer (pH 7.2) with 10 mM dithiothreitol (DTT). The cell pellet was then resuspended in 10 ml of 50 mM cold KPi buffer with 10 mM DTT, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 100 μl bacterial protease inhibitor cocktail (Sigma, St. Louis, MO). The cell suspension was passed through a French press twice at 19,000 lb/in2. The lysate was then centrifuged at 4,000 × g for 12 min, and the supernatant was recovered. This supernatant was then spun at 17,000 × g for 15 min at 4°C, and the resulting supernatant was saved for protein analysis. Protein concentrations were determined by the Bradford method (41), with bovine serum albumin as a standard. Precast 12% (wt/vol) Mini-PROTEAN TGX gels (Bio-Rad, Hercules, CA) were used to separate cellular proteins. Protein extracts were loaded onto the gels at 30 μg per lane in denaturing Laemmli loading buffer (42). Gels were run for 90 min at 100 V at RT in Tris-glycine SDS buffer consisting of 25 mM Tris (pH 8.3), 0.2 M glycine, and 3.5 mM SDS on the Mini-Protean system (Bio-Rad). Protein transfer onto a polyvinylidene difluoride membrane (Millipore, Billerica, MA) was performed overnight at 150 mA at 4°C in transfer buffer containing 25 mM Tris (pH 8.3), 192 mM glycine, and 20% (vol/vol) methanol. After transfer, membranes were washed briefly with Tris-buffered saline (20 mM Tris and 150 mM NaCl [pH 7.5]) with 0.2% (vol/vol) Tween 20 (TBST). Membranes were then blocked for 1 h in TBST plus 5% (vol/vol) goat serum (MP Biomedicals, Santa Ana, CA) at RT. After blocking, membranes were incubated with primary rabbit anti-HgcA at a dilution of 1:5,000 in TBST plus 5% (vol/vol) goat serum for 1 h at RT. Membranes were then washed 3 times for 5 min each in TBST and subsequently incubated for 1 h at RT with secondary goat anti-rabbit IgG-horseradish peroxidase (Sigma)-conjugated antibody at a 1:3,000 dilution in TBST plus 5% (vol/vol) goat serum. After secondary antibody incubation, membranes were washed three times for 5 min each in TBST. Chemiluminescence detection was performed with the Pierce ECL Western blotting substrate (Thermo Scientific, Rockford, IL) according to the manufacturer's instructions. Membranes were exposed to X-ray film (Thermo Scientific) for 3 to 45 s and developed in an X-ray film processor (SRX-101A; Kinoca Minolta, China). Denatured protein migration positions were compared to protein standards (Bio-Rad) to determine molecular weights. JWN1001, the ΔhgcAB strain, served as a negative control for HgcA protein expression.
RESULTS
The identification of two proteins essential for Hg methylation led to the proposal of a novel mechanism of methyl transfer by HgcA. Although the nature of the Hg substrate is not known, it was reasoned that a cysteine thiolate coordination to the Co of the cobamide cofactor would promote methyl transfer to Hg(II), and theoretical calculations have supported this proposal (30).
We tested the importance of the conserved residue Cys93 and the residues in the cap helix for the methylation of Hg (12, 30). Because five-coordinate methylcorrinoids favor carbocation transfer (43), we hypothesized that replacing Cys93 with a residue unable to coordinate Co would have a deleterious effect on Hg methylation. We mutated Cys93 to Ala93 or to Thr93 (the latter being found in CFeSP) and found that MeHg production was eliminated (Fig. 4 and Table 3). On the basis of quantum chemical calculations, it was previously predicted that an HgcA Cys93His mutant, which should allow “His-on” Co coordination, might be able to methylate Hg (30). Indeed, the His93 mutant produced MeHg but only at 5% of the level of wild-type strain ND132 (Fig. 4 and Table 3). When Trp92, also strictly conserved in HgcA homologs (Fig. 1), was changed to Ala92 or Phe92, MeHg production was reduced to 21% or 5 to 10%, respectively. These results show the importance of both Cys93 and its neighboring amino acid, Trp92, in the methylation reaction but that a large hydrophobic residue is not essential to position the cysteine for the methylation reaction (Fig. 4 and Table 3).
FIG 4.

(A) Alignment of 77 known HgcA orthologs found by an NCBI BLAST search as of November 2014. The degree of blue shading indicates the degree of amino acid conservation. The cap helix region has been enlarged, showing the amino acid sequence of ND132 HgcA. The 9-amino-acid cap helix is framed by a solid red box. (B) Hg(II) methylation potential of site-directed mutations of the cap helix region of HgcA in D. desulfuricans ND132. The predicted chemically important residue Cys93 is identified by a black arrow. MeHg production of the mutants is presented in the column on the right as a percentage of that of the D. desulfuricans ND132 wild-type control. The activity of amino acid substitutions across the putative cap helix is also depicted, with red indicating completely altered activity, orange indicating intermediately altered activity, and green indicating unaltered activity. Specific mutant assay data and statistics are presented in Table 3.
TABLE 3.
MeHg production in D. desulfuricans ND132 wild-type and mutant strains
| D. desulfuricans strain | Characteristic(s)a | Mean MeHg concn (pmol/ml culture) ± SD | Mean MeHg concn (pmol/mg protein) ± SD | % MeHg productionb (vs WT) | P value | Reference |
|---|---|---|---|---|---|---|
| ND132 | Wild type | 5.4 ± 1.9 | 78.1 ± 25.1 | 100 | This study | |
| JWN1001 | ND132 Δ(hgcAB)::(Pnpt-npt-upp) | <DL | <DL | <DL** | <0.005 | 12 |
| JWN1001 | ND132 Δ(hgcAB)::(Pnpt-npt-upp) | <DL | <DL | <DL** | <0.005 | This study |
| JWN1005 | HgcAΔ(Ala166 to TGA stop codon) | <DL | <DL | <DL** | <0.005 | This study |
| JWN1006 | HgcA(Cys93Thr) | <DL | <DL | <DL** | <0.005 | This study |
| JWN1016 | HgcA(Cys93Ala) | <DL | <DL | <DL** | <0.005 | This study |
| JWN1017 | HgcA(Trp92Ala) | 1.2 ± 0.2 | 16.8 ± 2.4 | 21.5 ± 3.1* | 0.014 | This study |
| JWN1018 | HgcA(Val91Ala) | 3.6 ± 0.5 | 55.8 ± 7.5 | 71.4 ± 9.6 | 0.214 | This study |
| JWN1019 | HgcA(Asn90Ala) | 7.0 ± 0.6 | 101 ± 9.2 | 129 ± 11.8 | 0.212 | This study |
| JWN1020 | HgcA(Ile89Ala) | 4.0 ± 0.5 | 61.3 ± 7.4 | 78.4 ± 9.5 | 0.329 | This study |
| JWN1021 | HgcA(Gly88Ala) | 3.4 ± 0.1 | 48.7 ± 1.3 | 62.3 ± 1.7 | 0.113 | This study |
| JWN1022 | HgcA(Arg87Ala) | 4.9 ± 0.8 | 73.2 ± 11.8 | 93.7 ± 15.1 | 0.775 | This study |
| JWN1024 | HgcA(Lys97Ala) | 4.9 ± 0.1 | 77.2 ± 2.0 | 98.8 ± 2.6 | 0.943 | This study |
| JWN1025 | HgcA(Gly98Ala) | 8.3 ± 0.5 | 118 ± 7.7 | 151 ± 9.8 | 0.058 | This study |
| JWN1026 | HgcA(Leu99Ala) | 3.3 ± 0.6 | 52.7 ± 9.0 | 67.4 ± 11.6 | 0.174 | This study |
| JWN1027 | HgcA(Phe100Ala) | 6.6 ± 0.4 | 89.1 ± 5.9 | 114 ± 7.6 | 0.501 | This study |
| JWN1028 | HgcA(Tyr101Ala) | 15.2 ± 0.8 | 192 ± 10 | 246 ± 12.8** | 0.002 | This study |
| JWN1029 | HgcA(Ala94Ser) | 8.7 ± 1.2 | 139 ± 19 | 178 ± 24.3* | 0.029 | This study |
| JWN1030 | HgcA(Ala95Ser) | 5.4 ± 1.6 | 91.7 ± 26.1 | 117 ± 33.4 | 0.551 | This study |
| JWN1031 | HgcA(Glu312Ala) | 8.9 ± 1.0 | 160 ± 19 | 205 ± 24.3* | 0.011 | This study |
| JWN1045 | HgcA(Cys93His) | 0.3 ± 0.2 | 3.8 ± 3.2 | 4.9 ± 4.1* | 0.007 | This study |
| JWN1064 | HgcA(Asn90Pro) | 1.7 ± 0.0 | 23.3 ± 0.4 | 29.8 ± 0.5* | 0.019 | This study |
| JWN1065 | HgcA(Val91Pro) | 0.4 ± 0.0 | 5.7 ± 0.7 | 7.3 ± 0.9* | 0.008 | This study |
| JWN1072 | HgcA(Ala94Pro) | <DL | <DL | <DL** | <0.005 | This study |
| JWN1067 | HgcA(Ala95Pro) | <DL | <DL | <DL** | <0.005 | This study |
| JWN1068 | HgcA(Trp92Pro) | <DL | <DL | <DL** | <0.005 | This study |
| JWN1069 | HgcA(Trp92Phe) | 0.4 ± 0.0 | 6.1 ± 0.2 | 7.8 ± 0.3* | 0.008 | This study |
| JWN1066 | HgcA(Glu187Stop) | <DL | <DL | <DL** | <0.005 | This study |
| JWN1073 | HgcA(Gly98Pro) | 3.6 ± 0.3 | 54.8 ± 5.3 | 70.1 ± 6.8 | 0.191 | This study |
| JWN1013 | HgcB(Cys94Ala) | 5.5 ± 0.4 | 85.2 ± 5.9 | 109 ± 7.6 | 0.066 | This study |
| JWN1014 | HgcB(Cys95Ala) | 6.6 ± 0.7 | 94.3 ± 10 | 121 ± 13 | 0.358 | This study |
| JWN1015 | HgcB(Cys94–95Ala) | 0.2 ± 0.1 | 3.4 ± 0.9 | 4.3 ± 1.1* | 0.007 | This study |
| JWN1081 | HgcB(Cys73Ala) | <DL | <DL | <DL** | <0.005 | This study |
Hg methylation potentials were determined from experiments performed with mutant strains of D. desulfuricans. The standard deviations from triplicate methylation assays are displayed for each value.
The average production of MeHg normalized to the protein concentration was used to estimate the percentage of MeHg produced by a mutant compared to the wild type (WT). MeHg data normalized to protein data were used for statistics. <DL denotes a value below the minimum detectable limit of 45 ± 20 fmol MeHg/ml. *, P value of <0.05; **, P value of <0.005 (by an unpaired t test) (means and standard deviations are from triplicate experiments).
To determine the contributions of residues in the cap helix region to Hg methylation, we systematically disrupted this region by alanine scanning. This approach has been shown to provide insights into the influence of amino acid side-chain chemistry on protein reactivity (44). Other than Cys93Ala and Trp92Ala, all other alanine mutants were found not to decrease the Hg(II) methylation capacity compared to that of wild-type strain ND132 (Fig. 4 and Table 3). Residues 94 and 95 are Ala in wild-type HgcA, so these residues were mutated separately to Ser94 and Ser95, respectively, and these mutants also showed no difference in MeHg production compared to the wild type (Fig. 4 and Table 3).
A proven technique to disrupt α-helical protein structure is to mutate a residue within the helix to a proline (45, 46). Site-directed proline mutations designed to disrupt the HgcA cap helix region near Cys93 were introduced singly into HgcA. The mutations Asn90Pro, Val91Pro, Trp92Pro, Ala94Pro, Ala95Pro, and Gly98Pro all led to decreases in the methylation capacity, ranging from below the detection limit when near Cys93 to as much as 70% of wild-type levels five residues away (Fig. 4 and Table 3). These results support the prediction of a helical structure for the sequence around Cys93. Additionally, these results strengthen the hypothesis that the helix plays a role in positioning Cys93 for ligation with the cobalt in the corrinoid cofactor. With the exception of Cys93, it appears that the maintenance of secondary structure in the cap helix is more important than amino acid residue chemistry.
To determine if the predicted cytoplasmic HgcA N-terminal domain could retain activity in the absence of the putative C-terminal transmembrane domain, we truncated the protein with two separate approaches. We first deleted the coding sequence of the HgcA C-terminal domain from amino acid 167 through the end of the protein (strain JWN1005) (Table 1). The resulting truncated mutant was unable to methylate Hg(II) (Table 3). However, through preliminary attempts to determine the transcriptional start sites (TSSs) of hgcA and hgcB (data not shown), we observed that the 3′ region of hgcA likely harbors a TSS of hgcB 144 bp upstream of the ATG start codon of hgcB and 130 bp upstream from the end of hgcA. If the primary TSS of hgcB were indeed located in the hgcA 3′-terminal region, hgcB would not be transcribed when that DNA was deleted, and the absence of HgcB would provide an established reason for the lack of methylation activity. Therefore, we engineered a translational stop site just after the HgcA N-terminal domain, replacing the Glu187 codon GAA with a TAA stop site, 477 bp upstream of the translational start of hgcB, retaining the putative TSS. This construct should produce a 186-amino-acid N-terminal HgcA fragment but should not eliminate the TSS for transcription of hgcB. When this mutation was successfully constructed (strain JWN1066) (Table 1), Hg methylation by the mutant strain was still not detected (Table 3).
Verification of expression of the HgcA soluble domain in the Glu187-to-TAA stop codon mutant was attempted by Western blot analysis with an antibody directed against amino acids 18 to 29 of HgcA. This analysis showed a band corresponding to full-length HgcA at ∼40 kDa in the wild-type ND132 controls, but in the mutant strain, there was no evidence of the truncated ∼20-kDa form of HgcA (Fig. 5). Therefore, the absence of Hg methylation could have resulted from the absence of the C-terminal transmembrane domain of HgcA or from rapid proteolytic degradation of this truncated N-terminal protein portion.
FIG 5.
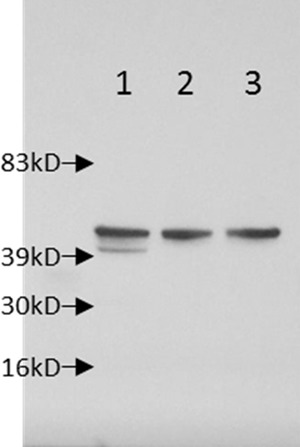
Western blot showing the expression of the full-length ∼40-kDa HgcA protein from wild-type strain ND132 (lane 1) and the lack of detection of truncated HgcA (expected 167-aa protein) (lane 3). Lane 2 is JWN1001, the ΔhgcAB strain used as a negative control. Note the nonspecific band above HgcA in all three samples showing equal loading of protein.
To explore the function of the ferredoxin-like protein HgcB, we chose to examine the highly conserved vicinal cysteines at its C terminus (12). Vicinal cysteines have been reported to have roles in Hg binding (47) or as redox switches in proteins (48, 49). In ND132, we replaced each of these two cysteines with alanine and then created a double mutant in which both cysteines were converted to alanines. When tested separately for Hg methylation, the Cys94Ala or Cys95Ala HgcB mutants showed no change in methylation from wild-type strain ND132, but mutation of both residues reduced methylation to 5% of wild-type levels (Table 3). Apparently, only one cysteine is required at the C terminus of HgcB for full function. This result also indicated that Hg binding by the vicinal cysteines together does not significantly contribute to methylation. However, another conserved cysteine, Cys73 in ND132, is present in HgcB (and in many of the HgcB proteins sequenced) but is apparently not part of a [4Fe-4S] cluster-binding motif. HgcB with Cys73 mutated to Ala showed no Hg methylation (Table 3).
DISCUSSION
To examine the function of conserved amino acids and protein regions of HgcA and HgcB, and to test predicted structural features of HgcA, site-directed mutations and specific deletions were made. This approach was facilitated by having a mutant of D. desulfuricans ND132 with both genes deleted and replaced by a selectable marker (11). Importantly, the first feature examined was the unusual cofactor coordination and chemistry predicted for the corrinoid protein HgcA. This protein was predicted to bind its cofactor in a Cys-on configuration in which the strictly conserved Cys93 residue is coordinated to Co in the lower axial position (11, 30). Additionally, it was proposed that HgcA may transfer a methyl carbanion to Hg(II), chemistry which has not yet been demonstrated for biological methyl transfer by a cobamide enzyme. To date, the corrinoid methyltransferase chemistry described in the literature has exclusively involved the transfer of a methyl carbocation (50). For these reasons, we targeted Cys93 for mutagenesis. The elimination of the methylation capacity of strains harboring mutations of Cys93 to Ala or Thr, but retention of limited methylation in the Cys93His mutant, was interpreted as supporting the hypothesis that this residue is indeed coordinated to Co.
It is interesting that Cys93, when mutated to His93, retains a small percentage of the methylating capacity. In many corrinoid proteins, the lower axial ligand to Co in the cofactor is a histidine (51). Using density functional theory calculations on a cluster model of HgcA, Zhou et al. (30) suggested that a histidine ligand to cobalt may facilitate the transfer of a methyl carbanion to a Hg(II) substrate. The mutagenesis results presented here cannot distinguish between a carbanion or radical transfer mechanism. However, when taken together with previously reported findings (12, 30), the results remain consistent with the proposed carbanion transfer mechanism.
Certain amino acids in the cap helix are more highly conserved than others (Fig. 1). The fact that a change of Trp92, a bulky, aromatic amino acid, to Ala92 resulted in a 70% reduction of the methylation capacity suggests that this residue, adjacent to Cys93, is functionally important. Additional structural studies will be needed to differentiate between possible roles of the Trp residue, but possibilities include positioning of Cys93 for access to the corrinoid or for providing protection to Co(I) through hydrophobic interactions with the corrinoid ring. Mutation of the other cap helix amino acids through alanine scanning or by the introduction of a helix-disrupting proline residue showed that the structure of this region is critical for methylation, supporting structural inferences from the sequence homology with CFeSP (11).
Although the identity of the cobamide involved in Hg methylation by ND132 is not known, preliminary experiments suggest that a modified corrinoid may be present (our unpublished data). To date, D. vulgaris Hildenborough is the only bacterium known to synthesize the rare guanylcobamide and hypoxanthylcobamide corrinoids and not pseudovitamin B12 (52).
Transfer of a methyl carbanion from HgcA to Hg would require two electrons to reduce Co(III) to Co(I) for continued catalytic turnover. HgcB is the logical electron donor, as was previously hypothesized (12), but the low redox potential of the Co(II)/Co(I) couple for free cobalamin (<−600 mV [53]) might be an impediment to the reduction. However, in HgcA, the corrinoid is predicted to be bound in a “base-off, Cys-on” configuration, which we expect would result in a Co(II)/Co(I) reduction potential of around −550 mV (54).
An interesting feature of HgcB is the vicinal cysteines at the C terminus. Mutagenesis showed that only one is required for full Hg methylation by ND132. Potential roles in electron transfer into, or out of, the [4Fe-4S] centers or delivery of Hg(II) to HgcA might be considered. It is known that vicinal cysteines poorly mediate redox reactions because of the strained stereochemistry between the two sulfurs (49). Juxtaposed cysteine residues have been shown to act as conformational redox switches, which, when oxidized, change the bond angle of the peptide backbone, thus altering the three-dimensional structure and possibly the function of the protein (48, 49). Vicinal cysteines have also been shown to selectively bind Hg (48). Of note, mutation of another cysteine (Cys73) in HgcB to alanine eliminated Hg methylation. The presence of a “fifth cysteine” adjacent to the [4Fe-4S] cluster of ferredoxin I from Azotobacter vinelandii has been shown to decrease the reduction potential of the cluster by ∼−50 mV (55). The possibility of vicinal cysteines (Cys94 and Cys95) working together with a separate cysteine (Cys73) to act on a Hg substrate has precedence. In the organomercurial lyase protein MerB, two vicinal cysteines act in coordination with a third cysteine to have maximal function (56). Mutation of one of the vicinal cysteines in this system led to only a slight reduction in activity (57). Future studies may determine whether Cys73 of HgcB functions in conjunction with either of the vicinal cysteines for electron movement, Hg binding, or structural integrity. Additional mutations of the cysteines predicted to be essential for the formation of the [4Fe-4S] clusters will also be needed to confirm their importance in Hg methylation.
In all but one of the HgcB homologs in Hg methylators identified to date, vicinal cysteines are present in the C-terminal region along with a cysteine analogous to Cys73 of HgcB in strain ND132. The exception is the known Hg methylator Dethiobacter alkaliphilus strain AHT 1 (DSM 19026) (11, 58), in which HgcB is truncated after Gly71. This residue corresponds to Gly72 in HgcB of ND132, which is just upstream of the expected position of Cys73 (Fig. 6A). We observed that HgcA of Dt. alkaliphilus has a 29-amino-acid insert in its N-terminal region containing two pairs of vicinal cysteines separated by 3 amino acids (Fig. 6B). It is tempting to speculate that these inserted cysteines might functionally replace the vicinal cysteines absent in HgcB. Future experiments reconstructing this arrangement of cysteines in the genetically accessible strain ND132 might allow testing of this hypothesis.
FIG 6.

Alignments of HgcB and HgcA of D. desulfuricans ND132 to those proteins of Dt. alkaliphilus. Strictly conserved amino acids are in black, while chemically similar amino acids are in gray. (A) Alignment of HgcB proteins of ND132 and Dt. alkaliphilus showing the locations of Cys73 and the vicinal cysteines (red), illustrating the truncation at Gly71 (which would be Gly72 in ND132) and the lack of Cys73 and the vicinal cysteines in Dt. alkaliphilus. The cysteines of the two 4Fe-4S clusters (CX2CX2CX3C) are shown in yellow. (B) Alignment of the full-length HgcA proteins of ND132 and Dt. alkaliphilus showing the 29-amino-acid insert in Dt. alkaliphilus 84 amino acids upstream of the cap helix (green) and the two pairs of vicinal cysteines (red) in the insert.
Our studies show that the putative transmembrane domain of HgcA is needed, at a minimum, for stability of the N-terminal corrinoid-binding region. The hydrophobic C-terminal region may protect the cofactor, localize the protein to the membrane (59), or facilitate Hg transport into, or MeHg transport out of, the cell. Lin et al. (60) showed that the hgcAB double-deletion mutant of G. sulfurreducens PCA exhibits decreased Hg(II) uptake by ∼30% relative to the wild type. The predicted transmembrane domain of HgcA has no detectable homology to any other known protein (12), limiting functional assignment of this region.
While this study sheds new light on the structural aspects and identifies key amino acid residues of HgcA and HgcB required for maximal Hg methylation, many questions remain unanswered. Our initial attempts to transfer Hg methylation capacity to a nonmethylating Desulfovibrio strain were unsuccessful (R. Bridou and S. D. Smith, unpublished data); thus, we infer that additional gene functions are needed, which are yet to be identified. It is hypothesized that HgcA and HgcB normally carry out a physiological role different from Hg methylation but that the unidentified role is not essential under laboratory culture conditions. The fact that some of the HgcA cap helix point mutations increased Hg methylation showed that HgcA, in its wild-type form, is nonoptimal for this activity. From this result, it is tempting to infer that the native function is something other than Hg methylation. The molecular mechanism of Hg(II) recognition and binding, the origin of the methyl group found in MeHg, the MeTr donating a methyl group to HgcA, and the electron donor for HgcB, believed to be essential for continued turnover, require further investigation. Identification of additional protein components participating in Hg methylation may also reveal any alternative physiological functions of HgcA and HgcB in anaerobic microbes.
ACKNOWLEDGMENTS
We thank Demian Riccardi and Jeremy C. Smith for suggesting the Cys93His mutant of HgcA and predicting its activity.
This work was supported by the U.S. Department of Energy (DOE) Office of Science, Biological and Environmental Research, Subsurface Biogeochemical Research (SBR) Program through grants DE SC0006809 and DE-FG02-07ER64396 and by subcontract number 40000099987 from the Oak Ridge National Laboratory (ORNL) Mercury Scientific Focus Area. ORNL is managed by UT-Battelle LLC for the U.S. DOE under contract number DE-AC05-00OR22725.
REFERENCES
- 1.Wood JM. 1974. Biological cycles for toxic elements in the environment. Science 183:1049–1052. doi: 10.1126/science.183.4129.1049. [DOI] [PubMed] [Google Scholar]
- 2.Driscoll CT, Mason RP, Chan HM, Jacob DJ, Pirrone N. 2013. Mercury as a global pollutant: sources, pathways, and effects. Environ Sci Technol 47:4967–4983. doi: 10.1021/es305071v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamborg CH, Hammerschmidt CR, Bowman KL, Swarr GJ, Munson KM, Ohnemus DC, Lam PJ, Heimburger L-E, Rijkenberg MJA, Saito MA. 2014. A global ocean inventory of anthropogenic mercury based on water column measurements. Nature 512:65–68. doi: 10.1038/nature13563. [DOI] [PubMed] [Google Scholar]
- 4.Hintelmann H. 2010. Organomercurials. Their formation and pathways in the environment. Met Ions Life Sci 7:365–401. doi: 10.1039/BK9781847551771-00365. [DOI] [PubMed] [Google Scholar]
- 5.Harris RC, Rudd JW, Amyot M, Babiarz CL, Beaty KG, Blanchfield PJ, Bodaly RA, Branfireun BA, Gilmour CC, Graydon JA, Heyes A, Hintelmann H, Hurley JP, Kelly CA, Krabbenhoft DP, Lindberg SE, Mason RP, Paterson MJ, Podemski CL, Robinson A, Sandilands KA, Southworth GR, St Louis VL, Tate MT. 2007. Whole-ecosystem study shows rapid fish-mercury response to changes in mercury deposition. Proc Natl Acad Sci U S A 104:16586–16591. doi: 10.1073/pnas.0704186104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vazquez M, Velez D, Devesa V. 2014. In vitro characterization of the intestinal absorption of methylmercury using a Caco-2 cell model. Chem Res Toxicol 27:254–264. doi: 10.1021/tx4003758. [DOI] [PubMed] [Google Scholar]
- 7.Gilmour CC, Henry EA, Mitchell R. 1992. Sulfate stimulation of mercury methylation in freshwater sediments. Environ Sci Technol 26:2281–2287. doi: 10.1021/es00035a029. [DOI] [Google Scholar]
- 8.Compeau GC, Bartha R. 1985. Sulfate-reducing bacteria: principal methylators of mercury in anoxic estuarine sediment. Appl Environ Microbiol 50:498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamelin S, Amyot M, Barkay T, Wang Y, Planas D. 2011. Methanogens: principal methylators of mercury in lake periphyton. Environ Sci Technol 45:7693–7700. doi: 10.1021/es2010072. [DOI] [PubMed] [Google Scholar]
- 10.Yu RQ, Reinfelder JR, Hines ME, Barkay T. 2013. Mercury methylation by the methanogen Methanospirillum hungatei. Appl Environ Microbiol 79:6325–6330. doi: 10.1128/AEM.01556-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilmour CC, Podar M, Bullock AL, Graham AM, Brown SD, Somenahally AC, Johs A, Hurt RA Jr, Bailey KL, Elias DA. 2013. Mercury methylation by novel microorganisms from new environments. Environ Sci Technol 47:11810–11820. doi: 10.1021/es403075t. [DOI] [PubMed] [Google Scholar]
- 12.Parks JM, Johs A, Podar M, Bridou R, Hurt RA Jr, Smith SD, Tomanicek SJ, Qian Y, Brown SD, Brandt CC, Palumbo AV, Smith JC, Wall JD, Elias DA, Liang L. 2013. The genetic basis for bacterial mercury methylation. Science 339:1332–1335. doi: 10.1126/science.1230667. [DOI] [PubMed] [Google Scholar]
- 13.Choi SC, Bartha R. 1993. Cobalamin-mediated mercury methylation by Desulfovibrio desulfuricans LS. Appl Environ Microbiol 59:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi SC, Chase T Jr, Bartha R. 1994. Enzymatic catalysis of mercury methylation by Desulfovibrio desulfuricans LS. Appl Environ Microbiol 60:1342–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi SC, Chase T, Bartha R. 1994. Metabolic pathways leading to mercury methylation in Desulfovibrio desulfuricans LS. Appl Environ Microbiol 60:4072–4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berman M, Chase T, Bartha R. 1990. Carbon flow in mercury biomethylation by Desulfovibrio desulfuricans. Appl Environ Microbiol 56:298–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goetzl S, Jeoung JH, Hennig SE, Dobbek H. 2011. Structural basis for electron and methyl-group transfer in a methyltransferase system operating in the reductive acetyl-CoA pathway. J Mol Biol 411:96–109. doi: 10.1016/j.jmb.2011.05.025. [DOI] [PubMed] [Google Scholar]
- 18.Svetlitchnaia T, Svetlitchnyi V, Meyer O, Dobbek H. 2006. Structural insights into methyltransfer reactions of a corrinoid iron-sulfur protein involved in acetyl-CoA synthesis. Proc Natl Acad Sci U S A 103:14331–14336. doi: 10.1073/pnas.0601420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bae HS, Dierberg FE, Ogram A. 2014. Syntrophs dominate sequences associated with the mercury methylation-related gene hgcA in the water conservation areas of the Florida Everglades. Appl Environ Microbiol 80:6517–6526. doi: 10.1128/AEM.01666-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu YR, Yu RQ, Zheng YM, He JZ. 2014. Analysis of the microbial community structure by monitoring an Hg methylation gene (hgcA) in paddy soils along an Hg gradient. Appl Environ Microbiol 80:2874–2879. doi: 10.1128/AEM.04225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaefer J, Kronberg RM, Morel F, Skyllberg U. 2014. Detection of a key Hg methylation gene, hgcA, in wetland soils. Environ Microbiol Rep 6:441–447. doi: 10.1111/1758-2229.12136. [DOI] [PubMed] [Google Scholar]
- 22.Ando N, Kung Y, Can M, Bender G, Ragsdale SW, Drennan CL. 2012. Transient B12-dependent methyltransferase complexes revealed by small-angle X-ray scattering. J Am Chem Soc 134:17945–17954. doi: 10.1021/ja3055782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ljungdahl LG, LeGall J, Lee JP. 1973. Isolation of a protein containing tightly bound 5-methoxybenzimidazolylcobamide (factor 3m) from Clostridium thermoaceticum. Biochemistry 12:1802–1808. doi: 10.1021/bi00733a022. [DOI] [PubMed] [Google Scholar]
- 24.Kung Y, Ando N, Doukov TI, Blasiak LC, Bender G, Seravalli J, Ragsdale SW, Drennan CL. 2012. Visualizing molecular juggling within a B12-dependent methyltransferase complex. Nature 484:265–269. doi: 10.1038/nature10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doukov T, Seravalli J, Stezowski JJ, Ragsdale SW. 2000. Crystal structure of a methyltetrahydrofolate- and corrinoid-dependent methyltransferase. Structure 8:817–830. doi: 10.1016/S0969-2126(00)00172-6. [DOI] [PubMed] [Google Scholar]
- 26.Ragsdale SW. 2008. Catalysis of methyl group transfers involving tetrahydrofolate and B12. Vitam Horm 79:293–324. doi: 10.1016/S0083-6729(08)00410-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banerjee R, Ragsdale SW. 2003. The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu Rev Biochem 72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 28.Hennig SE, Jeoung J-H, Goetzl S, Dobbek H. 2012. Redox-dependent complex formation by an ATP-dependent activator of the corrinoid/iron-sulfur protein. Proc Natl Acad Sci U S A 109:5235–5240. doi: 10.1073/pnas.1117126109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferguson T, Soares JA, Lienard T, Gottschalk G, Krzycki JA. 2009. RamA, a protein required for reductive activation of corrinoid-dependent methylamine methyltransferase reactions in methanogenic archaea. J Biol Chem 284:2285–2295. doi: 10.1074/jbc.M807392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou J, Riccardi D, Beste A, Smith JC, Parks JM. 2014. Mercury methylation by HgcA: theory supports carbanion transfer to Hg(II). Inorg Chem 53:772–777. doi: 10.1021/ic401992y. [DOI] [PubMed] [Google Scholar]
- 31.Bertini I, Donaire A, Feinberg BA, Luchinat C, Piccioli M, Yuan H. 1995. Solution structure of the oxidized 2[4Fe-4S] ferredoxin from Clostridium pasteurianum. Eur J Biochem 232:192–205. doi: 10.1111/j.1432-1033.1995.tb20799.x. [DOI] [PubMed] [Google Scholar]
- 32.Davy SL, Osborne MJ, Moore GR. 1998. Determination of the structure of oxidised Desulfovibrio africanus ferredoxin I by 1H NMR spectroscopy and comparison of its solution structure with its crystal structure. J Mol Biol 277:683–706. doi: 10.1006/jmbi.1998.1631. [DOI] [PubMed] [Google Scholar]
- 33.Bruschi M, Hatchikian EC. 1982. Non-heme iron proteins of Desulfovibrio: the primary structure of ferredoxin I from Desulfovibrio africanus. Biochimie 64:503–507. doi: 10.1016/S0300-9084(82)80166-1. [DOI] [PubMed] [Google Scholar]
- 34.Gilmour CC, Elias DA, Kucken AM, Brown SD, Palumbo AV, Schadt CW, Wall JD. 2011. Sulfate-reducing bacterium Desulfovibrio desulfuricans ND132 as a model for understanding bacterial mercury methylation. Appl Environ Microbiol 77:3938–3951. doi: 10.1128/AEM.02993-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zane GM, Yen H-CB, Wall JD. 2010. Effect of the deletion of qmoABC and the promoter-distal gene encoding a hypothetical protein on sulfate reduction in Desulfovibrio vulgaris Hildenborough. Appl Environ Microbiol 76:5500–5509. doi: 10.1128/AEM.00691-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li MZ, Elledge SJ. 2012. SLIC: a method for sequence- and ligation-independent cloning. Methods Mol Biol 852:51–59. doi: 10.1007/978-1-61779-564-0_5. [DOI] [PubMed] [Google Scholar]
- 37.Magis C, Taly JF, Bussotti G, Chang JM, Di Tommaso P, Erb I, Espinosa-Carrasco J, Notredame C. 2014. T-Coffee: tree-based consistency objective function for alignment evaluation. Methods Mol Biol 1079:117–129. doi: 10.1007/978-1-62703-646-7_7. [DOI] [PubMed] [Google Scholar]
- 38.Shapiro AL, Vinuela E, Maizel JV Jr. 1967. Molecular weight estimation of polypeptide chains by electrophoresis in SDS-polyacrylamide gels. Biochem Biophys Res Commun 28:815–820. doi: 10.1016/0006-291X(67)90391-9. [DOI] [PubMed] [Google Scholar]
- 39.Towbin H, Staehelin T, Gordon J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76:4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burnette WN. 1981. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem 112:195–203. [DOI] [PubMed] [Google Scholar]
- 41.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 42.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 43.Ragsdale SW, Lindahl PA, Munck E. 1987. Mossbauer, EPR, and optical studies of the corrinoid/iron-sulfur protein involved in the synthesis of acetyl coenzyme A by Clostridium thermoaceticum. J Biol Chem 262:14289–14297. [PubMed] [Google Scholar]
- 44.Morrison KL, Weiss GA. 2001. Combinatorial alanine-scanning. Curr Opin Chem Biol 5:302–307. doi: 10.1016/S1367-5931(00)00206-4. [DOI] [PubMed] [Google Scholar]
- 45.von Heijne G. 1991. Proline kinks in transmembrane alpha-helices. J Mol Biol 218:499–503. doi: 10.1016/0022-2836(91)90695-3. [DOI] [PubMed] [Google Scholar]
- 46.Nilsson I, Saaf A, Whitley P, Gafvelin G, Waller C, von Heijne G. 1998. Proline-induced disruption of a transmembrane alpha-helix in its natural environment. J Mol Biol 284:1165–1175. doi: 10.1006/jmbi.1998.2217. [DOI] [PubMed] [Google Scholar]
- 47.DeSilva TM, Veglia G, Porcelli F, Prantner AM, Opella SJ. 2002. Selectivity in heavy metal-binding to peptides and proteins. Biopolymers 64:189–197. doi: 10.1002/bip.10149. [DOI] [PubMed] [Google Scholar]
- 48.Carugo O, Cemazar M, Zahariev S, Hudaky I, Gaspari Z, Perczel A, Pongor S. 2003. Vicinal disulfide turns. Protein Eng 16:637–639. doi: 10.1093/protein/gzg088. [DOI] [PubMed] [Google Scholar]
- 49.Wouters MA, Fan SW, Haworth NL. 2010. Disulfides as redox switches: from molecular mechanisms to functional significance. Antioxid Redox Signal 12:53–91. doi: 10.1089/ars.2009.2510. [DOI] [PubMed] [Google Scholar]
- 50.Matthews RG. 2009. Cobalamin- and corrinoid-dependent enzymes. Met Ions Life Sci 6:53–114. doi: 10.1039/BK9781847559159-00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ludwig ML, Matthews RG. 1997. Structure-based perspectives on B12-dependent enzymes. Annu Rev Biochem 66:269–313. doi: 10.1146/annurev.biochem.66.1.269. [DOI] [PubMed] [Google Scholar]
- 52.Guimarães DH, Weber A, Klaiber I, Vogler B, Renz P. 1994. Guanylcobamide and hypoxanthylcobamide-corrinoids formed by Desulfovibrio vulgaris. Arch Microbiol 162:272–276. doi: 10.1007/BF00301850. [DOI] [Google Scholar]
- 53.Matthews RG. 2001. Cobalamin-dependent methyltransferases. Acc Chem Res 34:681–689. doi: 10.1021/ar0000051. [DOI] [PubMed] [Google Scholar]
- 54.Ferry JG. 1993. Fermentation of acetate, p 304–334. In Ferry JG. (ed), Methanogenesis: ecology, physiology, biochemistry & genetics. Chapman & Hall, New York, NY. [Google Scholar]
- 55.Iismaa SE, Vazquez AE, Jensen GM, Stephens PJ, Butt JN, Armstrong FA, Burgess BK. 1991. Site-directed mutagenesis of Azotobacter vinelandii ferredoxin I. Changes in [4Fe-4S] cluster reduction potential and reactivity. J Biol Chem 266:21563–21571. [PubMed] [Google Scholar]
- 56.Lafrance-Vanasse J, Lefebvre M, Di Lello P, Sygusch J, Omichinski JG. 2009. Crystal structures of the organomercurial lyase MerB in its free and mercury-bound forms: insights into the mechanism of methylmercury degradation. J Biol Chem 284:938–944. doi: 10.1074/jbc.M807143200. [DOI] [PubMed] [Google Scholar]
- 57.Pitts KE, Summers AO. 2002. The roles of thiols in the bacterial organomercurial lyase (MerB). Biochemistry 41:10287–10296. doi: 10.1021/bi0259148. [DOI] [PubMed] [Google Scholar]
- 58.Sorokin DY, Tourova TP, Mussmann M, Muyzer G. 2008. Dethiobacter alkaliphilus gen. nov. sp. nov., and Desulfurivibrio alkaliphilus gen. nov. sp. nov.: two novel representatives of reductive sulfur cycle from soda lakes. Extremophiles 12:431–439. doi: 10.1007/s00792-008-0148-8. [DOI] [PubMed] [Google Scholar]
- 59.Hagerhall C, Hederstedt L. 1996. A structural model for the membrane-integral domain of succinate:quinone oxidoreductases. FEBS Lett 389:25–31. doi: 10.1016/0014-5793(96)00529-7. [DOI] [PubMed] [Google Scholar]
- 60.Lin H, Hurt R Jr, Johs A, Parks JM, Morrell-Falvey JL, Liang L, Elias D, Gu B. 2014. Unexpected effects of gene deletion on mercury interactions with the methylation-deficient mutant ΔhgcAB. Environ Sci Technol Lett 1:271–276. doi: 10.1021/ez500107r. [DOI] [Google Scholar]
- 61.Thomsen MC, Nielsen M. 2012. Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res 40:W281–W287. doi: 10.1093/nar/gks469. [DOI] [PMC free article] [PubMed] [Google Scholar]