

Abstract
Plant pathogens cause major economic losses in the agricultural industry because late detection delays the implementation of measures that can prevent their dissemination. Sensitive and robust procedures for the rapid detection of plant pathogens are therefore required to reduce yield losses and the use of expensive, environmentally damaging chemicals. Here we describe a simple and portable system for the rapid detection of viral pathogens in infected plants based on immunofiltration, subsequent magnetic detection, and the quantification of magnetically labeled virus particles. Grapevine fanleaf virus (GFLV) was chosen as a model pathogen. Monoclonal antibodies recognizing the GFLV capsid protein were immobilized onto immunofiltration columns, and the same antibodies were linked to magnetic nanoparticles. GFLV was quantified by immunofiltration with magnetic labeling in a double-antibody sandwich configuration. A magnetic frequency mixing technique, in which a two-frequency magnetic excitation field was used to induce a sum frequency signal in the resonant detection coil, corresponding to the virus concentration within the immunofiltration column, was used for high-sensitivity quantification. We were able to measure GFLV concentrations in the range of 6 ng/ml to 20 μg/ml in less than 30 min. The magnetic immunoassay could also be adapted to detect other plant viruses, including Potato virus X and Tobacco mosaic virus, with detection limits of 2 to 60 ng/ml.
INTRODUCTION
Many plant viruses transmitted by vectors or direct contact cause billions of dollars of economic losses every year in the agricultural industry (1). Certain economically important plant viruses are widely used as models for the development of detection assays and countermeasures, e.g., Grapevine fanleaf virus (GFLV), Potato virus X (PVX), and Tobacco mosaic virus (TMV).
GFLV is the oldest known virus that infects grapevine plants, and it has a severe impact on grapevine cultivation worldwide, with losses of up to 80% (2–4). It belongs to subgroup A of the genus Nepovirus in the subfamily Comovirinae (5). GFLV is predominantly transmitted by nematodes in infected vineyard soils, where the virus can remain dormant for many years without any need for host plants (6). Furthermore, GFLV can be transmitted by grafting or the propagation of infected grapevine plants. The early identification and quantification of GFLV are therefore necessary to prevent the spread of disease and to plan and monitor measures, such as sanitary selection and soil disinfection with nematicides.
PVX is the type member of the genus Potexvirus in the family Flexiviridae (7). This virus infects members of the Solanaceae (e.g., potato, tomato, and tobacco), and in most cases it is transmitted mechanically from plant to plant (8). A new resistance-breaking isolate of PVX (PVX MS, also known as PVX fcaOl) that multiplies in genotypes carrying the Rx gene was detected in Argentina (9). TMV is the type member of the genus Tobamovirus in the family Virgaviridae. It was the first virus to be discovered, and it is one of the best characterized. TMV has a wide host range, but it predominantly infects members of the Solanaceae, causing stunted growth, leaf necrosis, and yield losses. Although many infected plants show no symptoms or mild symptoms, yield losses of up to 30% have been reported in tobacco (10).
Plant pathogens, such as GFLV, PVX, and TMV, can be detected using serological methods, such as the enzyme-linked immunosorbent assay (ELISA) (11–14), or molecular methods, such as PCR, which can be up to 1,000 times more sensitive than ELISA (12, 15). However, the PCR-based detection of RNA viruses is laborious because it is necessary to extract and reverse transcribe the RNA into cDNA before amplification; thus, ELISA-based detection remains more popular (15). A disadvantage shared by both methods is that samples must be collected in the field and sent to qualified laboratories for analysis, which is labor-intensive, time-consuming, and expensive, given that crops must be monitored regularly throughout the cultivation period. Farmers need to collect a small number of random samples to achieve a statistical analysis of the pathogen distribution, but in most cases farmers do not collect samples unless there is symptomatic evidence of disease, and by that time a viral infection may have already taken hold. Typical laboratory analysis may take days or weeks, so by the time that the farmer becomes aware of a field infection, it is likely to have spread further.
Field-based quantitative diagnostic tests with sensitivities comparable to those of laboratory assays are not currently available. Rapid, lateral-flow immunological assay formats, such as those that use dipsticks, which can achieve sensitivities comparable to the sensitivity of ELISA (16, 17), are available for several plant viruses (16–19), but this type of assay is not quantitative. Accordingly, there is great interest in the development of rapid and inexpensive quantitative tests that can be used in the field to test randomly selected samples, producing real-time data about viral infections.
We set out to develop a convenient and portable diagnostic assay for plant viruses using GFLV as a model system. GFLV-specific antibodies were combined with magnetic nanoparticles to detect the virus and measure the infection grade. A similar method based on magnetic nanoparticles linked to specific antibodies has been developed for the detection and quantification of human pathogens (20–22). An immunoassay approach was developed for the detection of bacterial and viral plant pathogens and achieves multiplex detection using different sets of fluorescence-coded microspheres (23). Our strategy of labeling biological samples with magnetic nanoparticles is advantageous because the bead-sample complex can easily be separated from raw samples using a magnetic field. A number of functionalized magnetic beads are commercially available, and these are routinely used for nucleic acid isolation and purification. Magnetic particles have also been combined with proteins in several applications, such as the isolation of antigens from serum (24) and the specific labeling of analytes before separation (25–27). These commercially available particles are usually 100 nm to 1 μm in diameter and comprise a superparamagnetic core surrounded by a polymer shell (e.g., starch or polyvinyl alcohol) suitable for additional functionalization. The superparamagnetic core allows the direct detection and quantification of the particles using a variety of methods (28–33).
Here we developed a method based on the detection and quantification of magnetic particles by frequency mixing (34, 35) using a customized handheld magnetic reader with a measuring head specially equipped for quantification. The nanoparticles enter a magnetic field comprising two different excitation frequencies, and the response signal (in mV) is detected at a third mixing frequency, allowing quantification over a large dynamic range of more than 4 orders of magnitude (34).
The functionalization of the outer shell allows the magnetic nanoparticles to be linked to specific antibodies, enabling the magnetic labeling of the corresponding antigens, as demonstrated with human pathogens (21, 22). Here we used this technique for the first time to detect plant pathogens and achieved the rapid and quantitative detection of GFLV for phytosanitary applications. However, the assay can easily be modified to detect and quantify other plant viruses, such as PVX and TMV, using the appropriate antibodies. This magnetic immunoassay therefore allows the rapid and inexpensive detection of viral pathogens in field settings with the same sensitivity (5 to 500 ng/ml) as laboratory-based ELISAs (36, 37).
MATERIALS AND METHODS
Propagation and purification of virus particles.
GFLV-containing sap from infected Chenopodium quinoa plants was purchased in a homogenized and lyophilized form from Bioreba AG (Reinbach, Switzerland). This preparation is routinely used as a positive control in commercial ELISAs for GFLV. The contents of one vial of lyophilized plant material were resuspended in 2.5 ml phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4·12 H2O, 1.5 mM KH2PO4, pH 7.4), and the concentration of the GFLV capsid protein was determined by capillary gel electrophoresis. A Bioanalyzer system was used with a high-sensitivity protein 250 kit according to the recommended Agilent protocol (Agilent Inc., Santa Clara, CA, USA) to determine the size and quantity of the proteins in the positive control. A distinguishable peak of 56 kDa was attributed to the GFLV capsid protein.
PVX and TMV particles were purified from infected Nicotiana benthamiana and Nicotiana tabacum plants, respectively. This infected plant material was harvested at 14 to 21 days postinfection, depending on the infection symptoms, and 50 g of plant material was used for virus purification following a modified protocol from the International Potato Center (CIP, Lima, Peru), as previously described (38). Polyethylene glycol precipitation was carried out as described in the original protocol, but the sucrose cushion centrifugation step was omitted due to the loss of viral particles. The pooled fractions of the sucrose gradient were centrifuged at 248,000 × g for at least 3 h. The virus concentration was determined by measuring the optical density at 260 nm (OD260) using the TMV extinction coefficient of 3.0, because the TMV concentration in the mixtures tended to be greater than the PVX concentration.
Antibody generation and purification.
GFLV capsid protein-specific murine monoclonal antibody mAbFL6 (39) was produced using a monoclonal hybridoma cell line and affinity purified by hydrophobic charge-inducing chromatography with mercaptoethylpyridine (MEP) ligand HyperCel matrix (Pall Corporation, Port Washington, NY, USA), followed by overnight dialysis against PBS at 4°C. Murine anti-PVX monoclonal antibody mAb80 (38) was used to capture and detect PVX particles, and murine anti-TMV monoclonal antibody mAb24 (40) was used to capture and detect TMV. Both antibodies were produced in hybridoma cell cultures and purified by affinity chromatography using the Affi-Gel Protein A MAPS II kit (Bio-Rad, Hercules, CA, USA), followed by dialysis against PBS. The purity and integrity of all monoclonal antibodies were verified by SDS-PAGE (41, 42) under native and denaturing conditions, followed by staining with Coomassie brilliant blue and Western blot analysis (43) with goat antimouse heavy and light chain-specific antibodies labeled with alkaline phosphatase (AP).
SPR spectroscopy.
Binding interactions were analyzed in real time by surface plasmon resonance (SPR) spectroscopy using a Biacore T100 instrument (Biacore AB, Uppsala, Sweden) with a CM5 sensor chip, where carboxymethylated dextran is covalently attached to a gold surface (Biacore AB, Uppsala, Sweden). The CM5 chip was coated with a rabbit anti-mouse IgG Fc antibody (RaM-Fc) and washed with HCl to remove impurities. Samples were diluted in HBS-EP buffer (150 mM NaCl, 10 mM HEPES [pH 7.4], 0.5 mM EDTA, 0.05% [vol/vol] Tween 20), and all measurements were taken at a flow rate of 30 μl/min.
Immunofiltration.
Immunofiltration was carried out using ABICAP immunofiltration columns (44–47), purchased from Senova GmbH (Weimar, Germany), with sintered polyethylene filters with a pore diameter of 50 μm. The columns were equilibrated in 96% (vol/vol) ethanol at 8 × 103 Pa in a desiccator for 20 min, and subsequent steps were carried out in flowthrough mode. The degassed columns were first washed with 750 μl ethanol-water (50/50), followed by 750 μl water and 750 μl immobilization buffer [15 mM Na2(CO3), 35 mM NaHCO3, pH 9.6]. The columns were then coated with 500 μl of the capturing antibody (10 μg/ml) and incubated for 1 h to allow self-organized hydrophobic adsorption. Finally, the columns were blocked using two 750-μl aliquots of 10 mg/ml bovine serum albumin (BSA) in PBS (pH 7.4) and stored at 4°C for up to 14 days before use. All immunofiltration steps were performed under gravity flow without centrifugation.
Antibody binding to magnetic beads.
Magnetic beads with a hydrodynamic diameter of 200 nm were obtained from Chemicell GmbH (Berlin, Germany). The fluidMAG-streptavidin beads had a superparamagnetic magnetite core covered with a hydrophilic polymer functionalized with covalently coupled streptavidin, allowing the binding of biotinylated molecules. Hence, biotinylated mAbFL6 antibodies were coupled to the streptavidin-coated magnetic beads before injection into the immunofiltration columns. Biotinylation of mAbFL6 was achieved using an EZ-Link biotinylation kit (Thermo Fisher Scientific, Waltham, MA, USA), with the conditions being adjusted according to the manufacturer's recommendations so that the number of biotin molecules could be statistically limited to 2 to 4 molecules per antibody. Biotinylation was verified by ELISA and SPR spectroscopy. Subsequently, 10 μg of biotinylated antibody and 180 μg of the streptavidin-coated beads were mixed in 1 ml PBS, and the mixture was incubated for 30 min at room temperature in an overhead shaker before the antibody-functionalized beads were magnetically separated and resuspended in 1 ml PBS.
Design and operating mode of the magnetic reader.
The magnetic particles were detected by frequency mixing as described previously (34, 35). The magnetic reader is a small handheld device with an intuitive touch screen allowing parameter adjustment and measurement control (Fig. 1). The portable magnetic reader measures 10 by 23 by 7 cm and can be connected to a conventional power outlet or battery, allowing the device to be used in remote locations without a power source if necessary. The detection head contains a set of four coaxial coils, one for excitation, one for driving, and two for detection. ABICAP columns containing immobilized superparamagnetic particles were inserted into a custom-made bore inside the detection head. The columns were positioned in such a way that the upper detection coil was adjacent to the magnetic particle-enriched matrix.
FIG 1.
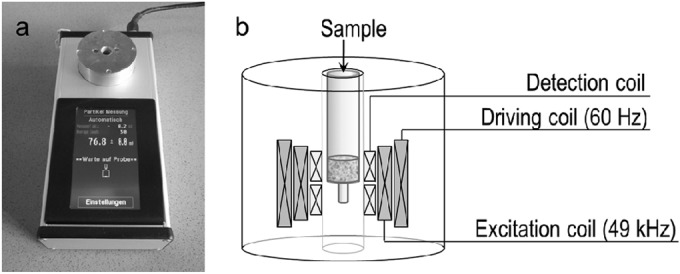
(a) Magnetic handheld reader device with intuitive touch display and an integrated detection head. (b) Schematic illustration of the measuring head, which comprises four coils arranged around the shaft containing the immunofiltration column. The excitation coil applies a magnetic field with a frequency of 49 kHz, and the outer driving coil applies a magnetic field with a frequency of 61 Hz. The resulting mixing frequency generated by the magnetic beads is detected by the inner detection coils.
During measurement, the superparamagnetic particles were driven to magnetic saturation by applying a strong low-frequency magnetic field to the driving coil, with the amplitude being in the millitesla range and the driving frequency (f2) being 61 Hz. Magnetic saturation of the superparamagnetic particles was achieved at each positive and negative extreme of the applied driving field, which alternated with a frequency of 2f2 of 122 Hz (34, 35).
The excitation coil was used to apply an additional magnetic field with a higher frequency (f1 = 49 kHz) to the magnetic beads inside the column, serving as a probe for the magnetization state. Although this additional field did not induce further magnetization, it caused strong particle magnetization according to the applied excitation field (f1) when the driving field fell to 0. The amplitude of the induced magnetization oscillation was thus modulated by twice the driving frequency (f1 ± 2f2). This mixing component was measured by the upper detection coil containing the sample. Direct induction from the excitation coil was minimized by differential winding of the detection coils, which consisted of two adjacent compartments with clockwise and counterclockwise rotation. On the basis of the arrangement of two detection coils with the sample residing in the upper coil alone, the magnetic sample induced a signal only in this coil and not in the lower coil. Therefore, the signal was not cancelled out.
The voltage induced in the detection coil was preamplified, and subsequently, the mixing component (f1 + 2f2) was demodulated by two-stage multiplication and low-pass filtering, enabling signal-dependent calibration using serially diluted samples with iron oxide concentrations of 0.12 to 1,300 mg/liter (34).
Magnetic immunodetection.
For magnetic immunodetection, 500 μl of analyte solution containing either known (calibration) or unknown concentrations of plant virus diluted in PBS was flushed by gravity flow through ready-to-use ABICAP columns coated with the capturing antibody, followed by incubation at room temperature for 30 min. Unbound material was removed by washing with 750 μl PBS, which took approximately 2 min to pass through the column by gravity flow. Then, 90 μg of streptavidin-coated magnetic beads suspended in 500 μl PBS and functionalized with 5 μg of the biotinylated detection antibody was added to the columns and allowed to bind the enriched virus particles (Fig. 2). After 30 min of incubation at room temperature, unbound antibody-bead complexes were removed by washing with 1.5 ml PBS. Finally, the ABICAP columns were inserted into the detection head of the reader and the signal (in mV) was recorded and displayed on the device screen (Fig. 1a).
FIG 2.
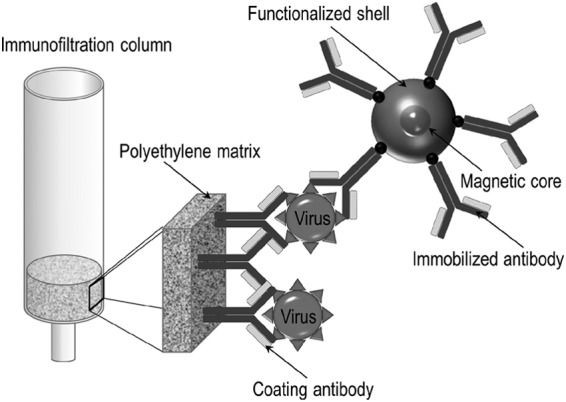
Schematic illustration of the magnetic immunofiltration assay. The capture antibody (e.g., mAbFL6) is immobilized on a polyethylene matrix in an ABICAP immunofiltration column with a pore diameter of 50 μm. The virus-containing solution is flushed through the column by gravity flow, and viral particles are enriched within the matrix. Finally, magnetic beads functionalized with the detection antibody (e.g., mAbFL6) are added to magnetically label the antigen in the column.
For optimized sequential magnetic immunodetection, incubation times were reduced and an additional binding step (binding of biotinylated antibody before the addition of streptavidin-coated magnetic beads) was introduced. Therefore, 500 μl of the analyte was added to the ready-to-use ABICAP columns, followed by 10 min of incubation at room temperature and washing with 750 μl PBS. After the virus particles bound to the ABICAP matrix, 500 μl of the biotinylated detection antibody (10 μg/ml in PBS) was flushed through the column and allowed to bind the captured virus particles. The incubation time was reduced to 5 min, and then an additional washing step (750 μl PBS) was performed. Subsequently, 500 μl PBS containing 90 μg of magnetic streptavidin-coated beads was passed through the column, followed by 5 min of incubation and a final wash with 1.5 ml PBS.
Enzyme-linked immunosorbent assay.
High-binding microtiter plates (Greiner Bio-One GmbH, Frickenhausen, Germany) were coated for 1 h at room temperature with 100 μl of the GFLV-, PVX-, or TMV-specific antibody (5 μg/ml) in PBS (pH 7.4). The wells were washed three times with 200 μl PBS containing 0.05% (vol/vol) Tween 20 (PBS-T) and then blocked with 200 μl 10 mg/ml BSA in PBS for 1 h. After blocking, the wells were washed three times with PBS-T before adding 100 μl of sample material (plant extract or diluted purified virus particles). PBS was used as the negative control. The plates were incubated for 1 h at room temperature. After three further washes, 100 μl of biotinylated virus-specific antibody (5 μg/ml) was added to the wells and the plates were incubated at room temperature for 1 h, followed by three washes with 200 μl PBS-T. Bound biotinylated antibodies were detected by adding 100 μl streptavidin conjugated to AP (Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA) diluted 1:1,000 in PBS to a final concentration of 1 μg/ml. ELISA readings (OD405) were taken after incubation for 30 min with AP substrate in SP buffer (0.1 mM diethanolamine, 1 mM MgCl2, pH 9.8) at 37°C.
RESULTS
Antibody-antigen binding specificity.
The magnetic immunodetection of plant viruses was demonstrated using GFLV as a model. GFLV-specific murine mAbFL6 was produced using a monoclonal hybridoma cell line (39), and its integrity was verified by SDS-PAGE, followed by staining with Coomassie brilliant blue, and by Western blot analysis. This revealed typical 150-kDa bands under native conditions and separate 55-kDa and 20-kDa bands representing the heavy and light chains, respectively, under denaturing conditions (data not shown). No contaminating bands were observed. Similar results were obtained following the purification of monoclonal antibodies against PVX and TMV (data not shown).
The selective detection of each target virus requires a specific antibody that can be used both to capture and to detect the virus particles (Fig. 2). Therefore, magnetic beads were functionalized with each specific antibody using the biotin-streptavidin system. Purified monoclonal antibodies against GFLV, PVX, and TMV were biotinylated using the EZ-Link biotinylation kit, and successful conjugation was verified by ELISA using AP-labeled streptavidin. For each antibody, a strong color change leading to strong absorption at 405 nm was detected, whereas the use of controls with nonbiotinylated antibodies did not lead to any change in absorption (data not shown).
Because the biotinylation reaction is based on the binding of random primary amine groups to the N-hydroxysuccinimide (NHS)-functionalized biotin molecule (48), it was necessary to ensure that epitope-binding activity was not significantly inhibited. Therefore, the binding efficiency and kinetic behavior of the biotinylated antibodies were evaluated by SPR spectroscopy (Fig. 3). Unmodified mAbFL6 and biotinylated mAbFL6 were flushed over a gold surface functionalized with a RaM-Fc, and the change in reflectivity was monitored over time. The nonspecific monoclonal antibody mAb54k was captured in parallel as a negative control (49).
FIG 3.

SPR spectroscopy is used to measure the binding of biotinylated and nonbiotinylated GFLV-specific mAbFL6 and nonspecific mAb54k as a negative control. The antibodies bound to the RaM-Fc-coated chip surface within the first 175 s. After washing with buffer, virus particles were added, resulting in an increment in reflectivity (response units) caused by antigen binding. Desorption behavior was analyzed following a subsequent washing step.
Both unmodified and biotinylated GFLV-specific mAbFL6 bound strongly to the RaM-Fc surface, as expected, without differences in binding behavior, as did the control antibody, mAb54k. Rinsing with buffer did not change the reflectivity, but rinsing with antigen solution (1.2 μg/ml) caused a significant increase in reflectivity for both unmodified and biotinylated GFLV-specific mAbFL6, as expected for affinity-based binding, whereas there was no change in the case of mAb54k. Biotinylated mAbFL6 showed an antigen binding efficiency (87%) slightly slower than that of its nonbiotinylated counterpart, as can be seen from the maximum difference in reflectivity change between the biotinylated and nonbiotinylated antibodies (Fig. 3). This result indicates that less than 15% of the molecules were biotinylated in such a way that the antigen binding site was blocked. In both cases, antibody-antigen binding occurred rapidly and resulted in near saturation within 3 min. Subsequent rinsing with buffer did not cause any change in reflectivity, confirming strong and irreversible antibody-antigen binding.
Magnetic immunodetection of GFLV.
The magnetic immunodetection of GFLV was assessed using GFLV-infected plant material with capsid protein concentrations of 12 to 60 ng/ml for calibration. mAbFL6 was immobilized on the polyethylene matrix of the immunofiltration column, and the biotinylated form was also linked to streptavidin-coated magnetic beads (21, 22). The magnetic immunodetection method was compared with a standard double-antibody sandwich ELISA over the same concentration range (Fig. 4a and b). In this assay, the microtiter plates were coated with the capture antibody, and streptavidin-labeled AP was used to detect biotinylated mAbFL6 binding to captured GFLV. In both assays, PBS was used as a negative control to provide blank readings. A further negative control consisting of resuspended lyophilized grapevine tissue from uninfected plants did not generate signals any stronger than the signal generated by the PBS control, indicating no cross-reaction between the antibody and plant proteins.
FIG 4.

(a) Comparison of magnetic immunoassay (squares, left y axis) and a standard double-antibody sandwich ELISA (circles, right y axis) using identical serial dilutions of positive-control material containing GFLV particles. Data points were fitted to the Hill-1 curve. Blank values are indicated by a solid line (magnetic immunoassay) and a dotted line (ELISA). (b) Linear range with the corresponding linear fit equation and coefficient of determination (R2). Data are shown as means ± SDs (n = 3).
In both assays, the calibration data points fitted ideally to a nonlinear Hill-1 curve (OriginPro, v8.1; OriginLab, Northampton, MA, USA) derived from the Hill equation (50, 51), leading to the typical saturation curve shown in equation 1.
| (1) |
where y is the output voltage of the magnetic reader, x is the concentration of the analyte, blank is the value for the blank, end is the saturation value for infinitely high concentrations, n is the Hill coefficient, and k is the concentration producing a half-maximum signal. The calculation of unknown sample concentrations is possible by adding the measuring value to the corresponding linear fit function shown in Fig. 4b and Table 1. The blank values in the ELISA (0.125 ± 0.0035 mV) were lower than those in the magnetic immunodetection assay (574.1 mV ± 78.4 mV). The detection limit was calculated from the blank value plus three times the standard deviation (SD) of the blank value, as shown in equation 2.
| (2) |
TABLE 1.
Parameters used to measure detection limit of magnetic immunoassays for viral pathogens compared to those used for a representative ELISAa
| Assay | Mean ± SD blank value (mV) | Linear fit function | R2 | Linear fit LOD (ng/ml) |
|---|---|---|---|---|
| ELISA | 0.125 ± 0.0035 | y = 0.262x + 0.108 | 0.962 | 105 |
| MID of GFLV by non-seq. | 574.1 ± 78.4 | y = 585.84x + 592.69 | 0.986 | 370 |
| MID of GFLV by seq. | 291.9 ± 6.4 | y = 816.79x + 306.24 | 0.994 | 6.0 |
| MID of TMV by seq. | 107.4 ± 0.9 | y = 777.48x + 108.40 | 0.993 | 2.2 |
| MID of PVX by seq. | 16.1 ± 2.3 | y = 48.85x + 20.27 | 0.974 | 56 |
LOD, limit of detection; ELISA, enzyme-linked immunosorbent assay; MID, magnetic immunodetection; non-seq., standard protocol; seq., optimized sequential protocol.
A detection limit of 0.1355, corresponding to a minimal detectable antigen concentration of 105 ng/ml, was calculated for the ELISA, and a detection limit of 809.3 mV, corresponding to a minimal detectable antigen concentration of 370 ng/ml, was calculated for the magnetic immunodetection assay. Unknown concentrations beyond the linear range of the calibration curve could be calculated using the nonlinear fit function.
Assay optimization.
The standard magnetic immunodetection assay was found to be three times less sensitive than the corresponding ELISA. The magnetic immunodetection assay was therefore optimized to achieve a lower detection threshold. Aggregates of biotinylated antibodies and streptavidin-coated beads were presumed to cause the high background signal as well as higher standard deviations between repeat measurements (Fig. 4). This disadvantage was overcome by adding the biotinylated antibody and streptavidin-coated magnetic beads to the column sequentially (Fig. 5). On the basis of the rapid binding kinetics detected by SPR spectroscopy (Fig. 3), the incubation time following analyte addition was also reduced from 30 to 10 min, and the mixture was incubated for 5 min after the application of the biotinylated antibody. The rapid binding kinetics between streptavidin and biotin (52–54) also allowed us to reduce the incubation time following the addition of the magnetic beads to just 5 min. The flowthrough time for a single wash was approximately 2 min. On the basis of these factors, the overall duration of the assay was reduced from 67 to 28 min (Fig. 5).
FIG 5.
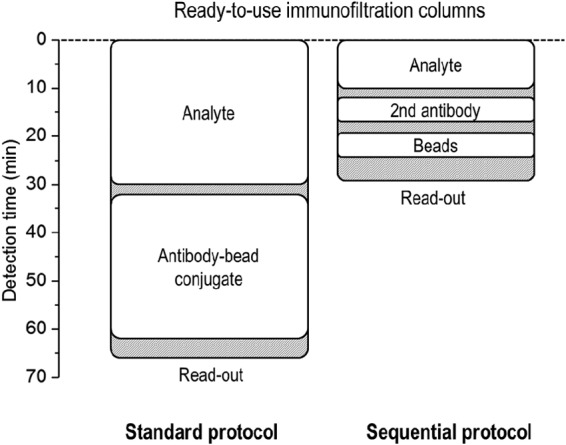
Time required for the standard protocol and optimized sequential protocol starting with ready-to-use precoated immunofiltration columns and finishing with the signal readout. White boxes represent incubation times, and gray-striped areas represent washing steps (∼2 min each) or double washing steps (∼4 min each).
The optimized assay parameters were verified by creating a new calibration curve using the same GFLV antigen concentrations used before (Fig. 6). A similar curve progression was achieved even with the much shorter incubation time. The signal-to-noise ratio also increased, and the blank values were approximately 50% lower than those recorded using the nonsequential standard protocol. The standard deviations were also lower, particularly at low antigen concentrations in the linear range, resulting in a more accurate linear fit, as shown by the R2 values (Table 1). The new detection limit was 6 ng/ml, which is 60 times more sensitive than the standard procedure and 17 times more sensitive than the ELISA (Table 1).
FIG 6.

(a) Comparison of GFLV magnetic immunodetection using the standard protocol (squares) and the sequential assay procedure (triangles). Data points were fitted to a nonlinear Hill-1 curve. Blank values are plotted as a solid line (standard protocol) and a dashed line (optimized protocol). (b) Linear range of calibration fitted using the linear fit function, with the coefficient of determination (R2) being shown. Data are shown as means ± SDs (n = 3).
Magnetic immunodetection of PVX and TMV.
The wider potential of magnetic immunodetection was demonstrated by adapting the protocol for the detection of PVX, using the anti-PVX antibody mAb80 (38), and for the detection of TMV, using the anti-TMV antibody mAb24 (40). Calibration curves were generated using the sequential protocol and dilutions of purified TMV (Fig. 7a and b) and PVX (Fig. 7c and d) particles of known concentrations, as described above for GFLV. Typical saturation curves with low blank values and standard deviations were obtained. The saturation curves were fitted according to equation 1, and the linear range of the calibration and the linear fit function were determined (Fig. 7b and d and Table 1). Detection limits were calculated using equation 2 and the linear fit functions, resulting in values of 56 ng/ml for PVX and 2.2 ng/ml for TMV.
FIG 7.

Calibration curves for the magnetic immunodetection of TMV (a) and PVX (c) and the linear range of calibration with the corresponding linear fits and coefficients of determination (R2) for TMV (b) and PVX (d). All experiments were carried out in triplicate using the sequential assay protocol. Data points in panels a and c were fitted to a nonlinear Hill-1 curve. Blank values are plotted as dashed lines. Data are shown as means ± SDs (n = 3).
DISCUSSION
The quantification of plant viruses is challenging, particularly when quantitative data must be acquired in the field during an ongoing infection. Quantitative laboratory tests for animal viruses are generally unsuitable for plant viruses because they rely on the preparation of cell cultures, e.g., plaque-based assays and endpoint dilution assays (55). Plant cell cultures are more difficult to establish and maintain than animal cell cultures, and the limited host range of many plant viruses would require a large and comprehensive panel of specific plant cell cultures. The severity of some virus infections can be determined using the local lesion method, where leaves are inoculated with the virus and the formation of lesions on the leaf surface is monitored and compared over time (56). This provides an indirect readout of the virus titer, but accurate quantitation is not possible because the statistical analysis of lesions often leads to errors. Electron microscopy can be used to measure virus titers directly in plant cells, but this is laborious and time-consuming and is therefore unsuitable for rapid pathogen quantitation. Rather than counting virions, it is easier to determine the concentration of the viral capsid protein, which increases in proportion to the severity of infection and can be quantified using calibration-based immunological assays, such as the ELISA (57).
Magnetic immunodetection combined with immunofiltration has numerous advantages over classic immunological assays, which remain the assays that are the most widely used for virus detection in plants. The speed, sensitivity, specificity, and convenience of immunofiltration-based assays have already been compared favorably to those of established detection techniques for the human pathogen Ebola virus (47). Typically, double-antibody sandwich ELISA protocols have detection limits of 10 to 500 ng/ml (36, 37). The sensitivity of such assays is strongly affected by factors such as host-virus combination, cultivar susceptibility, amplification of the detection signal, and assay incubation times (36). Some modified ELISA formats for plant viruses have detection thresholds below 10 ng/ml (58–61). However, more time is required to complete these assays, and the procedure is more complex because additional equipment and additional expertise are required, making these assays unsuitable for fieldwork. The ELISA is also based on a typical microtiter plate format with a limited binding surface. The matrix surface of the ABICAP immunofiltration columns is ∼40 times larger than the microcavity surface of a microtiter plate, so the analyte can be enriched more efficiently. Only 200 μl of analyte can be applied to each well of a microtiter plate, whereas the analyte volume flushed through the immunofiltration column can be increased to the milliliter range. However, a volume of 500 μl, which is used in the standard protocol, is sufficient for antigen detection in standard applications. The magnetic immunodetection protocol is simple; i.e., the analyte and bead solutions are mixed, and then there are two washing steps. Even the optimized protocol has only one additional step and three further washes. Samples can therefore be analyzed in less than 30 min, which is significantly faster than an ELISA. Most commercial ELISA kits for the detection of plant viruses require overnight incubation steps, and detection can therefore take up to 2 days. Even with abbreviated incubation times, ELISAs generally take at least 3 to 4 h and require laboratory-based equipment.
The magnetic immunodetection approach is therefore ideal for the rapid on-site detection of viruses. Immunofiltration columns prepared in advance can be used for analyte enrichment without special laboratory equipment. Analyte preparation can be achieved by squeezing or grinding leaf material, followed by placement of the processed material in buffer. All further assay steps are driven by gravity flow, and no special treatment of the columns is needed. Following the application and washing steps, the columns can be analyzed using the portable magnetic reader. If necessary, this can be powered using a battery, allowing rapid pathogen detection even in rural areas with limited power supplies.
The use of magnetic beads in an immunoassay format has numerous advantages over methods that rely on fluorescent labels or enzymatic reactions. Labeling the analyte with magnetic particles rather than fluorescent or adsorptive labels eliminates the risk of false-positive results caused by autofluorescence. Plant tissues often contain chromophores that interfere with the signals generated by fluorescent, chemiluminescent, and chromogenic ELISA substrates (62). Also, phosphatases and peroxidases in plant extracts interfere with alkaline phosphatase (AP) and horseradish peroxidase, leading to high ELISA background signals (63). The sensitivity of the frequency mixing technique is comparable to or even greater than that of other immunological methods (23). Furthermore, magnetic beads allow the separation of analyte molecules from a complex medium, followed by enrichment and active delivery, e.g., to a special sensor surface, by applying an external force. This can be used for novel and innovative biosensor approaches with low limits of detection (64).
Antibody-antigen binding characteristics determine the efficiency of all immunological assays. The antibody of choice should therefore bind strongly, rapidly, and irreversibly to the target antigen. For magnetic immunodetection, it is also necessary to confirm that biotinylation of the secondary antibody does not significantly inhibit the binding activity. SPR spectroscopy is the analytical method of choice to demonstrate that an antibody binds efficiently to a specific antigen, and we found that both the biotinylated and nonbiotinylated versions of mAbFL6 bound to the RaM-Fc-coated surface via the Fc region (Fig. 3). The addition of GFLV particles resulted in a visible change in reflectivity, which became saturated within ∼3 min, whereas the control antibody mAb54k had no effect. There was only a marginal kinetic difference between the biotinylated and nonbiotinylated antibodies, suggesting that both versions of the antibody are capable of rapid and efficient antigen binding and that biotinylation does not significantly inhibit antibody binding activity. Subsequent rinsing with buffer did not reduce the signal, confirming that antigen-antibody binding was strong and irreversible.
Concentration-dependent calibration curves for magnetic immunodetection resulted in typical saturation behavior similar to that of the ELISA (Fig. 3, 4, and 7). Binding between the secondary biotinylated antibody and the captured viral particles was confirmed in the double-antibody sandwich ELISA by adding streptavidin-labeled AP, allowing direct comparison with the magnetic immunodetection assay, where the equivalent step was represented by streptavidin-coated magnetic beads. ELISAs based on enzyme-labeled avidin and/or streptavidin binding to biotinylated secondary antibodies are 12 to 25 times more sensitive than corresponding double-antibody sandwich ELISAs using enzyme-labeled monoclonal antibodies (60, 61). The detection limits of the magnetic immunoassay and ELISA protocols were calculated using equation 2 and the fit function, describing the linear range of the calibration curve. The standard magnetic immunoassay protocol generated significantly higher blank values than the corresponding ELISA and was three times less sensitive. As well as detecting GFLV with concentrations above the detection limit, magnetic immunodetection also allowed the quantification of viruses on the basis of the calibration curve using the linear fit function. Concentrations beyond the linear range and below the level of saturation of the calibration curve can be determined using the Hill-1 fit according to equation 1. Magnetic immunodetection has a significantly greater dynamic range than the ELISA, leading to saturation at higher analyte concentrations. Magnetic immunodetection thus achieves virus quantification over a wider concentration range than the ELISA, which is consistent with the results of magnetic immunoassays for the detection of human pathogens (20–22).
The fast binding kinetics, confirmed by SPR spectroscopy (Fig. 3), allowed the magnetic immunodetection protocol to be optimized by reducing the duration of the incubation steps following the addition of the analyte and secondary biotinylated antibody to 10 and 5 min, respectively. The incubation time for the streptavidin-coated magnetic beads was reduced to 5 min, on the basis of the high dissociation rate constant and binding kinetics for biotin-streptavidin interactions (52).
The relatively high blank values observed in the standard magnetic immunodetection protocol can be explained by aggregation during the preparation of the antibody-functionalized magnetic beads. Biotinylation typically results in the conjugation of several biotin residues per antibody, and each bead offers multiple streptavidin molecules for biotin binding. Therefore, beads can be cross-linked by multiple biotinylated antibodies, similar to the aggregation of biotinylated gold nanoparticles and streptavidin (65). Larger aggregates either would remain in the column or would pass through slowly, resulting in a stronger background signal. To prevent aggregation, which leads to the formation of nonreproducible and undefined complexes, the antibody could be covalently attached to the magnetic beads. This can be achieved in a nonspecific manner (e.g., using NHS coupling chemistry) or in a site-specific manner (e.g., coupling the antibody by its glycan chains). Even in these cases, the reaction conditions must be adapted and optimized carefully. However, we chose a different optimization strategy by developing a sequential protocol in which the biotinylated secondary antibody was applied to the column without magnetic beads and the streptavidin-coated magnetic beads were applied to the column after a washing step. Free antibodies were therefore washed through the column, and those already fixed to the matrix were less likely to aggregate, resulting in a significantly lower background signal. The accuracy of calibration was also improved, as shown by the low standard deviation between repeat measurements. All these improvements increased the sensitivity of the assay, allowing the detection of viruses at concentrations of 2 to 10 ng/ml, which is sensitive enough to detect early-stage virus infections (23).
Having demonstrated that the magnetic immunodetection technology can be used to detect GFLV, we modified the assay for the detection of two additional model viruses (PVX and TMV) that also cause significant economic damage. In both cases, the sequential approach was used and calibration curves with low blank values were generated. The detection limit for TMV was ∼2 ng/ml, which is 20-fold lower than the limit for PVX (56 ng/ml), and indeed, all PVX signals were significantly weaker than those on the calibration curves for GFLV and TMV. This result may reflect the comparative binding activities of the three antibodies because slower binding kinetics would inhibit the capture of the virus and fewer magnetic beads would accumulate in the column. It may also reflect the composition of the virus capsid, because fewer epitopes or the presence of less-accessible epitopes would also inhibit virus capture. Nevertheless, reliable virus detection was possible in all three cases, with the magnetic immunodetection technology having a lower detection limit than the corresponding ELISAs. Furthermore, it was possible to quantify the virus on the basis of the calibration curve. Even without calibration, rough quantification was possible on the basis of signal intensity, allowing the grade of infection to be predicted.
Magnetic immunodetection was shown to be suitable for the rapid and sensitive detection of plant viruses in situ, and unlike current tests, it is also possible to quantify the concentration of viruses in plant tissues and therefore determine the grade of infection. The development and optimization of a sequential approach reduced the duration of the assay to 28 min, while they increased the sensitivity up to 17-fold compared to that of the corresponding ELISA. The magnetic immunodetection assay allows the detection of viruses in the ng/ml range; e.g., the detection limit is 6 ng/ml for GFLV and 2.2 ng/ml for TMV. Unlike the ELISA, the magnetic immunodetection assay does not require special laboratory equipment or skilled personnel. Additionally, the assay principle can easily be adapted to detect other plant viruses. We therefore intend to optimize the assay for fieldwork by simplifying the sample handling steps and by also using the magnetic beads for the magnetic separation of target pathogens from inhomogeneous and contaminated samples. The assay could also be adapted for the detection of a broad variety of other plant pathogens, including bacteria and fungi, and may therefore be transferable to many further applications in the fields of agricultural biosecurity and food safety. Antibodies against diverse pathogens are commercially available, and customized assays using these antibodies could be established with little effort. Additional fine-tuning and the inclusion of tailored monoclonal antibodies with high affinities for target pathogens could increase the sensitivity of the assay even further, reducing the limit of detection to approximately 0.5 ng/ml. Furthermore, the simultaneous detection of multiple plant pathogens, as recently reported (23), could be achieved by arranging consecutive matrices coated with different antibodies and adding a readout that discriminates between the different signals. In this context, the handheld device could be improved by designing an intuitive operating system that provides a direct readout of the pathogen and state of infection plus recommendations for suitable countermeasures. Farmers will be able to check their field plants routinely without sending samples to qualified laboratories because statistical sampling and subsequent on-site analysis provide real-time data showing the infection state of field plants.
ACKNOWLEDGMENTS
We thank Holger Spiegel (Fraunhofer Institute for Molecular Biology and Applied Ecology IME, Aachen, Germany) for technical help with SPR spectroscopy and Richard M. Twyman for editing the manuscript.
This work is supported by a MEF (Mittelständische Eigenforschung) of the Fraunhofer Gesellschaft.
REFERENCES
- 1.Zaitlin M, Palukaitis P. 2000. Advances in understanding plant viruses and virus diseases. Annu Rev Phytopathol 38:117–143. doi: 10.1146/annurev.phyto.38.1.117. [DOI] [PubMed] [Google Scholar]
- 2.Raski DJ, Goheen AC, Lider LA, Meredith CP. 1983. Strategies against grapevine fanleaf virus and its nematode vector. Plant Dis 67:335–339. doi: 10.1094/PD-67-335. [DOI] [Google Scholar]
- 3.Martelli GP, Savino V. 1991. Fanleaf degeneration, p 48–49. In Pearson RC, Goheen A (ed), Compendium of grape diseases. APS Press, St. Paul, MN. [Google Scholar]
- 4.Andret-Link P, Laporte C, Valat L, Ritzenthaler C, Demangeat G, Vigne E, Laval V, Pfeiffer P, Stussi-Garaud C, Fuchs M. 2004. Grapevine fanleaf virus: still a major threat to the grapevine industry. J Plant Pathol 86:183–195. doi: 10.4454/jpp.v86i3.987. [DOI] [Google Scholar]
- 5.Mayo MA, Robinson DJ. 1996. Nepoviruses: molecular biology and replication, p 139–185. In Harrison BD, Murant AF (ed), The plant viruses, polyhedal virions and bipartite RNA, vol 5 Plenum, New York, NY. [Google Scholar]
- 6.Demangeat G, Voisin R, Minot JC, Bosselut N, Fuchs M, Esmenjaud D. 2005. Survival of Xiphinema index in vineyard soil and retention of grapevine fanleaf virus over extended time in the absence of host plants. Phytopathology 95:1151–1156. doi: 10.1094/PHYTO-95-1151. [DOI] [PubMed] [Google Scholar]
- 7.Adams MJ, Antoniw JF, Bar-Joseph M, Brunt AA, Candresse T, Foster GD, Martelli GP, Milne RG, Zavriev SK, Fauquet CM. 2004. The new plant virus family Flexiviridae and assessment of molecular criteria for species demarcation. Arch Virol 149:1045–1060. doi: 10.1007/s00705-004-0384-x. [DOI] [PubMed] [Google Scholar]
- 8.Koenig R. 1971. Nucleic acids in Potato virus X group and in some other plant viruses—comparison of molecular weights by electrophoresis in acrylamide-agarose composite gels. J Gen Virol 10:111–114. doi: 10.1099/0022-1317-10-1-111. [DOI] [PubMed] [Google Scholar]
- 9.Tozzini AC, Ceriani MF, Cramer P, Palva ET, Hopp HE. 1994. PVX MS, a new strain of Potato virus that overcomes the extreme resistance gene RX. J Phytopathol 141:241–248. doi: 10.1111/j.1439-0434.1994.tb01467.x. [DOI] [Google Scholar]
- 10.Johnson CS, Main CE, Gooding GV. 1983. Crop loss assessment for flue-cured tobacco cultivars infected with tobacco mosaic virus. Plant Dis 67:881–885. doi: 10.1094/PD-67-881. [DOI] [Google Scholar]
- 11.Imada J, Narisawa N. 1984. Detection of Grapevine fanleaf virus by enzyme-linked immunosorbent assay ELISA. Bull Fruit Tree Res Stn Ser E (Akitsu) 5:71–76. [Google Scholar]
- 12.Liebenberg A, Freeborough M-J, Visser CJ, Bellstedt DU, Burger JT. 2009. Genetic variability within the coat protein gene of Grapevine fanleaf virus isolates from South Africa and the evaluation of RT-PCR, DAS-ELISA and ImmunoStrips as virus diagnostic assays. Virus Res 142:28–35. doi: 10.1016/j.virusres.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 13.Walter B, Kuszala J, Vuittenez A. 1979. Use of PALLAS and ELISA serological tests to detect Beet necrotic yellow vein virus and Grapevine fanleaf virus. Ann Phytopathol 11:568–569. [Google Scholar]
- 14.Engvall E, Perlmann P. 1971. Enzyme-linked immunosorbent assay (ELISA) quantitative assay of immunoglobulin G. Immunochemistry 8:871–874. doi: 10.1016/0019-2791(71)90454-X. [DOI] [PubMed] [Google Scholar]
- 15.Čepin U, Gutiérrez-Aguirre I, Balažic L, Pompe-Novak M, Gruden K, Ravnikar M. 2010. A one-step reverse transcription real-time PCR assay for the detection and quantitation of Grapevine fanleaf virus. J Virol Methods 170:47–56. doi: 10.1016/j.jviromet.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 16.Safenkova I, Zherdev A, Dzantiev B. 2012. Factors influencing the detection limit of the lateral-flow sandwich immunoassay: a case study with potato virus X. Anal Bioanal Chem 403:1595–1605. doi: 10.1007/s00216-012-5985-8. [DOI] [PubMed] [Google Scholar]
- 17.Salomone A, Mongelli M, Roggero P, Boscia D. 2004. Reliability of detection of citrus tristeza virus by an immunochromatographic lateral flow assay in comparison with ELISA. J Plant Pathol 86:43–48. doi: 10.4454/jpp.v86i1.936. [DOI] [Google Scholar]
- 18.Salomone A, Roggero P. 2002. Host range, seed transmission and detection by ELISA and lateral flow of an Italian isolate of pepino mosaic virus. J Plant Pathol 84:65–68. doi: 10.4454/jpp.v84i1.1088. [DOI] [Google Scholar]
- 19.Chirkov SN, Byzova NA, Sheveleva AA, Mitrofanova IV, Prikhod'ko YN, Dzantiev BB, Atabekov IG. 2012. Large-scale testing of lateral flow devices for the on-site Plum pox virus detection. Sel'skokhozyaistvennaya Biol 1:110–116. [Google Scholar]
- 20.Keusgen M, Meyer MHF, Krause HJ, Hartmann M, Miethe P, Oster J. 2007. Francisella tularensis detection using magnetic labels and a magnetic biosensor based on frequency mixing. J Magnetism Magn Mater 311:259–263. doi: 10.1016/j.jmmm.2006.10.1175. [DOI] [Google Scholar]
- 21.Meyer MHF, Hartmann M, Krause HJ, Blankenstein G, Mueller-Chorus B, Oster J, Miethe P, Keusgen M. 2007. CRP determination based on a novel magnetic biosensor. Biosens Bioelectron 22:973–979. doi: 10.1016/j.bios.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Meyer MHF, Stehr M, Bhuju S, Krause HJ, Hartmann M, Miethe P, Singh M, Keusgen M. 2007. Magnetic biosensor for the detection of Yersinia pestis. J Microbiol Methods 68:218–224. doi: 10.1016/j.mimet.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 23.Charlermroj R, Himananto O, Seepiban C, Kumpoosiri M, Warin N, Oplatowska M, Gajanandana O, Grant IR, Karoonuthaisiri N, Elliott CT. 2013. Multiplex detection of plant pathogens using a microsphere immunoassay technology. PLoS One 8:e62344. doi: 10.1371/journal.pone.0062344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen GD, Alberts CJ, Rodriguez W, Toner M. 2010. Concentration and purification of human immunodeficiency virus type 1 virions by microfluidic separation of superparamagnetic nanoparticles. Anal Chem 82:723–728. doi: 10.1021/ac9024522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dezfouli M, Vickovic S, Iglesias MJ, Nilsson P, Schwenk JM, Ahmadian A. 2014. Magnetic bead assisted labeling of antibodies at nanogram scale. Proteomics 14:14–18. doi: 10.1002/pmic.201300283. [DOI] [PubMed] [Google Scholar]
- 26.Wright DJ, Chapman PA, Siddons CA. 1994. Immunomagnetic separation as a sensitive method for isolating Escherichia coli O157 from food samples. Epidemiol Infect 113:31–39. doi: 10.1017/S0950268800051438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossi LM, Quach AD, Rosenzweig Z. 2004. Glucose oxidase-magnetite nanoparticle bioconjugate for glucose sensing. Anal Bioanal Chem 380:606–613. doi: 10.1007/s00216-004-2770-3. [DOI] [PubMed] [Google Scholar]
- 28.Brzeska M, Panhorst M, Kamp PB, Schotter J, Reiss G, Pühler A, Becker A, Brückl H. 2004. Detection and manipulation of biomolecules by magnetic carriers. J Biotechnol 112:25–33. doi: 10.1016/j.jbiotec.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 29.Matz H, Drung D, Hartwig S, Gross H, Kotitz R, Muller W, Vass A, Weitschies W, Trahms L. 1999. A SQUID measurement system for immunoassays. Appl Superconductivity 6:577–583. doi: 10.1016/S0964-1807(99)00014-9. [DOI] [Google Scholar]
- 30.Kriz K, Gehrke J, Kriz D. 1998. Advancements toward magneto immunoassays. Biosens Bioelectron 13:817–823. doi: 10.1016/S0956-5663(98)00047-5. [DOI] [PubMed] [Google Scholar]
- 31.Ejsing L, Hansen MF, Menon AK, Ferreira HA, Graham DL, Freitas PP. 2004. Planar Hall effect sensor for magnetic micro- and nanobead detection. Appl Phys Lett 84:4729–4731. doi: 10.1063/1.1759380. [DOI] [Google Scholar]
- 32.Graham DL, Ferreira HA, Freitas PP, Cabral JMS. 2003. High sensitivity detection of molecular recognition using magnetically labelled biomolecules and magnetoresistive sensors. Biosens Bioelectron 18:483–488. doi: 10.1016/S0956-5663(02)00205-1. [DOI] [PubMed] [Google Scholar]
- 33.Schotter J, Kamp PB, Becker A, Pühler A, Reiss G, Brückl H. 2004. Comparison of a prototype magnetoresistive biosensor to standard fluorescent DNA detection. Biosens Bioelectron 19:1149–1156. doi: 10.1016/j.bios.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Krause HJ, Wolters N, Zhang Y, Offenhausser A, Miethe P, Meyer MHF, Hartmann M, Keusgen M. 2007. Magnetic particle detection by frequency mixing for immunoassay applications. J Magnetism Magn Mater 311:436–444. doi: 10.1016/j.jmmm.2006.10.1164. [DOI] [Google Scholar]
- 35.Miethe P, Krause H, Zhang Y, Wolters N, Plaksin D. 2011. Apparatus for the selective detection of super paramagnetic and/or ferromagnetic particles, in bioassays to measure interactions, measures a frequency component at a mixed frequency in the magnetic fields. Patent applications WO2004077044-A1, DE10309132-A1, EP1597573-A1, and JP2006519366-W and patent US8071027-B2 (granted December 2011).
- 36.Narayanasamy P. 2001. Plant pathogen detection and disease diagnosis, 2nd ed CRC Press, New York, NY. [Google Scholar]
- 37.Voller A, Bartlett A, Bidwell DE, Clark MF, Adams AN. 1976. Detection of viruses by enzyme-linked immunosorbent assay (ELISA). J Gen Virol 33:165–167. doi: 10.1099/0022-1317-33-1-165. [DOI] [PubMed] [Google Scholar]
- 38.Uhde-Holzem K, Schloesser V, Viazov S, Fischer R, Commandeur U. 2010. Immunogenic properties of chimeric potato virus X particles displaying the hepatitis C virus hypervariable region I peptide R9. J Virol Methods 166:12–20. doi: 10.1016/j.jviromet.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 39.Nölke G, Cobanov P, Uhde-Holzem K, Reustle G, Fischer R, Schillberg S. 2009. Grapevine fanleaf virus (GFLV)-specific antibodies confer GFLV and Arabis mosaic virus (ArMV) resistance in Nicotiana benthamiana. Mol Plant Pathol 10:41–49. doi: 10.1111/j.1364-3703.2008.00510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fischer R, Liao YC, Drossard J. 1999. Affinity-purification of a TMV-specific recombinant full-size antibody from a transgenic tobacco suspension culture. J Immunol Methods 226:1–10. doi: 10.1016/S0022-1759(99)00058-7. [DOI] [PubMed] [Google Scholar]
- 41.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 42.Fairbanks G, Steck TL, Wallach DFH. 1971. Electrophoretic analysis of major polypeptides of human erythrocyte membrane. Biochemistry 10:2606–2617. doi: 10.1021/bi00789a030. [DOI] [PubMed] [Google Scholar]
- 43.Towbin H, Staehelin T, Gordon J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets—procedure and some applications. Proc Natl Acad Sci U S A 76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schell D, Erhardt C, Erhardt U. 1992. A new method for the fast quantification of monoclonal antibodies. Proc MoBBEL 6:91–93. [Google Scholar]
- 45.Hartmann H, Lubbers B, Casaretto M, Bautsch W, Klos A, Kohl J. 1993. Rapid quantification of C3A and C5A using a combination of chromatographic and immunoassay procedures. J Immunol Methods 166:35–44. doi: 10.1016/0022-1759(93)90326-3. [DOI] [PubMed] [Google Scholar]
- 46.Erhardt U, Erhardt C. 30 January 1997. Carrier material loadable by a through flow for solid phase assays. German patent WO/1997/028448.
- 47.Lucht A, Formenty P, Feldmann H, Gotz M, Leroy E, Bataboukila P, Grolla A, Feldmann F, Wittmann T, Campbell P, Atsangandoko C, Boumandoki P, Finke E-J, Miethe P, Becker S, Grunow R. 2007. Development of an immunofiltration-based antigen-detection assay for rapid diagnosis of Ebola virus infection. J Infect Dis 196:S184–S192. doi: 10.1086/520593. [DOI] [PubMed] [Google Scholar]
- 48.Strachan E, Mallia AK, Cox JM, Antharavally B, Desai S, Sykaluk L, O'Sullivan V, Bell PA. 2004. Solid-phase biotinylation of antibodies. J Mol Recognit 17:268–276. doi: 10.1002/jmr.669. [DOI] [PubMed] [Google Scholar]
- 49.Rasche S, Martin A, Holzem A, Fischer R, Schinkel H, Schillberg S. 2011. One-step protein purification: use of a novel epitope tag for highly efficient detection and purification of recombinant proteins. Open Biotechnol J 5:1–6. doi: 10.2174/1874070701105010001. [DOI] [Google Scholar]
- 50.Hill AV. 1910. The possible effects of the aggregation of the molecules of hemoglobin on its dissociation curves. Proc Physiol Soc 40:iv–vii. [Google Scholar]
- 51.Goutelle S, Maurin M, Rougier F, Barbaut X, Bourguignon L, Ducher M, Maire P. 2008. The Hill equation: a review of its capabilities in pharmacological modelling. Fundam Clin Pharmacol 22:633–648. doi: 10.1111/j.1472-8206.2008.00633.x. [DOI] [PubMed] [Google Scholar]
- 52.Piran U, Riordan WJ. 1990. Dissociation rate constant of the biotin-streptavidin complex. J Immunol Methods 133:141–143. doi: 10.1016/0022-1759(90)90328-S. [DOI] [PubMed] [Google Scholar]
- 53.Bayer EA, Wilchek M. 1990. Biotin-binding proteins: overview and prospects. Methods Enzymol 184:49–51. doi: 10.1016/0076-6879(90)84258-I. [DOI] [PubMed] [Google Scholar]
- 54.Chilkoti A, Stayton PS. 1995. Molecular origins of the slow streptavidin-biotin dissociation kinetics. J Am Chem Soc 117:10622–10628. doi: 10.1021/ja00148a003. [DOI] [Google Scholar]
- 55.Flint SJ, Enquist LW, Racaniello VR, Skalka AM. 2009. Principles of virology, 3rd ed John Wiley & Sons, Inc, Hoboken, NJ. [Google Scholar]
- 56.Smith KM. 1980. Virus assay, p 180–187. In Introduction to virology. Springer, Dordrecht, Netherlands. [Google Scholar]
- 57.Narayanasamy P. 2011. Detection of virus and viroid pathogens in plants, p 7–220. In Microbial plant pathogens—detection and disease diagnosis. Springer, Dordrecht, Netherlands. [Google Scholar]
- 58.Gerber M, Sarkar S. 1988. Comparison between urease and phosphatase for virus diagnosis with ELISA technique. J Plant Dis Prot 95:544–550. [Google Scholar]
- 59.Siitari H, Kurppa A. 1987. Time-resolved fluoroimmunoassay in the detection of plant viruses. J Gen Virol 68:1423–1428. doi: 10.1099/0022-1317-68-5-1423. [DOI] [Google Scholar]
- 60.Zrein M, Burckard J, Vanregenmortel MHV. 1986. Use of the biotin-avidin system for detecting a broad range of serologically related plant-viruses by ELISA. J Virol Methods 13:121–128. doi: 10.1016/0166-0934(86)90079-0. [DOI] [PubMed] [Google Scholar]
- 61.Sukhacheva E, Novikov V, Plaksin D, Pavlova I, Ambrosova S. 1996. Highly sensitive immunoassays for detection of barley stripe mosaic virus and beet necrotic yellow vein virus. J Virol Methods 56:199–207. doi: 10.1016/0166-0934(95)01962-6. [DOI] [PubMed] [Google Scholar]
- 62.Swanson C, D'Andrea A. 2013. Lateral flow assay with near-infrared dye for multiplex detection. Clin Chem 59:641–648. doi: 10.1373/clinchem.2012.200360. [DOI] [PubMed] [Google Scholar]
- 63.Swanson MM, Valand GB, Muniyappa V, Harrison BD. 1998. Serological detection and antigenic variation of two whitefly-transmitted geminiviruses: tobacco leaf curl and croton yellow vein mosaic viruses. Ann Appl Biol 132:427–435. doi: 10.1111/j.1744-7348.1998.tb05219.x. [DOI] [Google Scholar]
- 64.Morozov VN, Groves S, Turell MJ, Bailey C. 2007. Three minutes-long electrophoretically assisted zeptornolar microfluidic immunoassay with magnetic-beads detection. J Am Chem Soc 129:12628–12629. doi: 10.1021/ja075069m. [DOI] [PubMed] [Google Scholar]
- 65.Aslan K, Luhrs CC, Pérez-Luna VH. 2004. Controlled and reversible aggregation of biotinylated gold nanoparticles with streptavidin. J Phys Chem B 108:15631–15639. doi: 10.1021/jp036089n. [DOI] [Google Scholar]