

Abstract
Four Staphylococcus aureus-Escherichia coli shuttle vectors were constructed for gene expression and production of tagged fusion proteins. Vectors pBUS1-HC and pTSSCm have no promoter upstream of the multiple cloning site (MCS), and this allows study of genes under the control of their native promoters, and pBUS1-Pcap-HC and pTSSCm-Pcap contain the strong constitutive promoter of S. aureus type 1 capsule gene 1A (Pcap) upstream of a novel MCS harboring codons for the peptide tag Arg-Gly-Ser-hexa-His (rgs-his6). All plasmids contained the backbone derived from pBUS1, including the E. coli origin ColE1, five copies of terminator rrnB T1, and tetracycline resistance marker tet(L) for S. aureus and E. coli. The minimum pAMα1 replicon from pBUS1 was improved through either complementation with the single-strand origin oriL from pUB110 (pBUS1-HC and pBUS1-Pcap-HC) or substitution with a pT181-family replicon (pTSSCm and pTSSCm-Pcap). The new constructs displayed increased plasmid yield and segregational stability in S. aureus. Furthermore, pBUS1-Pcap-HC and pTSSCm-Pcap offer the potential to generate C-terminal RGS-His6 translational fusions of cloned genes using simple molecular manipulation. BcgI-induced DNA excision followed by religation converts the TGA stop codon of the MCS into a TGC codon and links the rgs-his6 codons to the 3′ end of the target gene. The generation of the rgs-his6 codon-fusion, gene expression, and protein purification were demonstrated in both S. aureus and E. coli using the macrolide-lincosamide-streptogramin B resistance gene erm(44) inserted downstream of Pcap. The new His tag expression system represents a helpful tool for the direct analysis of target gene function in staphylococcal cells.
INTRODUCTION
Staphylococci are commensals of human and animal skin. In some species, particularly in Staphylococcus aureus, community- and hospital-associated clones have acquired a multitude of virulence and antibiotic resistance mechanisms, representing a serious public health risk (1, 2). An increased understanding of the resistance and pathogenicity mechanisms of staphylococci is needed. Therefore, molecular tools for genetic analysis, including gene disruption and ectopic expression, have been developed (3). S. aureus-Escherichia coli shuttle vectors have been constructed using restriction fragments derived from small, natural high-copy-number S. aureus plasmids, such as pUB110, pT181, pE194, and pC194 (4–7). These plasmids propagate through rolling-circle replication (RCR) in staphylococci, requiring the replication initiator protein (Rep) and the double-strand origin (dso) to initiate leading-strand synthesis as well as the single-strand origin (sso) for subsequent efficient lagging-strand synthesis (8). The absence of a functional sso sequence in RCR plasmids has been associated with single-stranded DNA (ssDNA) accumulation, low copy numbers, and plasmid instability (9–11). While some sso sequences support replication only in the native host, other sequences are functional in a number of different Gram-positive bacteria (8). For example, pUB110 is a broad-host-range plasmid whose sso is recognized in both Staphylococcus and Bacillus (12). Related plasmids pBC16 and pAMα1 have also been isolated from Bacillus cereus and Enterococcus/Streptococcus, respectively. pAMα1 is a composite of two separable replicons that depends on the pAMα1Δ2 derivative for replication in Enterococcus/Streptococcus and the pAMα1Δ1 derivative, associated with pUB110/pBC16, for replication in Bacillus subtilis (13, 14). For simplicity, the pAMα1Δ1 replicon is referred to as the pAMα1 replicon in this and other studies. To circumvent plasmid replication via ssDNA intermediates, S. aureus-E. coli shuttle vectors, which replicate using a theta-mode mechanism in Staphylococcus, have been constructed. These constructs contain pSK1 or pI258-derived origins from natural low-copy-number S. aureus plasmids and might exhibit higher segregational and structural stability (15, 16). Furthermore, a series of cassette-based shuttle vectors was constructed to provide flexibility in use of Gram-positive replicons, selectable markers, and promoter regions for inducible or constitutive gene expression (16). Additionally, a number of regulated promoter systems were established for inducible gene expression from shuttle plasmids in S. aureus; among them are the xylose-inducible promoter Pxyl and the tetracycline-regulated hybrid promoter Pxyl-tetO (17–21), the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible hybrid promoter Pspac (18, 20), arsenic- and cadmium-inducible promoters (16, 22), and thermally regulated promoters (23). Although an increasing number of shuttle vectors to express genes in Staphylococcus have been developed, the number remains limited compared with the versatile systems established for E. coli. Particularly, the use of tag fusion systems to analyze gene function is not frequently encountered in staphylococci. Shuttle vectors for the regulated coexpression of green and red fluorescent fusion proteins have been recently constructed (20). However, there are no expression vectors for the generation of polyhistidine (His) tag fusion proteins directly from Staphylococcus cells. The small His tag does not typically interfere with protein activity, and fusion proteins can be isolated using metal affinity matrices or can be detected using antibody-based methods. Therefore, we anticipate that the production of His tag protein fusions directly in staphylococcal cells will facilitate the analysis of the molecular interactions of this opportunistic pathogen in biochemical experiments.
Therefore, we constructed S. aureus-E. coli shuttle vectors for the expression of target genes and Arg-Gly-Ser-hexa-His (rgs-his6) codon fusions. These vectors were derived from pBUS1 (24), which has frequently been used to clone genes in S. aureus (25–32), and comprise the Gram-positive replicon pAMα1 or pT181, the pBluescript II SK multiple cloning site (MCS) without a promoter, or a novel MCS with joinable rgs-his6 codons downstream of the strong S. aureus type 1 capsule gene 1A promoter (Pcap) (33). The shuttle vectors were tested for plasmid stability and copy number in S. aureus. The macrolide-lincosamide-streptogramin B (MLSB) resistance gene erm(44), cloned downstream of Pcap (26), was used to demonstrate the feasibility of rgs-his6 codon fusion and the production of Erm(44)-RGS-His6 protein in both S. aureus and E. coli.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains used in the present study are listed in Table 1. E. coli and S. aureus RN4220 strains were grown in Luria-Bertani (LB) broth with shaking at 220 rpm or on LB agar plates at 37°C. B. subtilis DSM4514(pUB110) was cultured in LB containing 20 μg/ml kanamycin at 30°C. Ultracompetent E. coli DH5α cells (34) and calcium chloride-competent E. coli AG100A or AG100 cells (35) were prepared and used for heat shock transformation as previously described. S. aureus RN4220 transformants were obtained through electroporation using the protocol of Schenk and Laddaga (36). E. coli and S. aureus transformants were selected on agar plates containing 10 μg/ml tetracycline, and this tetracycline concentration was also used to maintain the plasmids in the cells.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s) | Source or reference |
|---|---|---|
| Strains | ||
| S. aureus | ||
| RN4220 | NCTC8325-4 derivative, antibiotic susceptible, restriction deficient; sau1 hsdR (rK− mK+) | 49 |
| RN4220/pVPF5 | RN4220 containing plasmid pVPF5 from S. xylosus VF5 | 43 |
| E. coli | ||
| DH5α | K-12 strain; recA1 endA1 hsdR17 (rK− mK+) | Life Technologies |
| AG100 | K-12 strain | 50 |
| AG100A | AG100 ΔacrAB::aphA1 (Kmr) mutant; increased susceptibility to antibiotics | 44 |
| B. subtilis | ||
| DSM4514 | Derivative of strain 168 containing plasmid pUB110 | DSMZ |
| Plasmids | ||
| pUB110 | S. aureus plasmid, replicates in B. subtilis; aadD (Kmr Neor) | 51 |
| pVPF5 | S. xylosus pT181 family plasmid; tet(K) | 43 |
| pBUS1 | S. aureus-E. coli shuttle vector; pAMα1 minimum replicon, ColE1, MCS pBluescript II SK (Stratagene), (rrnB T1)5 tet(L) | 24 |
| pBUS1-Pcap | S. aureus-E. coli shuttle vector; pAMα1 minimum replicon, ColE1, promoter of S. aureus type 1 capsule gene 1A (Pcap) upstream of a new MCS with joinable rgs-his6 codons (Pcap-MCS-rgs-his6), (rrnB T1)5 tet(L) | This study |
| pBUS1-HC | S. aureus-E. coli shuttle vector; pAMα1 minimum replicon, sso oriL ColE1, MCS pBluescript II SK (Stratagene), (rrnB T1)5 tet(L) | This study |
| pBUS1-Pcap-HC | S. aureus-E. coli shuttle vector; pAMα1 minimum replicon, sso oriL ColE1 Pcap-MCS-rgs-his6 (rrnB T1)5 tet(L) | This study |
| pTSSC | S. aureus-E. coli shuttle vector; pT181 replicon, ColE1, MCS pBluescript II SK (Stratagene), (rrnB T1)5 tet(L) | This study |
| pTSSC-Pcap | S. aureus-E. coli shuttle vector; pT181 replicon, ColE1 Pcap-MCS-rgs-his6 (rrnB T1)5 tet(L) | This study |
| pTSSCm | pTSSC with two silent mutations in the repC sequence to remove HindIII and XbaI sites | This study |
| pTSSCm-Pcap | pTSSC-Pcap with two silent mutations in the repC sequence to remove HindIII and XbaI sites | This study |
| pBJW13 | S. aureus-E. coli shuttle vector; pAMα1 minimum replicon; ColE1 Pcap erm(44) (MLSBr) (rrnB T1)5 tet(L) | 26 |
| pBJW13-HC | S. aureus-E. coli shuttle vector; pAMα1 minimum replicon, sso oriL ColE1 Pcap erm(44) (rrnB T1)5 tet(L) | This study |
| pTJW13 | S. aureus-E. coli shuttle vector; pT181 replicon, ColE1 Pcap erm(44) (rrnB T1)5 tet(L) | This study |
| pBJW13-RGS-His | S. aureus-E. coli shuttle vector; pAMα1 minimum replicon, ColE1 Pcap erm(44)-rgs-his6 (rrnB T1)5 tet(L) | This study |
| pBJW13-HC-RGS-His | S. aureus-E. coli shuttle vector; pAMα1 minimum replicon, sso oriL ColE1 Pcap erm(44)-rgs-his6 (rrnB T1)5 tet(L) | This study |
| pTJW13-RGS-His | S. aureus-E. coli shuttle vector; pT181 replicon, ColE1 Pcap erm(44)-rgs-his6 (rrnB T1)5 tet(L) | This study |
DNA preparation, PCR, and sequencing.
Plasmid DNA and genomic DNA were isolated using peqGOLD Plasmid Miniprep Kit I and a peqGOLD bacterial DNA kit (Peqlab Biotechnologie GmbH, Erlangen, Germany), respectively. To improve lysis of S. aureus and B. subtilis, the cells were first incubated for 15 min at 37°C in solution I supplemented with 100 μg/ml lysostaphin (Sigma-Aldrich, St. Louis, MO) and 2 mg/ml lysozyme (Roche Diagnostics, Rotkreuz, Switzerland), respectively. Oligonucleotide primers were synthesized at Microsynth (Balgach, Switzerland). The primers used for plasmid construction are listed in Table 2. For analytical purposes, PCRs were performed using Taq DNA polymerase (Solis BioDyne, Tartu, Estonia). PCRs for plasmid construction were performed using high-fidelity DNA polymerases (such as the Pfu DNA polymerase [Promega, Madison, WI] or the Phusion Hot Start II High-Fidelity DNA polymerase [Thermo Scientific, Waltham, MA]) according to the manufacturers' instructions, unless otherwise specified. The PCR products were purified using a High Pure PCR Product Purification Kit (Roche Diagnostics, Rotkreuz, Switzerland) prior to incubation with restriction endonucleases and T4 DNA ligase (ExpressLink T4 DNA Ligase [Invitrogen, Carlsbad, CA]). All plasmid constructs were generated in E. coli DH5α cells, examined based on restriction digestion patterns, and sequenced using an ABI Prism 3100 genetic analyzer (Applied Biosystems, Foster City, CA). The plasmids are listed in Table 1.
TABLE 2.
Oligonucleotide primers and probes
| Function and name | Sequence (5′–3′ [modification])a | Target | Reference or source |
|---|---|---|---|
| Primers for plasmid construction | |||
| capHis-pBUS1-F | TGCAGCTAGCATATGATAAACCTCCTATTTTCCTTTCTTGTTTTCCATTATATATAATCCCCTGTATATTTTGCAAACTCTGGTACCACGCGTTGCGCTCAC | pBUS1 | This study |
| capHis-pBUS1-R | TTTATCATATGCTAGCTGCAGGAATTCTCTGACTAGTCGACCGAAAGCTTTGCTGGATCCGCATGCTCGAGAGGTTCTCATCACCATCACCATCACTAATCTAGAGCGGCCGCCACCG | pBUS1 | This study |
| pBUS1-oriL-F | AATGTTGTACAGAAAACCTCTGACACATGCAG | pBUS1, pBUS1-Pcap, pBJW13 | This study |
| pBUS1-oriL-R | AGTTAAGATCTAGGATCAATTTTGAACTCTCTCC | pBUS1, pBUS1-Pcap, pBJW13 | This study |
| oriL-BsrGI-F | TGAATTGTACACTTCCAAGTAAAGTATAACACAC | pUB110 | This study |
| oriL-BglII-R | TTAGAAGATCTGCGATTGCTGAATAAAAGATACG | pUB110 | This study |
| pBUS1-BsrGI-F | TTAGTTGTACAGGCCATATTGTTGTATAAGTGATG | pBUS1, pBUS1-Pcap, pBJW13 | This study |
| pBlueScript-BglII-R | AGTTAAGATCTATGACCAAAATCCCTTAACGTG | pBUS1, pBUS1-Pcap, pBJW13 | This study |
| pT181-BglII-F | AGATAAGATCTATAGAACATGCATTTATGCCGAG | pVPF5 | This study |
| pT181-BsrGI-R | TAATATGTACACTATTTCCAAAATTTAAATTCATG | pVPF5 | This study |
| mut-repC-F | AAAGCTCCACAGAAATTCCAGAACAAAATATAAGAATTTG | pTSSC, pTSSC-Pcap | This study |
| mut-repC-R | AATTTCTGTGGAGCTTTCCCCATTCTTCTTCATC | pTSSC, pTSSC-Pcap | This study |
| Primers and probes for qPCR | |||
| nuc263-F | AAAGCGATTGATGGTGATACGGTT | nuc | 38 |
| nuc355-R | TGCTTTGTTTCAGGTGTATCAACCA | nuc | 38 |
| nuc294-P | ATGTACAAAGGTCAACCAATGACATTCAGA (Cy5, BHQ2) | nuc | 38 |
| qPCR-tetL-F | GGCTTTCGTTCACCAAAACAGT | tet(L) | This study |
| qPCR-tetL-R | TGGTAAAGTTAAGCAAACTCATTCCA | tet(L) | This study |
| FAM-tetL-TAMRA | TTGTTTCAAGTAGCTTGAAACAGCAGGAAGCTG (FAM, TAMRA) | tet(L) | This study |
Bases binding to the template are underlined. Restriction sites used for cloning are in boldface italics. Complementary 5′ ends of cloning and mutagenesis primers are in italics. In mutagenesis primers, mismatched bases are in boldface. Cy5, cyanine fluorescein 5; BHQ2, black hole quencher 2; FAM, 6-carboxyfluorescein; TAMRA, tetramethylrhodamine.
Construction of pBUS1-Pcap.
The synthetic sequence containing both the S. aureus type 1 capsule gene 1A promoter Pcap and a new multiple cloning site with joinable rgs-his6 codons was inserted into the plasmid pBUS1 (24) using PCR-based QuikChange site-directed mutagenesis (Agilent Technologies, Santa Clara, CA). The PCR was performed with 40 ng of the DNA template pBUS1, 0.5 μM capHis-pBUS1-F primer, 0.3 μM capHis-pBUS1-R primer (Table 2), and 1U of Phusion Hot Start II High-Fidelity DNA polymerase in buffer HF (Thermo Scientific) supplemented with 5% glycerol. DNA amplification was performed for 30 cycles: 10 cycles with an annealing temperature of 55°C and an extension time of 2 min, followed by 20 cycles with an annealing temperature of 62°C and an extension time of 2 min. The PCR products were treated with the restriction enzyme DpnI and directly transformed into E. coli DH5α cells. The selected clones were analyzed for the correct Pcap-MCS-rgs-his6 insertion sequence through colony PCR and sequencing using primers pBUS1-F2 (5′-TTTACAAGCCCAGAGCTC) and pBUS1-R (5′-CTTTGAGTGAGCTGATAC). The plasmid DNA of one correct clone, referred to as pBUS1-Pcap, was completely sequenced (Table 1).
Construction of pBUS1-HC, pBUS1-Pcap-HC, and pBJW13-HC.
The oriL sequence was obtained from plasmid pUB110 through PCR amplification using primers oriL-BsrGI-F and oriL-BglII-R (Table 2) and introduced into vectors pBUS1, pBUS1-Pcap, and pBJW13 (Table 1). PCR was performed using Pfu polymerase and 80 ng of pUB110 template for 30 cycles, consisting of 4 initial cycles with an annealing temperature of 56°C and an extension time of 1 min, followed by 26 cycles with an annealing temperature of 60°C and an extension time of 1 min. The vector sequences were PCR amplified using 100 ng of pBUS1, pBUS1-Pcap, or pBJW13 as a DNA template, Pfu polymerase, and primers pBUS1-oriL-F and pBUS1-oriL-R (Table 2). PCRs were performed as described above but using an extension time of 11 min and an annealing temperature of 61°C. All amplicons were digested using BglII and BsrGI, whose restriction sites were incorporated into primer sequences to facilitate cloning. The vector amplicons were treated with alkaline phosphatase prior to ligation with the oriL fragment. The new constructs were named pBUS1-HC, pBUS1-Pcap-HC, and pBJW13-HC (where HC indicates high copy) (Table 1).
Construction of pTSSC, pTSSC-Pcap, and pTJW13.
The pAMα1 minimum replicon of pBUS1, pBUS1-Pcap, and pBJW13 was replaced with the pT181 replicon. To this end, pBUS1, pBUS1-Pcap, and pBJW13 sequences were amplified as described above for vector sequences but using the primers pBUS1-BsrGI-F and pBlueScript-BglII-R (Table 2). The pT181 replicon was amplified from pVPF5 (Table 1) using 25 ng of DNA template, Pfu polymerase, and the primers pT181-BglII-F and pT181-BsrGI-R (Table 2) using 30 amplification cycles with an annealing temperature of 56°C and an extension time of 3 min. Restriction digestion, dephosphorylation, and ligation were performed as described above, generating pTSSC, pTSSC-Pcap, and pTJW13 (Table 1).
Construction of pTSSCm and pTSSCm-Pcap.
The plasmids pTSSCm and pTSSCm-Pcap (Table 1) were generated through QuikChange site-directed mutagenesis using partially overlapping primers (37). The PCRs were performed using Phusion Hot Start II High-Fidelity DNA polymerase, 5 ng of pTSSC or pTSSC-Pcap as a DNA template, and the primers mut-repC-F and mut-repC-R (Table 2) for 30 cycles, including 3 initial cycles with an annealing temperature of 54°C and an extension time of 3.5 min, followed by 27 cycles with an annealing temperature of 65°C and an extension time of 3.5 min. The PCR products were DpnI digested and directly transformed into DH5α cells.
Generation of erm(44)-rgs-his6 fusion plasmids.
A total of 500 ng of pBJW13, pBJW13-HC, or pTJW13 was digested with BcgI, and 20 μM S-adenosylmethionine was added to the reaction mixture according to the manufacturer's instructions (New England BioLabs, Ipswich, MA). The DNA was purified using a High Pure PCR Product Purification Kit prior to ligation and transformation into E. coli DH5α. The transformants were confirmed for correct 3′ rgs-his6 codon fusion through colony PCR and sequencing using the primers pBUS1-F2 and contig11-F1 (5′-CCAACTCTTATTTTCATCC). The suffix RGS-His was appended to vector names for variants expressing erm(44)-rgs-his6 (Table 1).
Growth curve measurement.
Growth of cultures of RN4220 strains was monitored in LB broth every 30 min during a 16-h incubation at 37°C with 420 shakes per minute using a Varioskan Flash plate reader (Thermo Scientific) for automated measurement of the optical density at 600 nm (OD600). To obtain similar initial inocula, the RN4220 strains containing plasmids grown overnight on selective agar were first adjusted to a 0.5 McFarland turbidity standard in saline and then diluted 1:100 in LB broth containing 2 or 10 μg/ml tetracycline. Experiments were performed in triplicate in 96-well microtiter plates with culture volumes of 200 μl per well. Strain RN4220 without plasmid was inoculated in selective and nonselective medium to serve as a negative and positive control, respectively.
Plasmid stability test.
For segregational stability analysis, RN4220 cells containing plasmids were grown for 24 h in LB broth without selection and subcultured on LB agar with and without 10 μg/ml tetracycline, and subsequently colonies were enumerated. Briefly, a single plasmid-carrying RN4220 colony was inoculated overnight into 5 ml of LB broth containing 10 μg/ml tetracycline. The overnight culture was diluted 1:500 into fresh LB medium without selection and grown for 8 h, followed by further dilution at 1:1,000 and incubation for 16 h. Fifty microliters of the diluted bacteria (1:1,300,000 in saline), corresponding to approximately 100 CFU, was plated onto LB agar to count the colony number. For each plasmid, the segregational test was performed in triplicate.
Structural plasmid stability was assessed through the restriction analysis of plasmid DNA isolated from RN4220 cells grown under selective conditions using enzymes NotI, PvuII, and StuI in combination with SacI.
Determination of plasmid copy number using quantitative real-time PCR assay.
Multiplex real-time quantitative PCR (qPCR) was performed in a 7500 Real-Time PCR system (7500 software, version 2.0.5; Applied Biosystems) using 1× TaqMan Universal PCR master mix (Applied Biosystems). TaqMan primer and probe sets were specific for the chromosomal S. aureus nuclease gene nuc (primers, nuc263-F and nuc355-R; probe, nuc294-P) (38) and for the plasmidic gene tet(L) (primers, qPCR-tetL-F and qPCR-tetL-R; probe, FAM-tetL-TAMRA, where FAM is 6-carboxyfluorescein and TAMRA is carboxytetramethylrhodamine) (designed using Express Software, version 2.0; Applied Biosystems) (Table 2). DNA amplification was done with 2 μl of template in 25-μl reaction volumes with 300 nM primers and 200 nM probes and using the following cycling parameters: 2 min at 50°C, 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. For standard curve preparation, plasmid pBUS1-Pcap-HC isolated from DH5α and genomic DNA extracted from RN4220 strain without plasmid were used. Separate qPCR standard curves were done for nuc and tet(L) measuring 4-fold serial dilutions ranging from 20 ng (6,783,250 copies) to 76 fg (26 copies) and from 500 pg (80,926,176 copies) to 1.9 fg (309 copies), respectively. DNA copies were calculated using the following formula: DNA copies = (g of DNA) × (6.022 × 1023 copies mol−1)/(bp of DNA) × (665 g mol−1 bp−1). The size of the chromosome of RN4220 (2.67 Mb) was obtained from Nair et al. (39). DNA for standard curves was prepared using peqlab kits; dsDNA concentration was measured accurately using a Quantus fluorometer and QuantiFluor dsDNA dye (Promega). Plasmid DNA was linearized using NotI prior to measurement. DNA for plasmid copy number determination was obtained by crude lysis of three RN4220 colonies exhibiting similar sizes on selective agar (10 μg/ml tetracycline) after 18 to 24 h of incubation. The colonies were lysed in 50 μl Tris-EDTA (TE) containing 100 μg/ml lysostaphin for 10 min at 37°C. After addition of 200 μl lysis buffer (100 mM Tris-HCl, pH 8.5, 240 μg/ml proteinase K, 0.05% [vol/vol] Tween 20), the samples were incubated for 30 min at 60°C and for 10 min at 95°C. For qPCR, the lysates were diluted 1:10. Threshold cycle (CT) values of standards and samples were measured in triplicates with automatic threshold settings.
Antimicrobial susceptibility testing.
MICs were determined through the broth microdilution technique using 96-well microtiter plates with serial 2-fold dilutions of erythromycin (Sigma-Aldrich), which ranged from 256 μg/ml to 0.5 μg/ml (40). The MIC was defined as the lowest concentration of erythromycin with no visible growth after a 20-h incubation in Mueller-Hinton broth at 37°C.
Expression and purification of Erm(44)-RGS-His6.
RN4220, AG100A, or AG100 cells carrying the empty vector or erm(44) expression vector were grown to an OD600 of approximately 1 in LB broth containing 10 μg/ml tetracycline. Forty-five milliliters of the cultures was harvested through centrifugation and washed with 1 volume of phosphate-buffered saline (PBS; 137 mM NaCl, 2.5 mM KCl, 10 mM Na2HPO, 1.8 mM KH2PO4, pH 7.4). RN4220 cells were preincubated in 0.4 ml of NPI-10 buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0) supplemented with 100 μg/ml lysostaphin for 10 min at 37°C to digest the cell wall under native conditions. Subsequently, 1.2 ml of cDNPI-10 buffer (8 M urea, 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0) was added. The following steps were performed under denaturing conditions on ice or at 4°C. The buffers were supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and 0.2% (vol/vol) Triton X-100 (except for elution buffer DNPI-250). AG100A and AG100 cells were directly resuspended in 1.6 ml of DNPI-10 buffer (6 M urea, 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0). RN4220, AG100, and AG100A cells were disrupted using sonication (Branson Sonifier 250; duty cycle control, constant; output control, 3; three times for 15 s each). Unbroken cells and debris were removed through centrifugation at 16,000 × g for 10 min. The supernatants were used as total protein extracts for Western blot analysis or protein purification using a nickel-nitrilotriacetic acid (Ni-NTA)-agarose matrix (Qiagen, Hilden, Germany). To this end, 80 μl of the Ni-NTA-agarose suspension (washed once with 1.0 ml of DNPI-10 buffer) was incubated with 1.2 ml of total protein extract for 3 h on a lab rotator. The Ni-NTA-agarose beads were washed three times with 0.8 ml of DNPI-20 buffer (6 M urea, 50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0) and collected through centrifugation at 110 × g for 2 min. The proteins were eluted from the Ni-NTA-agarose matrix with 160 μl of DNPI-250 buffer (6 M urea, 50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0) for 5 min at room temperature with shaking at 850 rpm. The protein eluates were mixed with Laemmli buffer, boiled, and separated using 15% SDS-PAGE. The gels were stained with Coomassie brilliant blue R-250 (VWR International, Radnor, PA).
Western blot analysis.
Total protein extracts were prepared as described above. The protein concentration was measured according to the Bradford method (Micro Assay, Sigma-Aldrich) using bovine serum albumin (BSA) as a standard. Thirty micrograms of total protein extract was separated using 15% SDS-PAGE and transferred to nitrocellulose membrane (Bio-Rad, Hercules, CA). The membranes were probed with primary and phosphatase-labeled secondary antibodies in TBS-T buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% [vol/vol] Tween 20) containing 2.5% nonfat dry milk. The target proteins were detected using the chromogenic substrates NBT/BCIP (nitroblue tetrazolium/5-bromo-4-chloro-3-indolylphosphate) as recommended (Roche Diagnostics, Rotkreuz, Switzerland). The primary antibodies included a monoclonal mouse anti-RGS-His antibody (diluted 1:1,000) (5 Prime GmbH, Hilden, Germany) and a polyclonal rabbit antiserum specific for E. coli ribosomal protein L20ΔN (diluted 1:5,000) (41). The secondary alkaline phosphatase-labeled antibodies included goat polyclonal anti-mouse IgG (diluted 1:5,000) (KPL, Gaithersburg, MD) and goat polyclonal anti-rabbit IgG (diluted 1:5,000) (KPL).
Nucleotide sequence accession numbers.
The nucleotide sequences for pBUS1-Pcap, pBUS1-Pcap-HC, and pTSSCm-Pcap were deposited in the GenBank/ENA/DDBJ databases under the accession numbers LN609189, LN609190, and LN609191, respectively.
RESULTS
Construction of a plasmid expression system containing the S. aureus type 1 capsule gene 1A promoter and a new multiple cloning site with joinable rgs-his6 codons.
Four vectors derived from pBUS1 (24), pBUS1-HC, pBUS1-Pcap-HC, pTSSCm, and pTSSCm-Pcap, were constructed containing the ColE1 origin for replication in E. coli, the resistance marker tet(L) for plasmid selection in both E. coli and S. aureus, and a tandem array of five copies of the T1 transcriptional terminator from the E. coli rrnB gene [(rrnB T1)5], located between the tet(L) gene and the MCS, to prevent RNA polymerase read-through (Fig. 1). The vectors differ in their Gram-positive replicons and MCSs. Vectors pBUS1-HC and pTSSCm contain the pBluescript II SK MCS (Stratagene) and are suitable for cloning and expressing genes under the control of their own promoters (Fig. 1A). In contrast, the expression vectors pBUS1-Pcap-HC and pTSSCm-Pcap contain the strong constitutive promoter Pcap (33) upstream of a novel MCS encoding the RGS-His6 epitope for the production of tagged proteins and for high-specificity immunodetection (Fig. 1B). First, promoter Pcap and the new MCS were introduced into plasmid pBUS1, generating pBUS1-Pcap. For this, synthetic DNA containing the Pcap sequence including the ribosomal-binding site (RBS), a novel MCS with rgs-his6 codons and 15 unique sites for type II restriction endonucleases, and a BcgI site was designed (Fig. 1B). BcgI recognizes the CGA(N)6TGC site and cleaves DNA upstream and downstream to excise a defined 32-bp fragment with 2-base 3′ overhangs. The BcgI site was placed within the MCS to facilitate the linkage of rgs-his6 codons to cloned genes ending at the TGA stop codon of the MCS. BcgI digestion, followed by religation, was used to convert the TGA stop to a TGC codon, leading to the expression of a C-terminal His-tagged protein variant (Fig. 1B). The 160-bp Pcap-MCS-rgs-his6 sequence was inserted into plasmid pBUS1 using a modified QuikChange site-directed mutagenesis (Agilent Technologies) approach to replace the pBluescript II SK MCS. The PCR primers capHis-pBUS1-F and capHis-pBUS1-R contained the 5′ and 3′ regions, respectively, of the Pcap-MCS-rgs-his6 insert at the overhangs (Table 2). In addition, these primers overlapped 20 bp at the 5′ end to facilitate in vivo recombination.
FIG 1.

S. aureus-E. coli shuttle vectors for cloning genes with associated promoters (pBUS1-HC and pTSSCm) and for the constitutive expression of the target gene or the rgs-his6 codon fusion from promoter Pcap (pBUS1-Pcap, pBUS1-Pcap-HC, and pTSSCm-Pcap). The identical backbone segments in all plasmids include the E. coli origin ColE1 and the terminator sequence (rrnB T1)5, shown in gray, and the selectable marker for S. aureus and E. coli tet(L), in green. For propagation in Gram-positive bacteria, the plasmids contain either the pAMα1 replicon shown in violet or the pT181-family replicon in pink. The elements required for rolling-circle replication are indicated: the replication initiator protein gene (repB or repC), the double-strand origin (oriU or dso), and the single-strand origin (oriL or palA). (A) Plasmid maps of pBUS1-HC and pTSSCm containing the multiple cloning site (MCS) derived from pBluescript II SK (Stratagene) as the original pBUS1. (B) Plasmid maps of pBUS1-Pcap, pBUS1-Pcap-HC, and pTSSCm-Pcap containing a novel MCS and the strong promoter (Pcap) and ribosomal-binding site (RBS) of S. aureus type 1 capsule biosynthetic gene 1A. The MCS comprises 15 unique restriction sites and codons for the peptide tag Arg-Gly-Ser-hexa-His (RGS-His6), highlighted in black. The −35, −10 promoter sequences, RBS, and the start and stop codons useful for cloning are indicated in bold and underlined. Recognition and cleavage sites for the enzyme BcgI are indicated in red. Target genes inserted between the NdeI start and the TGA stop codon of the MCS (represented as a gray arrow) can be converted to a target gene-rgs-his6 variant through BcgI digest, followed by ligation. Thereby, the TGA stop codon is mutated to TGC (red underlining), which encodes a cysteine residue. The RGS-His6 tag is linked to the C-terminal end of the protein through two additional amino acids (CS). The plasmid maps were generated using SnapGene software (GSL Biotech, Chicago, IL).
The backbone of pBUS1-Pcap and the precursor pBUS1 contain the minimum pAMα1 replicon derived from the shuttle plasmid pHY300PLK (42), containing repB and dso oriU but lacking sso for S. aureus (Fig. 1B). To optimize the stability of the shuttle vectors pBUS1-Pcap and pBUS1, we introduced the sso oriL of the staphylococcal pUB110 plasmid (12) using a PCR-based strategy with primers that carry a BglII or BsrGI site. In the newly generated constructs, pBUS1-Pcap-HC and pBUS1-HC (HC for high copy), sso oriL was inserted downstream of tet(L) (Fig. 1), as originally situated in pBC16 and pAMα1 plasmids. In addition, we replaced the complete minimum pAMα1 replicon of pBUS1-Pcap and pBUS1 with the pT181 replicon (8) using a similar PCR-based strategy and the Staphylococcus xylosus pVPF5 plasmid (43) as a replicon source. The resulting plasmids, pTSSC-Pcap and pTSSC, contained restriction sites for HindIII and XbaI in the repC coding sequence of pT181, which are also present in the MCS. To obtain the final constructs pTSSCm-Pcap and pTSSCm, these sites were removed and replaced with two silent mutations using site-directed mutagenesis (Fig. 1).
Stability and copy number of the shuttle vectors.
The plasmid constructs were tested for stability and copy number in S. aureus. Differences among the constructs were visible in the RN4220 host colony morphology. Cells harboring vectors with complete Gram-positive replicons (pBUS1-Pcap-HC, pTSSC-Pcap, and pTSSCm-Pcap) displayed larger colonies than RN4220 carrying pBUS1 or pBUS1-Pcap on agar plates containing 10 μg/ml tetracycline (Fig. 2A). Growth retardation was also monitored in liquid cultures for RN4220 cells carrying plasmid pBUS1 or pBUS1-Pcap (Fig. 2B). The effect was, however, less pronounced when the tetracycline concentration was reduced from 10 to 2 μg/ml (Fig. 2B). Plasmids pBUS1-HC and pTSSCm displayed similar growth kinetics and colony sizes as their Pcap-carrying variants (data not shown). Segregational stability was quantified by determining the fractions of cells carrying plasmids after 24 h of growth in nonselective broth. Vectors pBUS1 and pBUS1-Pcap were maintained in less than 1% of RN4220 cells (pBUS1, 0.9%; pBUS1-Pcap, 0.8%). In contrast, vectors with a complemented pAMα1 replicon were present in almost half of the cells (pBUS1-Pcap-HC, 46%; pBUS1-HC, 48%), and vectors containing the pT181 replicon were maintained in at least 88% of the RN4220 cell populations (pTSSC-Pcap, 95%; pTSSCm-Pcap, 99%; pTSSCm, 88%). The plasmid yield was increased after the minimum pAMα1 replicon of pBUS1-Pcap was complemented with oriL and after the minimum replicon was replaced with the pT181 replicon. Analysis of plasmid extractions from equal numbers of RN4220 cells revealed a clear plasmid profile for pBUS1-Pcap-HC, pTSSC-Pcap, and pTSSCm-Pcap, while no bands were visible for pBUS1 and pBUS1-Pcap (Fig. 2C). The plasmid yield indicated that higher copy numbers were generated with the pT181 replicon than with the pAMα1 replicon. Plasmid extractions from RN4220 cells containing pBUS1-HC or pTSSCm were similar to extractions from pBUS1-Pcap-HC- and pTSSC-Pcap-/pTSSCm-Pcap-containing cells, respectively (data not shown). The absolute plasmid copy number in RN4220 transformants was determined by qPCR from strains grown under tetracycline selection by comparing the copy number ratio of the plasmidic tet(L) gene to that of the chromosomal nuc gene. Standard curves were prepared for each gene using a defined DNA concentration [PCR efficiency for nuc, 85.9%, R2 = 0.999; for tet(L), 86.2%, R2 = 0.997]. The estimated plasmid copy number per cell was around 16 to 20 for pBUS1-HC and pBUS1-Pcap-HC and 21 to 30 for vectors containing the pT181 replicon (Table 3). Structural plasmid stability was confirmed for the final vectors (pBUS1-Pcap-HC, pBUS1-HC, pTSSCm-Pcap, and pTSSCm) using restriction analysis (see Fig. S1 in the supplemental material).
FIG 2.

Analysis of cell morphology, growth curves, and plasmid profile of S. aureus RN4220 transformed with shuttle vector pBUS1, pBUS1-Pcap, pBUS1-Pcap-HC, pTSSC-Pcap, and pTSSCm-Pcap. (A) Colony size of RN4220 transformants after 16 h of growth on LB agar containing 10 μg/ml tetracycline. (B) Growth of RN4220 transformants in LB broth containing 10 μg/ml or 2 μg/ml tetracycline monitored through OD600 measurements during 16 h. Strain RN4220 without plasmid inoculated in selective (LB-tet RN4220) and nonselective (LB-RN4220) medium was used as a control. OD600 values are averages from triplicates. Note, OD600 values of 0.2 to 0.3 correspond to dense stationary-phase cultures; low values are due to measurement conditions, including short light path length. (C) Plasmid yield of the different vectors in RN4220. Plasmid DNA was isolated from similar 14-ml overnight cultures grown in selective LB broth and separated on 1% agarose gel. One-tenth of total plasmid extraction was loaded. M, 1 kb DNA ladder (Solis BioDyne).
TABLE 3.
Estimated plasmid copy numbers of S. aureus RN4220 transformants determined by quantitative real-time PCR
| Plasmid |
CT (avg ± SD)a |
Avg no. of copies (CV [%])b |
PCN (avg ± SD)c | ||
|---|---|---|---|---|---|
| nuc | tet(L) | nuc | tet(L) | ||
| None | 21.44 ± 0.03 | 3.12 × 105 (1.8) | |||
| pBUS1 | 21.52 ± 0.06 | 20.59 ± 0.11 | 2.97 × 105 (3.4) | 3.71 × 106 (6.9) | 12.48 ± 0.86 |
| pBUS1-Pcap | 22.04 ± 0.09 | 20.48 ± 0.10 | 2.16 × 105 (5.4) | 3.98 × 106 (6.4) | 18.45 ± 1.18 |
| pBUS1-HC | 21.56 ± 0.02 | 20.15 ± 0.03 | 2.91 × 105 (1.4) | 4.87 × 106 (1.9) | 16.74 ± 0.31 |
| pBUS1-Pcap-HC | 21.28 ± 0.05 | 19.55 ± 0.07 | 3.45 × 105 (3.3) | 7.12 × 106 (4.2) | 20.60 ± 0.86 |
| pTSSC | 21.00 ± 0.05 | 19.20 ± 0.02 | 4.12 × 105 (2.8) | 8.81 × 106 (1.5) | 21.41 ± 0.32 |
| pTSSC-Pcap | 20.77 ± 0.06 | 18.55 ± 0.10 | 4.73 × 105 (3.9) | 1.32 × 107 (6.1) | 27.93 ± 1.71 |
| pTSSCm | 20.98 ± 0.02 | 19.18 ± 0.03 | 4.17 × 105 (1.3) | 8.91 × 106 (2.1) | 21.39 ± 0.44 |
| pTSSCm-Pcap | 21.25 ± 0.02 | 19.06 ± 0.06 | 3.52 × 105 (1.2) | 9.59 × 106 (4.0) | 27.27 ± 1.06 |
n = 3.
CV, coefficient of variation (n = 3).
PCN, plasmid copy number (n = 3).
Analysis of RGS-His6 codon fusion and protein expression using the MLSB resistance gene erm(44).
We recently cloned the new MLSB resistance gene erm(44) into vector pBUS1-Pcap, generating plasmid pBJW13 (26). For that purpose, the open reading frame of erm(44) was inserted between the NdeI start codon and the TGA stop codon of the MCS and expressed from the strong promoter Pcap (Fig. 1B). Similar to vector pBUS1-Pcap, the plasmid stability of pBJW13 was optimized through the inclusion of the oriL sequence or the replacement of the minimum pAMα1 replicon with the pT181 replicon (Fig. 1B). The new erm(44) expression vectors were named pBJW13-HC (+oriL) and pTJW13 (pT181 replicon).
The conversion of erm(44) into erm(44)-rgs-his6 through BcgI cleavage and religation was tested for plasmids pBJW13, pBJW13-HC, and pTJW13. Analysis using colony PCR showed that at least 50% of the clones carried the expected BcgI-induced deletion (Fig. 1B). Three positive clones were sequenced for each construct, and all possessed the correct erm(44)-rgs-his6 fusion sequence. For these constructs, pBJW13-RGS-His, pBJW13-HC-RGS-His, and pTJW13-RGS-His, the expression of the erm(44)-rgs-his6 product in S. aureus and E. coli was determined through Western blotting. Erm(44)-RGS-His6 was detected with all three constructs in S. aureus RN4220 extracts (Fig. 3A). The effect of different plasmid yields was only partially apparent based on the amount of protein produced. A slight reduction in Erm(44)-RGS-His6 synthesis was detected for RN4220 cells harboring the plasmid pBJW13-RGS-His, which contains the pAMα1 replicon without sso (Fig. 3A). The S. aureus Pcap promoter was functional in E. coli, and AG100A or AG100 cells expressed equal Erm(44)-RGS-His6 protein levels with all constructs (Fig. 3B). The functional activity of Erm(44)-RGS-His6 protein was examined by measuring the MIC of erythromycin for S. aureus and E. coli. Erm(44) conferred high levels macrolide resistance in RN4220, regardless of the expression vector used and whether the protein was His tagged (see Table S1 in the supplemental material). Erm(44) with or without RGS-His6 also conferred erythromycin resistance in E. coli (see Table S1). As E. coli exhibits native resistance to macrolide antibiotics, we also transformed plasmids into the hypersensitive E. coli strain AG100A, an acrAB knockout mutant of AG100 (44). However, while vectors containing the pAMα1 replicon propagate without problems in AG100A, AG100A cells harboring shuttle vectors containing the pT181 replicon showed severe growth defects, necessitating the use of the wild-type AG100 strain for these constructs (see Table S1). To estimate protein overexpression from the gene under the control of Pcap in S. aureus and E. coli, a simple one-step purification was performed using Ni-NTA-agarose to immobilize His-tagged Erm(44). Total protein extracts and Ni affinity fractions were analyzed using SDS-PAGE, and proteins were visualized using Coomassie staining. Although Erm(44)-RGS-His6 was not clearly detected in RN4220 total protein extracts, this protein could be concentrated using Ni beads, representing the dominant band in the affinity fractions (Fig. 4A). Slightly larger amounts of Erm(44)-RGS-His6 protein were detected in the fractions from RN4220 extracts carrying optimized expression vectors (pBJW13-HC-RGS-His and pTJW13-RGS-His) than in those from RN4220 extracts carrying pBJW13-RGS-His. Erm(44)-RGS-His6 production was higher in E. coli and could be already detected in the total extract (Fig. 4B).
FIG 3.
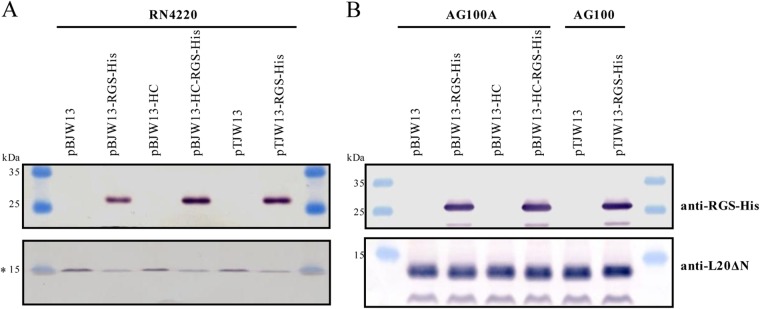
Analysis of Erm(44)-RGS-His6 protein synthesis in S. aureus RN4220 (A) and E. coli AG100A or AG100 (B) cells harboring the expression vector for erm(44) or erm(44)-rgs-his6. Thirty micrograms of total protein extract per sample was separated using 15% SDS-PAGE and analyzed through Western blotting. The upper part of the membrane was probed with anti-RGS-His antibody (5 Prime GmbH, Hilden, Germany) to detect tagged Erm(44), and the lower part of the membrane was probed with antiserum specific for the E. coli ribosomal protein L20ΔN (41), which served as a loading control. The asterisk at 15 kDa indicates the weak possible interaction of the antibody specific to E. coli L20ΔN with an S. aureus homolog. Prestained peqGOLD protein marker IV molecular size markers were used.
FIG 4.

One-step nickel affinity purification of Erm(44)-RGS-His6 from S. aureus RN4220 (A) and E. coli AG100A or AG100 (B). Cells carried one of the erm(44)-rgs-his6 expression vectors (pBJW13-RGS-His, pBJW13-HC-RGS-His, and pTJW13-RGS-His) or the empty pBUS1-Pcap-HC vector as a negative control. Total protein extracts (T) or nickel affinity fractions (Ni) were separated using 15% SDS-PAGE, and the gels were stained with Coomassie brilliant blue. For RN4220, 12 μl of total protein extracts and 30 μl of nickel affinity fractions were analyzed. For E. coli AG100A and AG100, 10 μl of total protein extracts and 25 μl of nickel affinity fractions were used. The position of Erm(44)-RGS-His6 on the gel is indicated with an asterisk. Prestained peqGOLD protein marker IV molecular size markers were used.
DISCUSSION
In Gram-positive bacteria, there are only a few tools available for protein affinity purification using small tag fusions. A pAM401-based shuttle vector has been developed for the expression of Strep-tag fusions in Enterococcus faecalis (45), and His8 tag or Strep-tag shuttle vectors (pHT254, pHT255, and pHT253) have been developed for the expression of fusion proteins in B. subtilis (MoBiTec GmbH, Goettingen, Germany); however, none of these expression systems has been optimized for Staphylococcus.
Thus, we established a His tag expression system for S. aureus. The S. aureus-E. coli shuttle vector pBUS1 (24) was therefore optimized for better stability in S. aureus through either the complementation of the minimum pAMα1 replicon with the sso oriL or the replacement of the replicon with the pT181 replicon. The vectors were supplemented with a novel MCS region containing a His tag and the promoter Pcap from S. aureus type 1 capsule biosynthetic gene 1A (33), a promoter used to regulate the strong expression of reporter genes in S. aureus (46, 47). Two S. aureus-E. coli expression vectors, pBUS1-Pcap-HC and pTSSCm-Pcap, were generated for the constitutive expression of target genes or rgs-his6 codon fusions. These vectors offer multifunctionality as a gene of interest can be inserted under the control of Pcap either directly as a fusion with rgs-his6 codons or as a wild-type gene with potential to subsequently become fused with the rgs-his6 codons through the BcgI-generated deletion of the stop codon and the linker sequence. Furthermore, these vectors are suitable for use in gene function studies in S. aureus and E. coli and for facilitating the purification of His-tagged fusion proteins from S. aureus or, with higher yield, from E. coli. In parallel, two S. aureus-E. coli cloning vectors were constructed, pBUS1-HC and pTSSCm, to study gene expression under the control of the native promoters.
The shuttle vectors constructed in the present study were based on Gram-positive RCR plasmids that replicate via ssDNA intermediates (8). The stability of RCR-based vectors is controversial (3, 11, 15). On some occasions, vector instability might reflect incomplete replicon sequences, as observed with pBUS1. As expected for RCR plasmids without functional sso sequences (9–11), the vectors pBUS1 and pBUS1-Pcap showed low plasmid yield and severe segregational instability in S. aureus. However, the plasmid copy numbers did not differ substantially between RN4220 containing pAMα1 plasmids with and without sso. In addition, RN4220 cells containing plasmids with the minimum pAMα1 replicon displayed growth retardation compared to cells harboring plasmids with complete Gram-positive replicons in both solid and liquid cultures under tetracycline selection. These results suggest that plasmids lacking sso display similar levels of leading-strand synthesis but impaired completion of plasmid replication, leading to both segregational instability and replication intermediates which are lost during plasmid purification. The new constructs (pBUS1-HC, pBUS1-Pcap-HC, pTSSCm, and pTSSCm-Pcap) did not exhibit structural instability. The shuttle vectors containing the pT181 replicon supported higher plasmid copy numbers and segregational stability than the vectors carrying the complemented minimum pAMα1 replicon. However, the plasmid yield of dsDNA observed in the agarose gel for vectors containing the pT181 replicon appeared to be higher than that of vectors carrying the pAMα1 replicon, suggesting a more efficient lagging-strand synthesis. The copy numbers of the pT181 constructs from this study were in the range of 20 to 25 copies, as published for the original pT181 plasmid in S. aureus (48). In a previous study, an inverse correlation between the size of the recombinant RCR plasmid and copy number/segregational stability was demonstrated with pUB110, suggesting that constructs larger than 9 kb become incompatible with RCR (11). Thus, the new shuttle vectors, ranging from 5,272 bp to 5,595 bp in size, should be suitable for cloning DNA fragments up to 3 kb. Notably, shuttle vectors containing the pT181 replicon induced a severe growth defect in E. coli AG100A but not in the wild-type AG100 strain or in DH5α. Although AG100A transformants containing shuttle vectors with the pAMα1 replicon formed large colonies after 16 h of growth on selective agar, tiny colonies were not visible until 20 to 24 h for vectors harboring the pT181 sequence. The dysfunction induced through the pT181 replicon sequence itself or its encoded products (the RepC protein and the CopA RNAs) in the AG100A strain remained unknown and might represent a side effect of the hypersensitive phenotype of this E. coli strain, which has an inactivated AcrAB transporter system (44). For phenotypic analysis, such as antibiotic resistance determination in AG100A, we therefore recommend the use of the pAMα1-based shuttle vectors pBUS1-Pcap-HC and pBUS1-HC.
The arrangement of the BcgI restriction site within the MCS of the His tag expression vectors pBUS1-Pcap-HC and pTSSCm-Pcap facilitates the generation of a C-terminal RGS-His6 translational fusion with a wild-type gene ending at the TGA stop codon of the MCS. The feasibility of converting a gene of interest to a 3′ rgs-his6 fusion using a simple cut-religation method was demonstrated with the MLSB resistance gene erm(44). The RGS-His6 tag did not affect the function of Erm(44) in either S. aureus or E. coli and facilitated affinity purification and the detection of methylase protein using an RGS-His4-specific antibody. The yields of His-tagged Erm(44) protein in S. aureus RN4220 were similar for both new expression vectors and were slightly higher than the yield observed with the original pBUS1-Pcap expression vector. Therefore, the amount of protein produced was only partially correlated with the plasmid yield of the different expression vectors. An increased amount of transcription from Pcap is expected (33); thus, a single plasmid copy might result in a substantial amount of transcript, whereas the amount of protein could be limited through other factors. Although, Erm(44)-RGS-His6 was not highly overexpressed in RN4220, the yield was sufficient for Ni-NTA affinity purification. In contrast, Erm(44)-RGS-His6 was highly expressed with the S. aureus promoter Pcap in E. coli. No sign of growth defect was observed for erm(44)-overexpressing cells, DH5α, AG100A, and AG100. The vectors presented herein might not be appropriate for the expression of genes with potential toxic effects. In such cases, the replacement of Pcap with an inducible promoter in E. coli and S. aureus, such as Pspac or Pxyl-tetO, should be considered.
In the present study, we established a plasmid-based expression system for S. aureus. This tool will facilitate the analysis of target gene function in biochemical experiments. For example, it could be used for the purification of fusion proteins directly from staphylococcal cells, the determination of translation initiation sites, posttranslational modifications, or even the examination of molecular interactions through the isolation of fusion protein complexes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Brigitte Berger-Bächi and Sibylle Burger (University of Zurich, Zurich, Switzerland) for providing the pBUS1 vector, Stuart B. Levy (Tufts University School of Medicine, Boston, MA) for E. coli strains AG100 and AG100A, Claude Chiaruttini (Université Denis Diderot, Paris, France) for antibodies specific for the E. coli ribosomal protein L20ΔN, and Corinne Ruppen (University of Bern, Bern, Switzerland) for help with and advice concerning the Varioskan Flash plate reader.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03803-14.
REFERENCES
- 1.David MZ, Daum RS. 2010. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev 23:616–687. doi: 10.1128/CMR.00081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindsay JA. 2014. Staphylococcus aureus genomics and the impact of horizontal gene transfer. Int J Med Microbiol 304:103–109. doi: 10.1016/j.ijmm.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 3.Prax M, Lee CY, Bertram R. 2013. An update on the molecular genetics toolbox for staphylococci. Microbiology 159:421–435. doi: 10.1099/mic.0.061705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruckner R. 1992. A series of shuttle vectors for Bacillus subtilis and Escherichia coli. Gene 122:187–192. doi: 10.1016/0378-1119(92)90048-T. [DOI] [PubMed] [Google Scholar]
- 5.Augustin J, Rosenstein R, Wieland B, Schneider U, Schnell N, Engelke G, Entian KD, Gotz F. 1992. Genetic analysis of epidermin biosynthetic genes and epidermin-negative mutants of Staphylococcus epidermidis. Eur J Biochem 204:1149–1154. doi: 10.1111/j.1432-1033.1992.tb16740.x. [DOI] [PubMed] [Google Scholar]
- 6.Sullivan MA, Yasbin RE, Young FE. 1984. New shuttle vectors for Bacillus subtilis and Escherichia coli which allow rapid detection of inserted fragments. Gene 29:21–26. doi: 10.1016/0378-1119(84)90161-6. [DOI] [PubMed] [Google Scholar]
- 7.Gaskill ME, Khan SA. 1988. Regulation of the enterotoxin B gene in Staphylococcus aureus. J Biol Chem 263:6276–6280. [PubMed] [Google Scholar]
- 8.Khan SA. 2005. Plasmid rolling-circle replication: highlights of two decades of research. Plasmid 53:126–136. doi: 10.1016/j.plasmid.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 9.Gruss AD, Ross HF, Novick RP. 1987. Functional analysis of a palindromic sequence required for normal replication of several staphylococcal plasmids. Proc Natl Acad Sci U S A 84:2165–2169. doi: 10.1073/pnas.84.8.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boe L, Gros MF, Riele TEH, Ehrlich SD, Gruss A. 1989. Replication origins of single-stranded-DNA plasmid pUB110. J Bacteriol 171:3366–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bron S, Luxen E, Swart P. 1988. Instability of recombinant pUB110 plasmids in Bacillus subtilis: plasmid-encoded stability function and effects of DNA inserts. Plasmid 19:231–241. doi: 10.1016/0147-619X(88)90041-8. [DOI] [PubMed] [Google Scholar]
- 12.Kramer MG, Espinosa M, Misra TK, Khan SA. 1999. Characterization of a single-strand origin, ssoU, required for broad host range replication of rolling-circle plasmids. Mol Microbiol 33:466–475. doi: 10.1046/j.1365-2958.1999.01471.x. [DOI] [PubMed] [Google Scholar]
- 13.Perkins JB, Youngman P. 1983. Streptococcus plasmid pAMα1 is a composite of two separable replicons, one of which is closely related to Bacillus plasmid pBC16. J Bacteriol 155:607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francia MV, Clewell DB. 2002. Amplification of the tetracycline resistance determinant of pAMα1 in Enterococcus faecalis requires a site-specific recombination event involving relaxase. J Bacteriol 184:5187–5193. doi: 10.1128/JB.184.18.5187-5193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grkovic S, Brown MH, Hardie KM, Firth N, Skurray RA. 2003. Stable low-copy-number Staphylococcus aureus shuttle vectors. Microbiology 149:785–794. doi: 10.1099/mic.0.25951-0. [DOI] [PubMed] [Google Scholar]
- 16.Charpentier E, Anton AI, Barry P, Alfonso B, Fang Y, Novick RP. 2004. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl Environ Microbiol 70:6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stary E, Gaupp R, Lechner S, Leibig M, Tichy E, Kolb M, Bertram R. 2010. New architectures for Tet-on and Tet-off regulation in Staphylococcus aureus. Appl Environ Microbiol 76:680–687. doi: 10.1128/AEM.02416-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, Fan F, Palmer LM, Lonetto MA, Petit C, Voelker LL, St John A, Bankosky B, Rosenberg M, McDevitt D. 2000. Regulated gene expression in Staphylococcus aureus for identifying conditional lethal phenotypes and antibiotic mode of action. Gene 255:297–305. doi: 10.1016/S0378-1119(00)00325-5. [DOI] [PubMed] [Google Scholar]
- 19.Corrigan RM, Foster TJ. 2009. An improved tetracycline-inducible expression vector for Staphylococcus aureus. Plasmid 61:126–129. doi: 10.1016/j.plasmid.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Brzoska AJ, Firth N. 2013. Two-plasmid vector system for independently controlled expression of green and red fluorescent fusion proteins in Staphylococcus aureus. Appl Environ Microbiol 79:3133–3136. doi: 10.1128/AEM.00144-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bateman BT, Donegan NP, Jarry TM, Palma M, Cheung AL. 2001. Evaluation of a tetracycline-inducible promoter in Staphylococcus aureus in vitro and in vivo and its application in demonstrating the role of sigB in microcolony formation. Infect Immun 69:7851–7857. doi: 10.1128/IAI.69.12.7851-7857.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corbisier P, Ji G, Nuyts G, Mergeay M, Silver S. 1993. luxAB gene fusions with the arsenic and cadmium resistance operons of Staphylococcus aureus plasmid pI258. FEMS Microbiol Lett 110:231–238. doi: 10.1111/j.1574-6968.1993.tb06325.x. [DOI] [PubMed] [Google Scholar]
- 23.Schofield DA, Westwater C, Hoel BD, Werner PA, Norris JS, Schmidt MG. 2003. Development of a thermally regulated broad-spectrum promoter system for use in pathogenic gram-positive species. Appl Environ Microbiol 69:3385–3392. doi: 10.1128/AEM.69.6.3385-3392.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rossi J, Bischoff M, Wada A, Berger-Bächi B. 2003. MsrR, a putative cell envelope-associated element involved in Staphylococcus aureus sarA attenuation. Antimicrob Agents Chemother 47:2558–2564. doi: 10.1128/AAC.47.8.2558-2564.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Senn MM, Giachino P, Homerova D, Steinhuber A, Strassner J, Kormanec J, Flückiger U, Berger-Bächi B, Bischoff M. 2005. Molecular analysis and organization of the σB operon in Staphylococcus aureus. J Bacteriol 187:8006–8019. doi: 10.1128/JB.187.23.8006-8019.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wipf JRK, Schwendener S, Perreten V. 2014. The novel macrolide-lincosamide-streptogramin B resistance gene erm(44) is associated with a prophage in Staphylococcus xylosus. Antimicrob Agents Chemother 58:6133–6138. doi: 10.1128/AAC.02949-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ender M, Berger-Bachi B, McCallum N. 2009. A novel DNA-binding protein modulating methicillin resistance in Staphylococcus aureus. BMC Microbiol 9:15. doi: 10.1186/1471-2180-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartmann T, Zhang B, Baronian G, Schulthess B, Homerova D, Grubmuller S, Kutzner E, Gaupp R, Bertram R, Powers R, Eisenreich W, Kormanec J, Herrmann M, Molle V, Somerville GA, Bischoff M. 2013. Catabolite control protein E (CcpE) is a LysR-type transcriptional regulator of tricarboxylic acid cycle activity in Staphylococcus aureus. J Biol Chem 288:36116–36128. doi: 10.1074/jbc.M113.516302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schulthess B, Bloes DA, Berger-Bächi B. 2012. Opposing roles of σB and σB-controlled SpoVG in the global regulation of esxA in Staphylococcus aureus. BMC Microbiol 12:17. doi: 10.1186/1471-2180-12-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwendener S, Perreten V. 2011. New transposon Tn6133 in methicillin-resistant Staphylococcus aureus ST398 contains vga(E), a novel streptogramin A, pleuromutilin, and lincosamide resistance gene. Antimicrob Agents Chemother 55:4900–4904. doi: 10.1128/AAC.00528-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwendener S, Perreten V. 2012. New MLSB resistance gene erm(43) in Staphylococcus lentus. Antimicrob Agents Chemother 56:4746–4752. doi: 10.1128/AAC.00627-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schulthess B, Meier S, Homerova D, Goerke C, Wolz C, Kormanec J, Berger-Bächi B, Bischoff M. 2009. Functional characterization of the σB-dependent yabJ-spoVG operon in Staphylococcus aureus: role in methicillin and glycopeptide resistance. Antimicrob Agents Chemother 53:1832–1839. doi: 10.1128/AAC.01255-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ouyang S, Lee CY. 1997. Transcriptional analysis of type 1 capsule genes in Staphylococcus aureus. Mol Microbiol 23:473–482. doi: 10.1046/j.1365-2958.1997.d01-1865.x. [DOI] [PubMed] [Google Scholar]
- 34.Inoue H, Nojima H, Okayama H. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 96:23–28. doi: 10.1016/0378-1119(90)90336-P. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 36.Schenk S, Laddaga RA. 1992. Improved method for electroporation of Staphylococcus aureus. FEMS Microbiol Lett 73:133–138. [DOI] [PubMed] [Google Scholar]
- 37.Zheng L, Baumann U, Reymond JL. 2004. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang HY, Kim S, Kim J, Park SD, Uh Y, Lee H. 2014. Multiplex real-time PCR assay for rapid detection of methicillin-resistant staphylococci directly from positive blood cultures. J Clin Microbiol 52:1911–1920. doi: 10.1128/JCM.00389-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nair D, Memmi G, Hernandez D, Bard J, Beaume M, Gill S, Francois P, Cheung AL. 2011. Whole-genome sequencing of Staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. J Bacteriol 193:2332–2335. doi: 10.1128/JB.00027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clinical and Laboratory Standards Institute. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 9th ed Approved standard M07-A9 CLSI, Wayne, PA. [Google Scholar]
- 41.Guillier M, Allemand F, Graffe M, Raibaud S, Dardel F, Springer M, Chiaruttini C. 2005. The N-terminal extension of Escherichia coli ribosomal protein L20 is important for ribosome assembly, but dispensable for translational feedback control. RNA 11:728–738. doi: 10.1261/rna.7134305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishiwa H, Shibahara-Sone H. 1986. New shuttle vectors for Escherichia coli and Bacillus subtilis. IV. The nucleotide sequences of pHY300PLK and some properties in relation to transformation. Jpn J Genet 61:515–528. [Google Scholar]
- 43.Perreten V, Giampà N, Schuler-Schmid U, Teuber M. 1998. Antibiotic resistance genes in coagulase-negative staphylococci isolated from food. Syst Appl Microbiol 21:113–120. doi: 10.1016/S0723-2020(98)80014-3. [DOI] [PubMed] [Google Scholar]
- 44.Okusu H, Ma D, Nikaido H. 1996. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J Bacteriol 178:306–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujimoto S, Ike Y. 2001. pAM401-based shuttle vectors that enable overexpression of promoterless genes and one-step purification of tag fusion proteins directly from Enterococcus faecalis. Appl Environ Microbiol 67:1262–1267. doi: 10.1128/AEM.67.3.1262-1267.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Neill AJ, Miller K, Oliva B, Chopra I. 2004. Comparison of assays for detection of agents causing membrane damage in Staphylococcus aureus. J Antimicrob Chemother 54:1127–1129. doi: 10.1093/jac/dkh476. [DOI] [PubMed] [Google Scholar]
- 47.Murray RW, Melchior EP, Hagadorn JC, Marotti KR. 2001. Staphylococcus aureus cell extract transcription-translation assay: firefly luciferase reporter system for evaluating protein translation inhibitors. Antimicrob Agents Chemother 45:1900–1904. doi: 10.1128/AAC.45.6.1900-1904.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Projan SJ, Carleton S, Novick RP. 1983. Determination of plasmid copy number by fluorescence densitometry. Plasmid 9:182–190. doi: 10.1016/0147-619X(83)90019-7. [DOI] [PubMed] [Google Scholar]
- 49.Kreiswirth BN, Löfdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 50.George AM, Levy SB. 1983. Amplifiable resistance to tetracycline, chloramphenicol, and other antibiotics in Escherichia coli: involvement of a non-plasmid-determined efflux of tetracycline. J Bacteriol 155:531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McKenzie T, Hoshino T, Tanaka T, Sueoka N. 1986. The nucleotide sequence of pUB110: some salient features in relation to replication and its regulation. Plasmid 15:93–103. doi: 10.1016/0147-619X(86)90046-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.