

Abstract
Little is known about the molecular and physiological function of co-occurring microbes within freshwater cyanobacterial harmful algal blooms (cHABs). To address this, community metatranscriptomes collected from the western basin of Lake Erie during August 2012 were examined. Using sequence data, we tested the hypothesis that the activity of the microbial community members is independent of community structure. Predicted metabolic and physiological functional profiles from spatially distinct metatranscriptomes were determined to be ≥90% similar between sites. Targeted analysis of Microcystis aeruginosa, the historical causative agent of cyanobacterial harmful algal blooms over the past ∼20 years, as well as analysis of Planktothrix agardhii and Anabaena cylindrica, revealed ongoing transcription of genes involved in microcystin toxin synthesis as well as the acquisition of both nitrogen and phosphorus, nutrients often implicated as independent bottom-up drivers of eutrophication in aquatic systems. Transcription of genes involved in carbon dioxide (CO2) concentration and metabolism also provided support for the alternate hypothesis that high-pH conditions and dense algal biomass result in CO2-limiting conditions that further favor cyanobacterial dominance. Additionally, the presence of Microcystis-specific cyanophage sequences provided preliminary evidence of possible top-down virus-mediated control of cHAB populations. Overall, these data provide insight into the complex series of constraints associated with Microcystis blooms that dominate the western basin of Lake Erie during summer months, demonstrating that multiple environmental factors work to shape the microbial community.
INTRODUCTION
Freshwater ecosystems are considered among the most endangered in the biosphere (1). One threat to fresh waters around the world is the now nearly annual occurrence of blooms of toxic cyanobacteria. Molecular signatures of cyanobacterial harmful algal blooms (cHABs) have been identified on all seven continents, and these blooms have been occurring with increased frequency and duration in recent years (2–4). The accumulation of bloom biomass has been associated with fish, avian, and mammal intoxication (5, 6); the formation of hypoxic zones (7); the production of taste and odor compounds (8, 9); and even human liver failure in extreme cases (10).
The Laurentian Great Lakes are an important freshwater resource, holding ∼18% of the world's potable water and ∼84% of the surface waters in North America (11). cHABs have had a persistent presence in Lake Erie and other Laurentian Great Lakes for several decades (3, 12, 13). Nuisance biomass and toxin production associated with cHABs in the Great Lakes have had a detrimental effect not only on ecosystem health but also on the economic health of the surrounding communities; indeed, in August 2014, a bloom event led to the shutdown of the water supply for some ∼500,000 residents in the region of the city of Toledo, OH (14). To date, research efforts have broadly identified nutrient inputs and temperature as contributing factors in the development of such blooms, but little focus, due primarily to methodological limitations, has been allocated to the study of the cellular physiology and metabolism of the entire bloom community. Not only do bloom-forming organisms such as Microcystis aeruginosa live in competition with other phytoplankton species, they also live in concert with heterotrophic bacteria, which can attach to the mucilaginous matrix produced by M. aeruginosa colonies (15, 16). There is a dearth of information concerning the functional role that co-occurring bacteria may play in bloom dynamics, although recent studies suggest that heterotrophs may play an important role in the metabolism of some nutrients (17, 18).
As metagenomic surveys have provided new insight into the dynamics of microbial communities, a growing number have suggested that microbial community function is independent of the identity of the member organisms, including one study of a Lake Erie cHAB community (18–20). These studies, however, allow only for inference about community functional potential, as they are based on the analysis of DNA. Here we applied RNA sequencing to multiple bloom sites across Lake Erie to test the hypothesis that cHAB community function is independent of taxonomic identity. We also compared the transcript profiles of native Microcystis, Planktothrix, and Anabaena populations to those of well-characterized laboratory isolates (M. aeruginosa NIES 843, P. agardhii NIVA CYA 126/8, and A. cylindrica PCC 7122) to identify physiological differences among important toxic cHAB organisms and possible important ecological controls across the western basin of Lake Erie during the 2012 summer bloom season.
MATERIALS AND METHODS
Sample collection and survey of environmental conditions.
Samples were collected from three survey stations in the western basin of Lake Erie during August 2012 (Table 1) from onboard the C.C.G.S. Limnos. All samples were collected during daylight hours and within a 24-h period. To concentrate the cyanobacterial community, the sample was collected by using a Nytex plankton net with a 20-μm pore size. Samples from multiple net tows at each site were homogenized and then divided into 4-ml technical triplicates at each station and flash-frozen in liquid nitrogen for storage and transport to the laboratory. Samples were stored at −80°C prior to processing.
TABLE 1.
Station locations and conditions for sample collection in August 2012
| Parameter | Value for indicated stationb |
||
|---|---|---|---|
| 973 | 882 | 1163 | |
| Latitude (N) | 41°47′30″ | 41°46′00″ | 41°28′10″ |
| Longitude (W) | 83°19′58″ | 83°18′30″ | 82°43′00″ |
| Mo/day, time of sample collectiona | 08/15, 14:45 | 08/15, 11:25 | 08/14, 19:10 |
| Surface temp (°C) | 23.7 | 23.4 | 22.8 |
| pH | 8.51 | 8.32 | 8.06 |
| Microcystin concn (μg/liter) | 0.37 | 0.11 | 0.61 |
| Anatoxin-a concn (μg/liter) | None detected | None detected | 0.77 |
| Cylindrospermopsin concn (μg/liter) | None detected | None detected | None detected |
| Mean chlorophyll a concn (μg/liter) ± SD | 25.4 ± 0.3 | 16.1 ± 0.1 | 15.6 ± 0.3 |
| Cyanobacterial counts (no. of cells/ml) | |||
| Microcystis spp. | 25,152 | 12,310 | 4,027 |
| Planktothrix spp. | ND | 6,839 | 267,626 |
| Anabaena-Dolichospermum spp. | 63,829 | 15,632 | 8,359 |
| Cylindrospermopsis-Raphidiopsis spp. | ND | ND | 15,121 |
Times are given as Eastern Standard Time.
ND, not detected.
Chlorophyll a (Chl a) concentrations for each field station sample were determined by using the nonacidification method (21, 22) and served as a proxy for total phytoplankton biomass. A 50-ml volume of surface water from each station was filtered through 0.2-μm-nominal-pore-size polycarbonate membrane filters (25-mm diameter; Millipore) and stored in a 2.0-ml Cryovial (Corning, NY) at −80°C until analysis. Chl a concentrations were determined after extraction into 90% acetone overnight (24 h) at −20°C and measured by using a calibrated Turner Designs model 10-AU fluorometer (Turner Designs, Sunnyvale, CA, USA). Onboard ship Environment Canada personnel provided pH and surface temperature measurements for each station, determined at the time of sampling. Toxin analyses were completed as previously described (23).
Phytoplankton enumeration and identification in environmental samples were performed on Lugol's iodine-preserved samples and based upon microscopic morphometrics (24, 25). A 50-ml volume of each sample was preserved by adding 0.5 ml of Lugol's iodine before storage at 4°C. Fixed samples were analyzed on a Micromaster light microscope (ThermoFisher) using a 1-ml Sedgwick-Rafter counting slide; a total of 40 fields were visualized for each sample. Due to difficulties in sometimes distinguishing cellular morphology, Anabaena-Dolichospermum and Cylindrospermopsis-Raphidiopsis populations are reported in groups.
Nucleic acid extraction, sequencing, and processing.
Total RNA was extracted with the MoBio RNA PowerSoil Total RNA Isolation kit (MoBio, Carlsbad, CA, USA). To remove genomic DNA contamination, samples were treated with Ambion Turbo DNA Free (Ambion, Grand Island, NY, USA). Samples were confirmed to be free of bacterial DNA by PCR using primers 27f and 1522r (26). The absence of amplification, as assessed by gel electrophoresis, was considered the metric that the sample was DNA free. Total RNA concentrations and quality were assessed spectrophotometrically by using a NanoDrop 1000 instrument. rRNA reduction was done by using the Epicentre Ribo-Zero rRNA removal kit (Epicentre, Madison, WI, USA). Secondary quality analysis and sequencing were done on the Illumina HiSeq platform for 50-bp paired-end sequencing by the HudsonAlpha Genomic Services Laboratory (Huntsville, AL, USA).
Bioinformatic and statistical analyses.
Sequences were imported into CLC Genomics Workbench version 7.5 (CLC Bio, Cambridge, MA, USA), with failed reads (based on default settings for quality scores) removed. To better resolve putative transcript function, reads were assembled de novo into contigs in CLC Genomics Workbench with a minimum contig length of 200 bp, with paired distances taken into account. To assess the degree to which contigs represented individual small reads, these 50-bp reads were subsequently mapped back to assembled contigs with default parameters in CLC Genomics Workbench. For specific analyses of cyanobacterial populations, RNA sequencing data were mapped to the chromosomal sequences of M. aeruginosa NIES 843 (GenBank accession number NC_010296.1), Planktothrix agardhii NIVA CYA 126/8 (accession number CM002803.1), and Anabaena cylindrica PCC 7122 (accession number NC_019771.1) (27). Default mapping stringency parameters were increased to a mismatch cost of 3, a length fraction of 0.9, and a similarity fraction of 0.9. Expression values were normalized and calculated as total counts per million (TCPM) by using the CLC Genomics Workbench Transcriptomics module. All reads which did not map to a cyanobacterial genome were mapped to the genome of Microcystis phage Ma-LMM01 (GenBank accession number NC_008562.1) using default mapping parameters in CLC Genomics Workbench and normalized by the number of input sequences (28).
Taxonomic identification of community members was performed by using the MetaPhlAn metagenomic profiler with the “Very Sensitive Local” option (29). Functional annotation of contigs was performed with the MG-RAST metagenomics analysis server (30, 31). Assemblies were categorized by function using the Subsystems (SEED) database with default settings (30, 32). Statistical comparisons of individual normalized gene expression (TCPM) values were performed using the CLC Genomics Workbench Transcriptomics module with Baggerly's test, and P values were corrected for the false discovery rate (FDR) (33).
Nucleotide sequence accession numbers.
Contig sequences were deposited in MG-RAST and can be found under project identification number 8691 (https://metagenomics.anl.gov/?page=MetagenomeProject&project=8691) for analysis and/or download; individual assembly identification numbers are listed in Table S1 in the supplemental material. Raw sequences analyzed in this project are available through the NCBI Sequence Read Archive under BioProject no. PRJNA277855.
RESULTS
Sample collection and sequencing output.
Water samples were collected from three sites in Lake Erie during August 2012 (Table 1). The stations are historical Environment Canada survey sites, denoted station 973 (located in open waters of the western basin of the lake), 882 (located in the western basin at the outflow of the Maumee River), and 1163 (located within Sandusky Bay, OH). Chlorophyll a levels exceeded the World Health Organization (WHO)-recommended limit of 10 μg/liter at each station. Microcystis cells (4,027 to 25,152 cells/ml) and Anabaena-Dolichospermum cells (8,359 to 15,673 cells/ml) were present and abundant at all stations, although Planktothrix populations dominated at Sandusky Bay (station 1163), with >150,000 cells/ml (Table 1).
RNA extraction yielded averages of 263.3 ng/μl (standard error [SE], ±51.1 ng/μl), 59.68 ng/μl (SE, ±21.0 ng/μl), and 163.7 ng/μl (SE, ±12.5 ng/μl) of total RNA for stations 973, 882, and 1163, respectively. Reads were filtered based on quality upon import into CLC Genomics Workbench, with failed reads removed based on default import parameters. Metatranscriptome libraries were sequenced in triplicate, resulting in a total of 384 million reads after quality control (QC) filtration. On average, 94.2% of reads passed QC for each library (see Table S1 in the supplemental material). The post-QC phred score was on average 37.15, and the percentage of ambiguous bases in each read was on average 0.057%. Each library was comprised of an average of 42.7 million, 50-bp paired-end sequences. Short-read paired-end (50-bp) libraries assembled into an average of ∼9,700 contigs (>200 bp) (see Table S1 in the supplemental material).
Identification of the active Lake Erie bloom community structure from metatranscriptomes.
A total of 32 bacterial and archaeal phyla were detected, with bacterial RNA representing >98% of all sequences (Fig. 1). Community analysis of short-read libraries showed a predominance of Cyanobacteria in the samples collected from all three sites: >88% of bacterial reads at stations 973 and 1163 were identified as Cyanobacteria (Fig. 1) and at the phylum level were similar in composition to that of a previously reported Lake Erie cyanobacterial bloom metagenome (18). The bacterial community structure at station 882 was more varied, with Cyanobacteria comprising ∼42% of the community and Proteobacteria comprising ∼35%. An examination of the cyanobacterial sequences revealed differences in the transcriptionally active cyanobacterial community structures among the three sites surveyed (Fig. 1). At the order level, Nostocales (∼76%) (i.e., Anabaena, Nostoc, and Cylindrospermopsis) were dominant at station 973, and Oscillatoriales (∼75%) (i.e., Planktothrix, Oscillatoria, and Lyngbya) were dominant at station 1163. Chroococcales (∼15%) (i.e., Microcystis), Nostocales (∼15%), and Oscillatoriales (∼10%) were evenly represented at station 882.
FIG 1.

Overview of community makeup based on transcript abundance. Bar graphs show the metatranscriptome community structure of the particle-associated (>20-μm) bacterial population at the phylum level. Pie charts show the order-level community structure of the cyanobacterial population associated with each station. N, Nostocales; O, Oscillatoriales; C, Chroococcales; S, Synechococcales; G, Gloeobacterales. All values reported are means for triplicate libraries.
Active biological functions of the Lake Erie bloom communities.
In contrast to differences in community structure, functional profiles of assembled community sequences were similar across all sites (Fig. 2). The population at station 973 had reductions (<4% of annotated contigs) in “photosynthesis,” “protein metabolism,” and “respiration” SEED categories and increases in the “amino acid metabolism” and “miscellaneous functions” categories in comparison to those of stations 882 and 1163 (Fig. 2). Multidimensional scaling (MDS) analysis of Bray-Curtis similarity among the three sites confirms this observed functional similarity among station populations, with all samples clustering within 75% similarity to one another (Fig. 3). Triplicates clustered more closely to each other than between sites (95%); however, stations 882 and 1163 also clustered closely (90%), suggesting stronger functional similarity at these two sites (Fig. 3) despite the differences in community structure (Fig. 1). To verify that functional profiles generated from assembled contigs were representative of short-read libraries, 50-bp sequences were recruited back to contigs, with an average of 69.9% (±10.1%) of short reads mapping to assembled contigs (see Table S1 in the supplemental material).
FIG 2.

Community functional analysis of each library assembly. Functional annotation is based on a Subsystems (SEED) database analysis performed via the MG-RAST server. To demonstrate reproducibility, individual replicates are shown separately.
FIG 3.
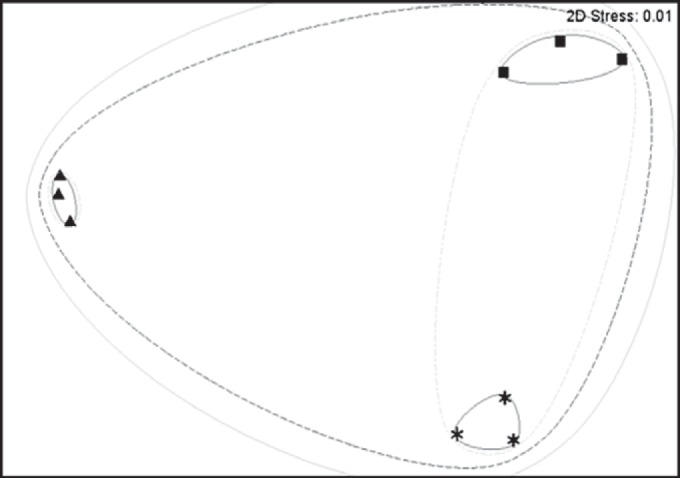
MDS analysis of a Bray-Curtis similarity matrix of functional assembly profiles for each station. Triangles represent station 973, squares represent station 882, and asterisks represent station 1163. The outer continuous gray line indicates 50% similarity, the outer dashed black line represents 75% similarity, the inner dashed gray lines indicate 90% similarity, and the inner solid gray lines represent 95% similarity between samples. 2D, two dimensional.
Cyanobacterial populations.
Members of the order Chroococcales, including Microcystis spp., comprised between 4.2% (±0.1%) and 15.1% (± 1.8%) of the total mRNA reads from Lake Erie during our August 2012 survey (Fig. 1). Among these, 0.8% to 7.3% of total sequence reads recruited nonredundantly to the genome of M. aeruginosa NIES 843, with wide variation among stations (Fig. 4A). On average, 68.5% (±1.5%) of genes within the NIES 843 genome were transcribed at station 973. Transcription of 64.8% (±1.4%) and 47.7% (±1.2%) of the NIES 843 genome was detected at stations 882 and 1163 (Fig. 4B). Members of the order Nostocales, including Anabaena spp., were dominant at station 973 (Fig. 1), with an average of 12.8% of all sequences recruiting to the genome of A. cylindrica PCC 7122, representing 22.0% of the genes in that genome (Fig. 4). This value was substantially reduced at stations 882 and 1163, with only 1.4% and 0.6% of reads, respectively, recruiting to the Anabaena genome (Fig. 4A). Oscillatoriales comprised a relatively minor component of the active communities at stations 973 and 882 but were dominant at station 1163, representing >75% of the total community mRNA (Fig. 1). This corresponded to an average of 23.5% of reads from station 1163 recruiting to the genome of P. agardhii NIVA CYA 126/8, with 98.5% of genes being transcribed at that station (Fig. 4).
FIG 4.

Summary of cyanobacterial mRNA profiles from August 2012 across the western basin of Lake Erie. (A) Mean percentages of individual sequence libraries that recruited nonredundantly to the genomes of M. aeruginosa NIES 843 (black), P. agardhii NIVA CYA 126/8 (white), and A. cylindrica PCC 7122 (gray). (B) Mean percentages of chromosomal genes for which transcripts were detected in each sequence library at each station for the genomes of M. aeruginosa NIES 843 (black), P. agardhii NIVA CYA 126/8 (white), and A. cylindrica PCC 7122 (gray). Bars represent standard deviations from triplicate libraries.
To determine which members of the cyanobacterial population may have contributed to microcystin toxin production detected at each station (Table 1), transcription of the M. aeruginosa NIES 843 and P. agardhii NIVA CYA 126/8 mcy toxin gene cassettes was assessed. mcyABCD and mcyG were transcribed by M. aeruginosa at stations 973 and 882, with more limited expression patterns at station 1163 (Fig. 5A). Transcription of all genes in the P. agardhii mcy cassette was detected at station 1163, corresponding to the genome-wide expression patterns detected here (Fig. 5B).
FIG 5.
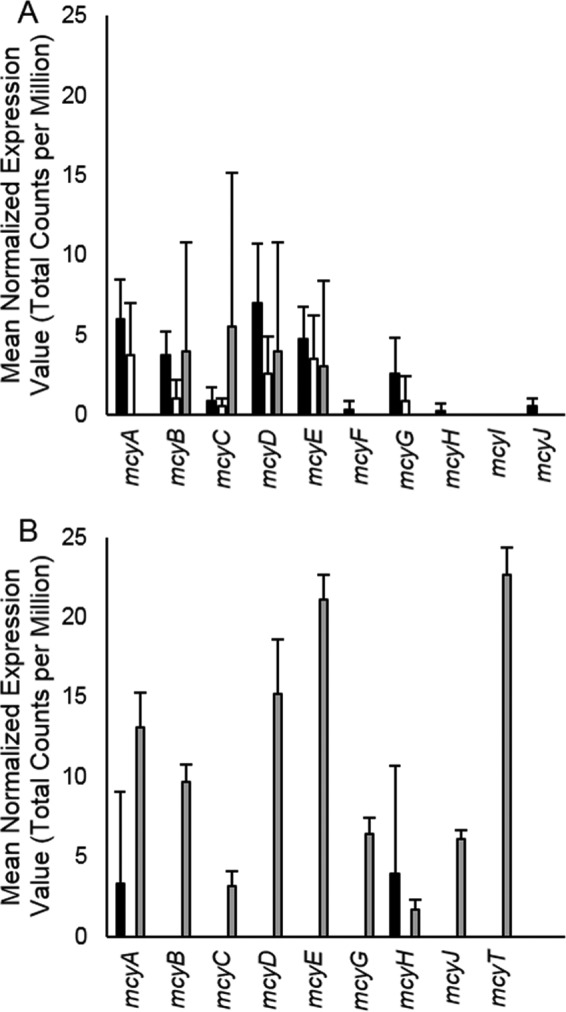
Calculated expression values (TCPM) for microcystin toxin synthesis genes. (A) mcyA to mcyJ mean gene expression values for M. aeruginosa NIES 843 at stations 973 (black), 882 (white), and 1163 (gray). (B) mcyA to mcyJ and mcyT mean gene expression values for P. agardhii NIVA CYA 126/8 at stations 973 (black), 882 (white), and 1163 (gray). Bars represent standard deviations from triplicate libraries.
Given the importance of nutrient abatement programs in managing the formation and persistence of cHABs (3, 35), we also examined the expression profiles of several key genes involved in nitrogen metabolism and transport for Lake Erie populations of M. aeruginosa NIES 843. Twenty-six genes representing metabolism and transport of ammonia (glnA, glnN, MAE_12590, MAE_17690, and MAE_40010 to MAE_40020), nitrate and nitrite (narB, nirA, and ntcABCD), and urea (ureABCDEFG and MAE_06180 to MAE_06220) were compared among sites to identify station-specific patterns in nitrogen metabolism (Fig. 6). The transcription levels of genes involved in nitrate/nitrite metabolism were significantly higher (P < 0.05) at station 973 than at station 882, including narB, nirA, ntcC, and the nitrate transport protein gene MAE_14800 (Fig. 6A). The transcription levels of genes involved in the transport of ammonia (MAE_40010), nitrate (ntcB and ntcC), and urea (MAE_06180 and MAE_06210) were significantly higher (P < 0.05) for Microcystis cells at station 1163 than for those at station 973 or 882 (Fig. 6B and C). Transcription of the global nitrogen regulator ntcA was significantly reduced at station 882 compared to the other sites in the western basin.
FIG 6.

Volcano plots depicting fold changes and P values (corrected for false discovery rate [FDR]) for statistical comparisons among all stations for M. aeruginosa NIES 843 genes involved in nitrogen metabolism and acquisition. Genes surveyed include those involved in ammonia (glnA, glnN, MAE_125900, MAE_17690, MAE_40010, and MAE_40020) (red), nitrate/nitrite (MAE_14800, narB, nirA, and ntcBCD) (blue), and urea (MAE_06180 to MAE_06220 and ureA to ureG) (green) pathways and the global nitrogen regulator ntcA (purple). Black points represent those genes that showed no significant difference (P > 0.05) in expression values between stations. Colored points represent those genes whose expression values were significantly different (P < 0.05) between stations 882 and 973 (A), 1163 and 973 (B), and 1163 and 882 (C).
In addition to nitrogen metabolism, gene markers for inorganic carbon uptake metabolism were also examined for M. aeruginosa NIES 843 (Fig. 7). M. aeruginosa populations at stations 973 and 1163 exhibited significantly higher (P < 0.01) transcription levels of the carbon-concentrating-mechanism genes ccmA, ccmK2, ccmL, ccmM, and ccmN than did the M. aeruginosa population at station 882 (Fig. 7). Transcription levels of rbcL (ribulose bisphosphate carboxylase [RuBisCO] large subunit), rbcS (RuBisCO small subunit), and rbcX (RuBisCO chaperonin) were also significantly (P < 0.01) lower at station 882 than at station 973 or 1163 (Fig. 7).
FIG 7.

Comparison of M. aeruginosa NIES 843 transcriptional patterns for genes involved in inorganic carbon acquisition across the western basin of Lake Erie. ccmA to ccmO encode carbon-concentrating-mechanism proteins. rbcL and rbcS encode the large and small subunits of RuBisCO, respectively, and rbcX encodes a RuBisCO chaperonin. Black points represent those genes whose expression levels are not significantly different (P > 0.05) between stations. Gray points are ccm genes which have significantly different (P < 0.05) expression levels between stations, and white points represent rbc genes which have significantly different (P < 0.05) expression levels between stations. Points in the circle all represent rbcL. Log10 mean expression values from triplicate libraries for each station are reported.
DISCUSSION
Application of deep sequencing technologies to the study of microbial community structure and function has resulted in the observation that variability in microbial community composition does not necessarily reflect differences in the functional potential of that community (19, 36, 37). Here we tested this hypothesis using RNA sequencing to identify the transcriptionally active microbial community members and the function of the genes being transcribed. Our data suggest that the microbial community structure of Lake Erie blooms varies across the western basin (Fig. 1), but the biological functions of these communities remain relatively static (Fig. 2 and 3). To better resolve this issue, we examined historically cited factors that shape cyanobacterial communities (nutrient cycling) as well as more recently hypothesized constraints (cyanophage activity). We couch our observations within the confines of a community-based sequencing approach to provide new insight into the complex nature of microbial populations and cyanobacterial blooms.
Lake Erie Microcystis blooms in particular have garnered much attention in recent months, with the August 2014 microcystin event that rendered the city of Toledo, OH, without potable water for several days (38). In our 2012 survey, microcystin concentrations were below the World Health Organization's drinking water limit of 1 μg/liter; however, this toxin was detected at all three sites (Table 1). We thus performed a targeted analysis of Microcystis gene transcription using the genome of M. aeruginosa NIES 843 as the model strain, as this is the only strain for which a fully closed Microcystis genome is available (27). Analysis of genes involved in microcystin toxin synthesis (mcy cassette) revealed transcription patterns suggesting that cyanobacterial populations responsible for toxin production differed across the western basin. At station 973, transcription of the mcy genes was performed predominately by M. aeruginosa (Fig. 5A). At station 1163, however, mcy transcripts were detected primarily from P. agardhii (Fig. 5B). The neurotoxin anatoxin-a was also detected at station 1163. The presence of anatoxin-a has been documented in several of the nearshore embayments of Lake Ontario and in Sandusky Bay in prior surveys (39). However, at this time, we do not know enough about the biosynthetic pathway to identify the causative organism. While Microcystis and Oscillatoria species have been reported to produce anatoxin-a, it is likely that the producer in Sandusky Bay is a member of the Anabaena-Dolichospermum clade (40). Cylindrospermopsis was also detected at station 1163 (and in visual examinations of the net tow samples), but the toxins cyclindrospermopsis and deoxycylindrospermopsis were not, suggesting that these species may be a nontoxic variant.
Our analyses also revealed a potentially important role for Microcystis spp. in nutrient cycling within the bloom-associated community (>20.0 μm), as genes involved in nitrogen and inorganic carbon acquisition and metabolism were being differentially transcribed across the western basin at the time of sample collection (Fig. 6 and 7). The transcriptional patterns of specific genes involved in N and P metabolism provide insight into the physiology and contribution of Microcystis to nutrient cycling in Lake Erie during August 2012. Transcripts for genes involved in nitrate and nitrite metabolism and acquisition (narB, nirA, MAE_14800, ntcB, and ntcC) were significantly increased at stations 973 and 1163 over those at station 882 (Fig. 5A and C). The global nitrogen regulator ntcA was also more highly transcribed at station 973 and 1163, as were genes involved in ammonia metabolism (glnA) and urea metabolism and acquisition (ureB, MAE_06180, MAE_06210, and MAE_06220) (Fig. 5A and C). Increased mRNA production of this suite of genes suggests that Microcystis populations at stations 973 and 1163 were more nitrogen stressed than those at station 882 during August 2012. Many of these same genes had significantly increased transcription at station 1163 compared to station 973 (Fig. 5B), suggesting that cells at station 1163 were experiencing the most nitrogen stress of the stations surveyed in this study. As the cyanobacterial population was predominantly composed of Planktothrix spp. at the time of sampling, this may perhaps indicate increased competition for N resources for minority members of the population at this station. In contrast, alkaline phosphatase (MAE_16640), a classical marker for P-limited plankton growth, exhibited more limited transcription by Microcystis populations at stations 882 and 1163, with significantly increased levels at station 973, suggesting potential P limitation at this site. Synthesis of N and P gene transcription patterns indicates that nitrogen and phosphorus limitation may have been variable across the western basin of Lake Erie in August 2012. To determine whether variable transcription patterns of genes involved in nitrogen metabolism by M. aeruginosa were indicative of site-specific N or P limitation across the western basin, we examined expression patterns of nifH, a common marker for nitrogen fixation, for A. cylindrica PCC 7122. While there was no significant expression level difference among stations, it was transcribed at all three sites sampled. Additionally, concurrent microcosm efforts have demonstrated that primary producers were most likely N limited during August 2012, although variability in sampling locations prevents direct comparisons of our study sites (41).
Far fewer studies have been dedicated to resolving the role of CO2 availability relative to N and P dynamics in cHAB formation and persistence. We detected transcripts for 12 genes involved in carbon concentration at each of the three stations surveyed (Fig. 7). Their increased transcription levels at stations 973 and 1163 over those at station 882 suggest that Microcystis cells were investing significantly more resources at these stations to acquire CO2, a potential indicator of CO2 limitation at these sites, than at station 882. Enhanced photosynthesis associated with dense algal blooms is commonly accompanied by a increase in pH, creating the potential for growth limitation of phototrophs by dissolved inorganic carbon supplies, and indeed, all three sites surveyed had a pH in excess of 8 (common to Lake Erie) at the time of sampling (42, 43). In such situations, organisms with efficient carbon-concentrating mechanisms are thought to have a competitive advantage (44). The evidence presented here supports the hypothesis that these conditions exist in the eutrophic western basin of Lake Erie, at least for Microcystis populations. Because little is known regarding the role of CO2 limitation in bloom community speciation and function (45), this hypothesis warrants further testing in the field. There have been recent calls in the literature for further study of this phenomenon, especially as atmospheric CO2 levels are projected to increase over the next century (46).
While significant attention has been directed toward understanding bottom-up controls of cHABs (i.e., nutrients and temperature), top-down controls such as grazing and phage infection can also play an important role in bloom dynamics (47). To quantify active phage infections, we recruited short reads unmapped to the NIES 843, NIVA CYA 126/8, or PCC 7122 genome to the genome of Microcystis phage Ma-LMM01 (28). The recruitments provided ∼9,800 to 11,000 reads mapped to the phage genome, suggesting that despite sampling of only the bloom-associated (>20-μm size class) population, transcriptionally active phages were detectable. While additional study is required to resolve the importance of phages in the natural control of cHABs in large lake systems, our observations provide preliminary evidence for transcriptionally active Microcystis phages within Lake Erie bloom communities. Furthermore, these observations suggest that extraction of virus gene sequences from particle-associated transcriptomes may be a useful way to better resolve the activity of DNA viruses in natural systems.
In addition to the known methodological constraints on environmental metatranscriptomes (for a current discussion, see reference 48), field sampling also has inherent limitations that must be considered (48). With that, an important caveat to our results concerns the difference in the time of sample collection. It is well documented that the expression of genes by cyanobacteria (49, 50) and specifically Microcystis (51) is impacted by the time of day. However, it should also be noted that in previously reported studies of cyanobacterial expression over the light-dark cycle, the most extreme differences occurred at the dark-light transitions (49, 51). The samples examined here were collected during the daylight hours prior to sunset to reduce the confounding influence of such cyclical transcription patterns. Additional consideration should also be given to the “snapshot” nature of such survey sampling. Collection of samples at a single time point during a bloom event that extends over months captures only a small subset of bloom populations. While sampling would ideally have occurred simultaneously and continuously across the summer season at all sites, due to the inherent limitations of field sampling efforts, the difference in the time of day and the “snapshot” nature of collection must be considered when interpreting differences between transcript profiles.
Conclusions.
Application of RNA sequencing technologies to the study of cHAB-associated microbial communities provides robust evidence that microbial community function can occur independent of taxonomic structure. Despite variable taxonomies within these bacterial communities, functional profiles were 90% similar across the western basin of Lake Erie (Fig. 1 to 3). M. aeruginosa, an important toxin producer in the lake, was an active member of all sites surveyed, as transcripts for genes associated with nitrogen metabolism, including urea degradation, and phosphorus metabolism were identified at all stations. Specific transcriptional patterns of these genes varied between stations, suggesting differences in N and P availability across the western basin of Lake Erie during August 2012. Of note, transcriptional profiles also indicate that potential CO2 limitation was occurring, particularly at stations 973 and 1163. As CO2 levels climb toward projected levels, the role of CO2 in driving phototroph community speciation may become increasingly important and warrants further study (46). In conjunction with Microcystis, other toxic cyanobacteria (Anabaena and Planktothrix) were detected across the western basin. In fact, Planktothrix appeared to dominate at station 1163, located in Sandusky Bay. Transcriptional evidence for microcystin toxin production by mixed populations of both Microcystis and Planktothrix occurred at all stations, suggesting that a single organism is not solely responsible for the toxic nature of these blooms. Additionally, initial transcriptional evidence of an active infection by a Microcystis cyanophage was identified as a potential top-down control. As it is unlikely that infection by only a single cyanophage occurred throughout the population, this observation highlights the potential importance of top-down in situ virus controls in cHAB dynamics. Taken together, this study provides new insight into the multiple possible constraints on cHAB communities across the western basin of Lake Erie under bloom conditions. The data provide a foundation for biomolecular modeling efforts that may ultimately build on the existence of “bloom warning” models to couple environmental conditions with cellular biochemistry across populations, thus including conditions that favor the production of microcystin as well as other cyanotoxins (52). This new information may be an important consideration for management practices, as a key factor in the construction of robust management strategies is a scientifically based understanding of the function of microbial populations which make up cHAB communities.
Supplementary Material
ACKNOWLEDGMENTS
We thank the captain and crew of the C.C.G.S. Limnos, the shipboard technical operations technicians, as well as Shawn Levy and colleagues at HudsonAlpha. We also thank Helena Pound and Katherine Perri for their assistance in the field. We also thank the three anonymous reviewers for their constructive insight.
This project was supported by grants from the National Science Foundation (IOS-0841918, DEB-1240870, and CBET-1230543) and a UT/ORNL Science Alliance JDRD award to S.W.W. Support was provided to M.M.S. via a Wallace-Dean fellowship from The University of Tennessee.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.04101-14.
REFERENCES
- 1.Dudgeon D, Arthington AH, Gessner MO, Kawabata Z-I, Knowler DJ, Lévêque C, Naiman RJ, Prieur-Richard A-H, Soto D, Stiassny MLJ, Sullivan CA. 2006. Freshwater biodiversity: importance, threats, status and conservation challenges. Biol Rev 81:163–182. doi: 10.1017/S1464793105006950. [DOI] [PubMed] [Google Scholar]
- 2.Michalak AM, Anderson EJ, Beletsky D, Boland S, Bosch NS, Bridgeman TB, Chaffin JD, Cho K, Confesor R, Daloğlu I, DePinto JV, Evans MA, Fahnenstiel GL, He L, Ho JC, Jenkins L, Johengen TH, Kuo KC, LaPorte E, Liu X, McWilliams MR, Moore MR, Posselt DJ, Richards RP, Scavia D, Steiner AL, Verhamme E, Wright DM, Zagorski MA. 2013. Record-setting algal bloom in Lake Erie caused by agricultural and meteorological trends consistent with expected future conditions. Proc Natl Acad Sci U S A 110:6448–6452. doi: 10.1073/pnas.1216006110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steffen MM, Belisle BS, Watson SB, Boyer GL, Wilhelm SW. 2014. Status, causes and controls of cyanobacterial blooms in Lake Erie. J Great Lakes Res 40:215–225. doi: 10.1016/j.jglr.2013.12.012. [DOI] [Google Scholar]
- 4.Hitzfeld BC, Lampert CS, Spaeth N, Mountfort D, Kaspar H, Dietrich DR. 2000. Toxin production in cyanobacterial mats from ponds on the McMurdo Ice Shelf, Antarctica. Toxicon 38:1731–1748. doi: 10.1016/S0041-0101(00)00103-3. [DOI] [PubMed] [Google Scholar]
- 5.Lindholm T, Öhman P, Kurki-Helasmo K, Kincaid B, Meriluoto J. 1999. Toxic algae and fish mortality in a brackish-water lake in Åland, SW Finland. Hydrobiologia 397:109–120. doi: 10.1023/A:1003667728458. [DOI] [Google Scholar]
- 6.Chen J, Zhang D, Xie P, Wang Q, Ma Z. 2009. Simultaneous determination of microcystin contaminations in various vertebrates (fish, turtle, duck and water bird) from a large eutrophic Chinese lake, Lake Taihu, with toxic Microcystis blooms. Sci Total Environ 407:3317–3322. doi: 10.1016/j.scitotenv.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Charlton M, Milne J. 2004. Review of thirty years of change in Lake Erie water quality. NWRI Environment Canada contribution no. 04-167. National Water Research Institute, Burlington, ON, Canada. [Google Scholar]
- 8.Tabachek J-AL, Yurkowski M. 1976. Isolation and identification of blue-green algae producing muddy odor metabolites, geosmin, and 2-methylisoborneol, in saline lakes in Manitoba. J Fish Res Board Can 33:25–35. doi: 10.1139/f76-004. [DOI] [Google Scholar]
- 9.Watson SB, Ridal J, Boyer GL. 2008. Taste and odour and cyanobacterial toxins: impairment, prediction, and management in the Great Lakes. Can J Fish Aquat Sci 65:1779–1796. doi: 10.1139/F08-084. [DOI] [Google Scholar]
- 10.Jochimsen EM, Carmichael WW, An J, Cardo DM, Cookson ST, Holmes CEM, Antunes MB, de Melo Filho DA, Lyra TM, Barreto VST, Azevedo SMFO, Jarvis WR. 1998. Liver failure and death after exposure to microcystins at a hemodialysis center in Brazil. N Engl J Med 338:873–878. doi: 10.1056/NEJM199803263381304. [DOI] [PubMed] [Google Scholar]
- 11.Fuller K, Shear H, Wittig J. 1995. The Great Lakes: an environmental atlas and resource book. US EPA, Chicago, IL. [Google Scholar]
- 12.Brittain SM, Wang J, Babcock-Jackson L, Carmichael WW, Rinehart KL, Culver DA. 2000. Isolation and characterization of microcystins, cyclic heptapeptide hepatotoxins from a Lake Erie strain of Microcystis aeruginosa. J Great Lakes Res 26:241–249. doi: 10.1016/S0380-1330(00)70690-3. [DOI] [Google Scholar]
- 13.Rinta-Kanto JM, Ouellette AJA, Boyer GL, Twiss MR, Bridgeman TB, Wilhelm SW. 2005. Quantification of toxic Microcystis spp. during the 2003 and 2004 blooms in western Lake Erie using quantitative real-time PCR. Environ Sci Technol 39:4198–4205. doi: 10.1021/es048249u. [DOI] [PubMed] [Google Scholar]
- 14.Department of Public Utilities, City of Toledo. 2014. Microcystin event preliminary summary. http://toledo.oh.gov/media/132055/Microcystin-Test-Results.pdf Accessed 5 March 2015.
- 15.Brunberg A-K. 1999. Contribution of bacteria in the mucilage of Microcystis spp. (Cyanobacteria) to benthic and pelagic bacterial production in a hypereutrophic lake. FEMS Microbiol Ecol 29:13–22. doi: 10.1111/j.1574-6941.1999.tb00594.x. [DOI] [Google Scholar]
- 16.Shen H, Niu Y, Xie P, Tao MIN, Yang XI. 2011. Morphological and physiological changes in Microcystis aeruginosa as a result of interactions with heterotrophic bacteria. Freshw Biol 56:1065–1080. doi: 10.1111/j.1365-2427.2010.02551.x. [DOI] [Google Scholar]
- 17.Jiang L, Yang L, Xiao L, Shi X, Gao G, Qin B. 2007. Quantitative studies on phosphorus transference occurring between Microcystis aeruginosa and its attached bacterium (Pseudomonas sp.), p 161–165. In Qin B, Liu Z, Havens K (ed), Eutrophication of shallow lakes with special reference to Lake Taihu, China. Developments in hydrobiology, vol 194 Springer, Dordrecht, Netherlands. [Google Scholar]
- 18.Steffen MM, Li Z, Effler TC, Hauser LJ, Boyer GL, Wilhelm SW. 2012. Comparative metagenomics of toxic freshwater cyanobacteria bloom communities on two continents. PLoS One 7:e44002. doi: 10.1371/journal.pone.0044002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burke C, Steinberg P, Rusch D, Kjelleberg S, Thomas T. 2011. Bacterial community assembly based on functional genes rather than species. Proc Natl Acad Sci U S A 108:14288–14293. doi: 10.1073/pnas.1101591108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saxton MA, Morrow EA, Bourbonniere RA, Wilhelm SW. 2011. Glyphosate influence on phytoplankton community structure in Lake Erie. J Great Lakes Res 37:683–690. doi: 10.1016/j.jglr.2011.07.004. [DOI] [Google Scholar]
- 21.Wetzel RG, Likens GE. 2000. Limnological analyses, 3rd ed Springer, New York, NY. [Google Scholar]
- 22.Welschmeyer NA. 1994. Fluorometric analysis of chlorophyll a in the presence of chlorophyll b and pheopigments. Limnol Oceanogr 39:1985–1992. doi: 10.4319/lo.1994.39.8.1985. [DOI] [Google Scholar]
- 23.Boyer GL. 2007. The occurrence of cyanobacterial toxins in New York lakes: lessons from the MERHAB-Lower Great Lakes program. Lake Reserv Manag 23:153–160. doi: 10.1080/07438140709353918. [DOI] [Google Scholar]
- 24.Auinger BM, Pfandl K, Boenigk J. 2008. Improved methodology for identification of protists and microalgae from plankton samples preserved in Lugol's iodine solution: combining microscopic analysis with single-cell PCR. Appl Environ Microbiol 74:2505–2510. doi: 10.1128/AEM.01803-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saxton MA, Arnold RJ, Bourbonniere RA, McKay RM, Wilhelm SW. 2012. Plasticity of total and intracellular phosphorus quotas in Microcystis aeruginosa cultures and Lake Erie algal assemblages. Front Microbiol 3:3. doi: 10.3389/fmicb.2012.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edwards U, Rogall T, Blöcker H, Emde M, Böttger EC. 1989. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res 17:7843–7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaneko T, Nakajima N, Okamoto S, Suzuki I, Tanabe Y, Tamaoki M, Nakamura Y, Kasai F, Watanabe A, Kawashima K, Kishida Y, Ono A, Shimizu Y, Takahashi C, Minami C, Fujishiro T, Kohara M, Katoh M, Nakazaki N, Nakayama S, Yamada M, Tabata S, Watanabe MM. 2007. Complete genomic structure of the bloom-forming toxic cyanobacterium Microcystis aeruginosa NIES-843. DNA Res 14:247–256. doi: 10.1093/dnares/dsm026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshida T, Nagasaki K, Takashima Y, Shirai Y, Tomaru Y, Takao Y, Sakamoto S, Hiroishi S, Ogata H. 2008. Ma-LMM01 infecting toxic Microcystis aeruginosa illuminates diverse cyanophage genome strategies. J Bacteriol 190:1762–1772. doi: 10.1128/JB.01534-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. 2012. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods 9:811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyer F, Paarmann D, D'Souza M, Olson R, Glass E, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards R. 2008. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glass EM, Wilkening J, Wilke A, Antonopoulos D, Meyer F. 2010. Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb Protoc 2010:pdb.prot5368. doi: 10.1101/pdb.prot5368. [DOI] [PubMed] [Google Scholar]
- 32.Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang H-Y, Cohoon M, de Crécy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Rückert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, Vonstein V. 2005. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baggerly KA, Deng L, Morris JS, Aldaz CM. 2003. Differential expression in SAGE: accounting for normal between-library variation. Bioinformatics 19:1477–1483. doi: 10.1093/bioinformatics/btg173. [DOI] [PubMed] [Google Scholar]
- 34.Steffen MM, Dearth SP, Dill BD, Li Z, Larsen KM, Campagna SR, Wilhelm SW. 2014. Nutrients drive transcriptional changes that maintain metabolic homeostasis but alter genome architecture in Microcystis. ISME J 8:2080–2092. doi: 10.1038/ismej.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paerl HW, Xu H, McCarthy MJ, Zhu G, Qin B, Li Y, Gardner WS. 2011. Controlling harmful cyanobacterial blooms in a hyper-eutrophic lake (Lake Taihu, China): the need for a dual nutrient (N & P) management strategy. Water Res 45:1973–1983. doi: 10.1016/j.watres.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 36.Fan L, Reynolds D, Liu M, Stark M, Kjelleberg S, Webster NS, Thomas T. 2012. Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc Natl Acad Sci U S A 109:E1878–E1887. doi: 10.1073/pnas.1203287109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frossard A, Gerull L, Mutz M, Gessner MO. 2012. Disconnect of microbial structure and function: enzyme activities and bacterial communities in nascent stream corridors. ISME J 6:680–691. doi: 10.1038/ismej.2011.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson EK. 2014. Danger from microcystins in Toledo drinking water unclear. Chem Eng News 92:9. [Google Scholar]
- 39.Boyer GL, Yang X, Thomas S. 2008. The occurrence of anatoxin-a and other cyanobacterial toxins in Lake Erie and Lake Ontario: it is more than just microcystins, p 102 Abstr 43rd Can Symp Water Qual Res, Burlington, ON, Canada. [Google Scholar]
- 40.Fristachi A, Sinclair JL, Habrook-Berkman JA, Boyer GL, Burkholder J, Burns J, Carmichael W, du Four A, Frazier W, Morton SL, O'Brien E, Walker S. 2008. Occurrence of Cyanobacterial Harmful Algal Blooms Workgroup report. Adv Exp Med Biol 619:45–103. doi: 10.1007/978-0-387-75865-7_3. [DOI] [PubMed] [Google Scholar]
- 41.Chaffin JD, Bridgeman TB, Bade DL. 2013. Nitrogen constrains the growth of late summer cyanobacterial blooms in Lake Erie. Adv Microbiol 3:16–26. doi: 10.4236/aim.2013.36A003. [DOI] [Google Scholar]
- 42.Ibelings BW, Maberly SC. 1998. Photoinhibition and the availability of inorganic carbon restrict photosynthesis by surface blooms of cyanobacteria. Limnol Oceanogr 43:408–419. doi: 10.4319/lo.1998.43.3.0408. [DOI] [Google Scholar]
- 43.Sandrini G, Matthijs HCP, Verspagen JMH, Muyzer G, Huisman J. 2014. Genetic diversity of inorganic carbon uptake systems causes variation in CO2 response of the cyanobacterium Microcystis. ISME J 8:589–600. doi: 10.1038/ismej.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shapiro J. 1997. The role of carbon dioxide in the initiation and maintenance of blue-green dominance in lakes. Freshw Biol 37:307–323. doi: 10.1046/j.1365-2427.1997.00164.x. [DOI] [Google Scholar]
- 45.Van de Waal DB, Verspagen JMH, Finke JF, Vournazou V, Immers AK, Kardinaal WEA, Tonk L, Becker S, Van Donk E, Visser PM, Huisman J. 2011. Reversal in competitive dominance of a toxic versus non-toxic cyanobacterium in response to rising CO2. ISME J 5:1438–1450. doi: 10.1038/ismej.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Neil JM, Davis TW, Burford MA, Gobler CJ. 2012. The rise of harmful cyanobacteria blooms: the potential roles of eutrophication and climate change. Harmful Algae 14:313–334. doi: 10.1016/j.hal.2011.10.027. [DOI] [Google Scholar]
- 47.Tucker S, Pollard P. 2005. Identification of cyanophage Ma-LBP and infection of the cyanobacterium Microcystis aeruginosa from an Australian subtropical lake by the virus. Appl Environ Microbiol 71:629–635. doi: 10.1128/AEM.71.2.629-635.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pascault N, Loux V, Derozier S, Martin V, Debroas D, Maloufi S, Humbert J-F, Leloup J. 13 September 2014. Technical challenges in metatranscriptomic studies applied to the bacterial communities of freshwater ecosystems. Genetica doi: 10.1007/s10709-014-9783-4. [DOI] [PubMed] [Google Scholar]
- 49.Zinser ER, Lindell D, Johnson ZI, Futschik ME, Steglich C, Coleman ML, Wright MA, Rector T, Steen R, McNulty N, Thompson LR, Chisholm SW. 2009. Choreography of the transcriptome, photophysiology, and cell cycle of a minimal photoautotroph, Prochlorococcus. PLoS One 4:e5135. doi: 10.1371/journal.pone.0005135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mohr W, Intermaggio MP, LaRoche J. 2010. Diel rhythm of nitrogen and carbon metabolism in the unicellular, diazotrophic cyanobacterium Crocosphaera watsonii WH8501. Environ Microbiol 12:412–421. doi: 10.1111/j.1462-2920.2009.02078.x. [DOI] [PubMed] [Google Scholar]
- 51.Straub C, Quillardet P, Vergalli J, de Marsac NT, Humbert J-F. 2011. A day in the life of Microcystis aeruginosa strain PCC 7806 as revealed by a transcriptomic analysis. PLoS One 6:e16208. doi: 10.1371/journal.pone.0016208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wynne TT, Stumpf RP, Tomlinson MC, Fahnenstiel GL, Dyble J, Schwab DJ, Joshi SJ. 2013. Evolution of a cyanobacterial bloom forecast system in western Lake Erie: development and initial evaluation. J Great Lakes Res 39(Suppl 1):90–99. doi: 10.1016/j.jglr.2012.10.003. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.