

Abstract
Large sulfur-oxidizing bacteria in the family Beggiatoaceae are important players in the global sulfur cycle. This group contains members of the well-known genera Beggiatoa, Thioploca, and Thiomargarita but also recently identified and relatively unknown candidate taxa, including “Candidatus Thiopilula” spp. and “Ca. Thiophysa” spp. We discovered a population of “Ca. Thiopilula” spp. colonizing cold seeps near Barbados at a ∼4.7-km water depth. The Barbados population consists of spherical cells that are morphologically similar to Thiomargarita spp., with elemental sulfur inclusions and a central vacuole, but have much smaller cell diameters (5 to 40 μm). Metatranscriptomic analysis revealed that when exposed to anoxic sulfidic conditions, Barbados “Ca. Thiopilula” organisms expressed genes for the oxidation of elemental sulfur and the reduction of nitrogenous compounds, consistent with their vacuolated morphology and intracellular sulfur storage capability. Metatranscriptomic analysis further revealed that anaerobic methane-oxidizing and sulfate-reducing organisms were active in the sediment, which likely provided reduced sulfur substrates for “Ca. Thiopilula” and other sulfur-oxidizing microorganisms in the community. The novel observations of “Ca. Thiopilula” and associated organisms reported here expand our knowledge of the globally distributed and ecologically successful Beggiatoaceae group and thus offer insight into the composition and ecology of deep cold seep microbial communities.
INTRODUCTION
The family Beggiatoaceae contains the sulfur-oxidizing genera Beggiatoa, Thioploca, and Thiomargarita, including the largest known bacterium, Thiomargarita namibiensis, which can have cell diameters of >750 μm (1, 2). Certain members of the Beggiatoaceae store nitrate in a large central vacuole, which supplies an electron acceptor to fuel anaerobic sulfur oxidation when oxygen is unavailable (2–4). As sulfide-oxidizing lithotrophs, members of the Beggiatoaceae thrive in diverse sulfidic environments (5, 6). Biomats constructed by these microorganisms are a conspicuous feature at oxygen minimum zones, hydrocarbon seeps, hydrothermal vents, and whale falls (see reference 7 and references therein), where they fill important roles in the global carbon and sulfur cycles. For example, the Beggiatoaceae are often primary producers in sulfidic marine sediments, where they act to conserve some of the potential energy present in sulfide that is produced during the anaerobic remineralization of organic carbon. As much as 80 to 95% of the sulfide produced during sulfate reduction escapes burial and is reoxidized, and much of this reoxidation occurs in the shallow sediment zone (8, 9). In addition to catalyzing important sulfur and nitrogen transformations, marine representatives of the Beggiatoaceae may even contribute to phosphate mineral formation (10–12). Furthermore, because of their large size and morphological complexity, Beggiatoa- and Thiomargarita-like cells have been identified in the fossil record and offer clues to microbial sulfur cycling earlier in Earth's history (13–17).
Recently, our knowledge of the Beggiatoaceae was dramatically expanded by systematic single-cell rRNA gene sequencing (1). Newly proposed candidate taxa include multiple clades of marine Beggiatoa-like organisms (“Candidatus Maribeggiatoa,” “Ca. Isobeggiatoa,” and “Ca. Parabeggiatoa”) as well as two groups of small, Thiomargarita-like organisms (“Ca. Thiophysa” and “Ca. Thiopilula”). Very little is known about “Ca. Thiophysa” and “Ca. Thiopilula.” Like other large, vacuolated, sulfur-oxidizing bacteria, both candidate genera remain uncultivated. “Ca. Thiophysa” and “Ca. Thiopilula” are spherical, vacuolated bacteria with elemental sulfur inclusions and either a completely sessile lifestyle or some limited rolling or twitching motility (1). “Ca. Thiophysa” organisms are solitary spherical cells with diameters of 56 to 90 μm. “Ca. Thiopilula” organisms are somewhat smaller, with reported diameters of between 14 and 67 μm, and occur as clusters of cells bound in thick biofilm matrices or enclosed in diatom frustules. “Ca. Thiopilula” spp. were first identified in oxygen minimum zone sediments in Namibia (1) and have also been found at cold seeps in the Barents Sea (18).
Here we report the occurrence of “Ca. Thiopilula” spp. in deep cold seeps near Barbados. Cold seeps occur in the Barbados accretionary prism, where sediment compaction and tectonic processes facilitate the movement of methane-rich fluids to the sediment-water interface. These hydrocarbon fluxes stimulate sulfate reduction and the anaerobic oxidation of methane (AOM), which produce sulfide-enriched sediments that in turn support chemosynthetic communities of free-living microorganisms and symbiotic invertebrates (19, 20). We used microscopy and rRNA methods to describe a Barbados microbial mat community containing abundant “Ca. Thiopilula”-like bacteria and applied metatranscriptomic analysis to explore gene expression by Barbados “Ca. Thiopilula” organisms when exposed to anoxic, sulfidic conditions. To our knowledge, this represents the first description of “Ca. Thiopilula” spp. that occur as solitary cells as well as the first metatranscriptomic information for members of this candidate genus and one of the first metatranscriptomic data sets from cold seep environments in general.
MATERIALS AND METHODS
Sample collection and microscopy.
Samples for this study were retrieved during a research cruise to the Barbados accretionary prism aboard the R/V Atlantis in June 2012 (Fig. 1). Cold seep sediments (13°46.65′N, 57°32.25′W; 4,743-m water depth) in the vicinity of the Manon mud volcano were collected by using push core (∼15 to 30 cm long) with the ROV Jason. Cold seep sediments, adjacent to Calyptogena clam colonies, were covered with a white microbial mat at the sediment-water interface (Fig. 2A). Immediately after collection, biofilm and sediment were observed and imaged with an Olympus SZX-16 stereomicroscope equipped with a Canon EOS T2i digital camera (Olympus, Japan). Samples of the biofilm for DNA extraction were fixed in RNAlater (Ambion, USA) and stored at −80°C, samples for fluorescence microscopy were preserved in 4% paraformaldehyde (PFA), and sediment samples for incubation experiments and live analysis were stored and transported at 4°C. Core top waters were syringe filtered (0.2 μm), acidified to pH 1 with HCl, and stored at 4°C until analysis.
FIG 1.

Map and geologic setting of the study site. Samples were collected from cold seeps in the Manon area of the Barbados accretionary complex (13°46.65′N, 57°32.25′W; 4,743-m water depth). (Bathymetry data courtesy of L. Brothers and the U.S. Geological Survey.)
FIG 2.

Images of cold seeps and Barbados “Ca. Thiopilula” cells. (A) Push core collection of cold seep sediments at a 4,743-m water depth. (B) Low-magnification image of the white biomat in panel A showing large spherical cells (5 to 40 μm in diameter) in the biofilm. (C to E) Photomicrographs of “Ca. Thiopilula” cells and associated microorganisms in the biofilms. (C) Phase-contrast image showing intracellular sulfur inclusions; (D) DAPI-stained cells; (E) FISH photomicrograph of postincubation sediments using the general bacterial probe EUBMIX with both 6-carboxyfluorescein (green) and cy5.5 (red) filter sets.
rRNA gene methods.
DNA was extracted from the biofilm and sediment by using the MoBio PowerSoil DNA isolation kit (MoBio Laboratories, USA). Extractions were performed according to the manufacturer's instructions, except that aliquots of each sample were bead beaten for 5, 10, and 15 min and recombined to reduce DNA extraction bias. rRNA gene libraries were generated from DNA extracts by amplifying the V3 and V5-V6 hypervariable regions of the 16S rRNA gene. V3 libraries were created by using bar-coded adaptor primers described previously (21), except that the priming sequence of the reverse primer was modified slightly (new V3 reverse primer, TTACCGCGGCTGCTGG) and a degenerate sequence of 4 to 7 nucleotides (e.g., NNNN) was added to each reverse primer (in addition to the forward primer) to improve cluster identification. Amplification by PCR was performed with HotStarTaq Plus polymerase (Qiagen, USA) under the following conditions: an initial denaturation step at 95°C for 5 min; 25 cycles of denaturation (95°C for 1 min), annealing (50°C for 1 min), and elongation (72°C for 1 min); and a final elongation step at 72°C for 7 min. Following extraction, PCR products were purified by using the Zymoclean gel DNA recovery kit (Zymo Research, USA), and libraries were pooled and sequenced at the University of Minnesota Genomics Center (UMGC) by using the Illumina MiSeq system (150 cycles, paired end). V5-V6 libraries were generated by using the automated amplicon sequencing service at the UMGC, with primers V5_F (RGGATTAGATACCC) and V6_R (CGACRRCCATGCANCACCT) (V6_R is 1064R [22], and V5_F is F784 [23]). V5-V6 primers included tail sequences to allow subsequent bar-coding (forward primer TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG and reverse primer GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG). Samples were amplified at the UMGC with Kapa HiFidelity Hot Start polymerase (Kapa Biosystems, USA), with an initial denaturation step at 95°C for 5 min and 25 cycles of denaturation (98°C for 20 s), annealing (55°C for 15 s), and elongation (72°C for 1 min). Barcodes and Illumina sequencing adaptors were attached with a second amplification step according to the recommended amplicon sequencing protocols (Illumina), and libraries were size selected (Caliper LabChip XT; PerkinElmer, USA), pooled, and sequenced by using the Illumina MiSeq system (300-bp paired end).
We used 4′,6-diamidino-2-phenylindole (DAPI) staining to image the cells and fluorescence in situ hybridization (FISH) to verify that the large sulfur-oxidizing bacteria were alive after the incubation experiments. FISH and DAPI staining were performed on paraformaldehyde-fixed sediment samples as described previously (24), using the general bacterial FISH probe EUBMIX (1:2 mixture of GCTGCCTCCCGTAGGAGT and GCWGCCACCCGTAGGTGT [25]). Cells were imaged with an Olympus BX61 compound microscope equipped with an Olympus DP72 camera running the Olympus CellSens Dimensions software package (Olympus, Japan). Cell diameters were measured from photomicrographs of fixed and live cells.
To verify the identity of the “Ca. Thiopilula”-like organisms, four individual cells were picked from the cold-seep sediments by a pipette and pooled for DNA extraction. DNA was extracted from the picked cells as described above, and 16S rRNA genes were amplified by using GoTaq Green master mix (Promega, USA) with Beggiatoaceae-specific primers VSO233Fmod (CCTATGYCRGATTAGCTW) and ITSReub (GCCAAGGCATCCACC). Primer VSO233Fmod was modified from previously described primer VSO233F (1), and the PCR amplification protocol was performed according to protocols described previously (1), except that the annealing temperature was lowered to 52°C and the total number of cycles was increased to 30. A second amplification was necessary to generate sufficient product for sequencing. The PCR product was directly sequenced with primers VSO233Fmod and VSO1300Rmod (ATCCGGACTACGAGWAR) at the UMGC with a capillary ABI 3730xl sequencer and ABI BigDye Terminator version 3.1 chemistry (Applied Biosystems, USA).
Incubation experiments.
Sediment incubations were performed as part of a larger experiment on phosphorus cycling, and the phosphorus-related results will be reported in detail elsewhere. Sediment samples for incubations were initially spiked with 0.5 mM phosphate (pH 8) after collection and stored in sealed conical tubes with minimal headspace at 4°C. Prior to incubation, sediment samples were gently washed three times by adding and removing 25 ml of phosphate-free artificial seawater. Sediment samples were then incubated under anoxic conditions for 6 h, in a fashion similar to that for experiments described previously (11). Two grams of sediment was added to 20 ml of anoxic artificial seawater (pH 7.3) on the surface of an agar plug with 1 mM Na2S (pH 7.3). The pH of the seawater solution was 7.3 before and after incubation, and the redox indicator resazurin was used to ensure that sediment samples remained anoxic for the duration of the experiment. After 6 h, incubation sediment samples were immediately preserved in RNAlater under anoxic conditions and stored at −80°C until RNA extractions were performed. Aliquots (1.5 ml) of the supernatant were collected at the start and at the end of the incubation experiment. The supernatants were syringe filtered (0.2 μm), stored frozen (−20°C), and later analyzed for major anions and cations by ion chromatography (IC) and inductively coupled plasma optical emission spectroscopy (ICP-OES), respectively, at the University of Minnesota Analytical Geochemistry Laboratory.
RNA extraction and cDNA library construction.
RNA was extracted from postincubation sediment samples by using the MoBio PowerBiofilm RNA isolation kit, according to the manufacturer's instructions (MoBio Laboratories, USA). DNA was removed with two DNase treatments: an on-column treatment with DNase I (MoBio, USA) and a second treatment with the Turbo DNase enzyme (Ambion, USA). The RNA extract was purified and concentrated with the RNA Clean & Concentrator kit (Zymo, USA). The absence of DNA from the RNA product was verified by attempting to amplify 16S rRNA genes with 35 PCR cycles and primers 27f and 1492r (94). RNA quality was checked by using a Bioanalyzer assay (Agilent Technologies, USA) as well as by successful amplification of 16S rRNA transcripts by reverse transcription (RT)-PCR (Sensiscript; Qiagen, USA) with primer pair 27f-1492r.
A cDNA library was generated from the RNA extract with the Ovation RNA-Seq system V2 (NuGEN Technologies, USA), according to the manufacturer's instructions. cDNA was purified with the MinElute Reaction Cleanup kit (Qiagen, USA), quantified with a NanoDrop instrument (Thermo Scientific, USA), and submitted for library preparation (TruSeq; Illumina, USA) and paired-end sequencing on MiSeq and HiSeq 2000 instruments (Illumina, USA) at the UMGC.
Bioinformatic analyses.
Analysis of 16S rRNA gene amplicon libraries was performed by using mothur (26) and QIIME (27). First, raw sequences were quality trimmed and filtered (average quality of 30 and minimum length of 80 bp) by using Sickle (https://github.com/najoshi/sickle), sequences containing residual adaptors were removed with cutadapt (95), and paired-end sequences were assembled with PEAR (28). Operational taxonomic units (OTUs) were defined at 97% similarity by using default parameters in QIIME, sequences were classified by using the Silva reference sequences and taxonomic outline (29), and the phylogenetic positions of certain representative sequences were determined by importing them into ARB (30). Putative chimeric sequences, as determined by UCHIME (31), were excluded from analyses.
Raw metatranscriptomic sequences were quality trimmed and filtered by using Sickle (average minimum quality of 28 and minimum length of 50 bp), screened for residual adaptors with cutadapt, dereplicated (identical forward and reverse reads) with an in-house script, and checked for quality with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Due to the presence of overrepresented kmers at the 5′ end, the first 10 bp were clipped from all sequences. Most rRNA sequences were removed prior to assembly with riboPicker (32) and then with additional BLASTN screening (33) following assembly. Transcripts were assembled with IDBA-UD (34), with kmer sizes of 45, 55, and 65. Read mapping was performed with the Burrows-Wheeler aligner (BWA) (35) using default parameters, and assembly quality was verified visually by using the Integrative Genome Viewer (IGV) (36). Contig coverage was determined from BWA mapping.
Contigs were annotated by using the standard bioinformatics pipeline at the MG-RAST server (37) and via the Rapid Analysis of Multiple Metagenomes with Clustering and Annotation Pipeline (RAMMCAP) (38) at the Community Cyberinfrastructure for Advanced Microbial Ecology Research and Analysis (CAMERA) online workbench (39). We identified Beggiatoaceae sequences with PhymmBL (40), using a confidence score cutoff of 0.65 (as determined by simulated taxonomic classification [see below; see also Fig. S1 in the supplemental material]) and a custom database amended with contigs from the following draft genomes: Beggiatoa alba B18LD (NCBI BioProject number 62137), “Beggiatoa orange Guaymas” (41, 42), “Beggiatoa PS” and “Beggiatoa SS” (43), Thioploca araucae Tha-CCL (NCBI BioProject number 19383), and an unpublished Thiomargarita nelsonii genome. The taxonomic composition of the mRNA subset was determined by PhymmBL classification of a subset of 31 conserved universal phylogenetic marker genes (44). We assessed the accuracy of PhymmBL taxonomic assignments by fragmenting and classifying four microbial genomes that were not represented in our PhymmBL database. When fragmented in silico to 1,000 bp, genome fragments were correctly assigned with a PhymmBL confidence cutoff of 0.65 at rates of >85% and >70% at the class and family levels, respectively, and assignment accuracy increased with increasing PhymmBL confidence scores (see Fig. S1a and S1b in the supplemental material) (40). Fewer than 50% of the total simulated sequences could be assigned with this cutoff value (see Fig. S1c and S1d in the supplemental material).
Small-subunit (SSU) rRNA transcripts from the metatranscriptome were assembled with EMIRGE (45) on a data set of 1 million randomly selected read pairs from the raw data set. Raw reads were quality screened as described above, except that the minimum average read quality cutoff in Sickle was increased to 30. Nearly full-length EMIRGE transcripts were aligned by using the SINA aligner (46) and manually corrected in ARB (30). Only EMIRGE contigs that represent >0.1% of rRNA transcripts were analyzed. Phylogenetic analysis of rRNA sequences included representatives of major groups in the Beggiatoaceae (1). Prior to phylogenetic analysis, alignments were end clipped so that all sequences were of equal length, with the exception of the shorter 16S rRNA gene sequence directly amplified from handpicked cells (950 characters), and positions with >50% gaps were removed (final alignment, 1,122 positions). Maximum likelihood analysis was performed with RAxML v.8 (47), using the general time-reversible substitution model with gamma rate variation, as selected by the corrected Akaike information criterion (AICc) with jModelTest v.2.1.5 (48). Base frequencies, proportions of invariant sites, and shape parameter (α) values were estimated from the data. Bootstrapping was performed with 1,000 replicates by using the RAxML rapid bootstrap algorithm. Hydroxylamine oxidoreductase (HAO)-like sequences were aligned with the Expresso algorithm in T-Coffee (49), and dissimilatory sulfite reductase (DsrA) sequences were aligned in ARB by using a database described previously (50). Alignments were trimmed with the automated1 option in TrimAL (51), for final lengths of 293 and 308 amino acids for the DsrA and HAO alignments, respectively. Maximum likelihood analyses were performed with RAxML with 100 rapid bootstrap replicates, using the LG amino acid substitution model (52), as determined by the AICc in ProtTest v.2.4 (53). Proportions of invariant sites and alpha parameter values were estimated from the data.
Nucleotide sequence accession numbers.
The partial 16S rRNA gene from the picked “Ca. Thiopilula” cells was deposited in GenBank under accession number KM279319. Amplicon libraries and the raw metatranscriptomic data set were deposited in the Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession number SRP045208, and assembled contigs can be accessed at MG-RAST (https://metagenomics.anl.gov) under ID 4556388.3.
RESULTS AND DISCUSSION
Observations of cold seeps and large sulfur-oxidizing bacteria.
Sulfide-oxidizing bacteria commonly form extensive microbial mats at cold seeps. These mats generally form near the sediment-water interface, where hydrogen sulfide, produced by the anaerobic oxidation of methane (AOM) and/or sulfate reduction, encounters dissolved oxygen or nitrate. At these interfaces, a variety of sulfide-oxidizing taxa, including Beggiatoa, Arcobacter, Thiothrix, and Thiomargarita, are known to form microbial mats, commonly with certain taxa being enriched within a certain biogeochemical niche or microenvironment (54, 55). Macrofaunas associated with mud volcanoes of the Barbados accretionary prism were previously reported (19, 20), but little work has been done to characterize the microbial communities at these deep-water methane seep sites (56).
Manon is one of several mud volcanoes found eastward of the accretionary prism deformation front (Fig. 1). Manon is a conical mud volcano ∼180 m in height and ∼1,000 m in diameter (57). Calyptogena clam colonies and microbial mats are present on the upper slope and summit (19). In our study, patches of microbial mat and black-stained sediment were found adjacent to beds of Calyptogena clams. Push core samples were collected from mats that consisted of white tufts and pustules (up to 1 mm thick) overlying gray-black sulfide-stained sediments that had an appearance distinct from that of the surrounding buff-colored sediment (Fig. 2). Examination of the white biomat at the sediment-water interface revealed large spherical cells (Fig. 2A and B). Cell diameters of the spherical organisms ranged from 5 to 40 μm, with smaller cells, between 5 and 10 μm in diameter, being much more abundant than larger cells. Cells were solitary and divided by binary fission (Fig. 2D). These cells morphologically resembled solitary Thiomargarita cells that are found at methane seeps but with much smaller cell diameters. The Barbados cells contained internal S globules, which were identified by their high relief and distinctive appearance by phase-contrast microscopy (Fig. 2C) and by the observation of inclusions rapidly dissolving when cells were dehydrated in ethanol (58). A central vacuole was observed under the microscope (Fig. 2). No evidence of motility was observed. The cells were maintained in cold seep sediments at 4°C and observed over a 3- to 5-week period, during which time their internal sulfur globules were slowly consumed and the cells became increasingly fragile and easily disrupted when handpicked via a pipette.
Identification of “Ca. Thiopilula.”
Although microscopic observations showed that large, vacuolated, sulfur-oxidizing bacteria were abundant in the cold seep sediments (Fig. 2), sequences affiliated with the family Beggiatoaceae were either exceedingly rare or absent in 16S rRNA gene amplicon libraries despite the use of two different primer sets (see below). Initial attempts at amplifying rRNA sequences from single cells using Beggiatoaceae-specific primers (1) were unsuccessful. However, following EMIRGE assembly of small-subunit (SSU) rRNA transcripts in the metatranscriptome, we identified a full-length 16S rRNA sequence representing 2.5% of SSU rRNA transcripts that shares 99% nucleotide identity with “Ca. Thiopilula aggregata.” This sequence is the eighth most abundant SSU rRNA transcript in the data set. Phylogenetic analysis places it with “Ca. Thiopilula” sequences in the Beggiatoaceae, along with a second full-length EMIRGE transcript that represents 0.8% of SSU rRNA transcripts (Fig. 3). This second transcript shares 95% identity with “Ca. Thiopilula aggregata.” The only other sequence affiliated with the Beggiatoaceae in the EMIRGE assembly was a sequence that shares 97% identity with Thiomargarita namibiensis and represents 0.19% of total sequences.
FIG 3.

Maximum likelihood phylogeny of abundant EMIRGE-assembled 16S rRNA transcripts from the Beggiatoaceae clade. Sequences in boldface type are the Barbados rRNA transcripts, and the associated value indicates the relative abundance of these sequences with respect to other SSU rRNA transcripts in the data set. Numbers indicate bootstrap values for nodes with >50% bootstrap support. GenBank accession numbers are in parentheses.
Upon inspection of the Barbados “Ca. Thiopilula” sequences retrieved from the EMIRGE assembly, we found that Beggiatoaceae-specific primers had mismatches with the Barbados sequences. We then modified forward primer VSO233F to also hit the Barbados “Ca. Thiopilula” rRNA transcripts (new primer VSO233Fmod). Using this new forward primer, we verified the identity of the large, vacuolated bacteria by successfully amplifying a partial “Ca. Thiopilula” 16S rRNA gene (973 bp) from a DNA extract from handpicked cells. The 16S rRNA gene retrieved from the picked cells shares 99% identity with “Ca. Thiopilula aggregata” and groups with the clade containing the Barbados “Ca. Thiopilula” sequences (Fig. 3).
“Ca. Thiopilula aggregata” is a small Thiomargarita-like bacterium with elemental sulfur inclusions and a large central vacuole (1, 6). “Ca. Thiopilula” bacteria were previously identified in the Namibian shelf and at cold seeps near Norway, at depths of 100 to 200 and 720 m, respectively (1, 18). “Ca. Thiopilula aggregata” cells from Namibia are somewhat larger than the Barbados “Ca. Thiopilula” cells (minimum cell diameter of 5 μm for the Barbados population versus 14 μm for Namibian “Ca. Thiopilula” populations), but their morphologies are otherwise consistent. Unlike the Namibia “Ca. Thiopilula aggregata” cells, which occur primarily as aggregates (1), the Barbados organisms were solitary although still associated with biofilm-like material (Fig. 2). The Barbados “Ca. Thiopilula” organisms did not appear to be motile.
We cannot rule out the possibility that some of the large cells in the sample belong to other clades within the Beggiatoaceae or that there is genetic heterogeneity among the cells. However, the morphology of the Barbados cells is consistent with that previously reported for “Ca. Thiopilula.” Because of this, and given the abundance of “Ca. Thiopilula” rRNA transcripts, we are confident in the conclusion that most of the Barbados cells are “Ca. Thiopilula.”
“Ca. Thiopilula” sequences were exceedingly rare or absent from 16S rRNA gene amplicon libraries despite their apparent numerical abundance in the mat and in the metatranscriptome. Numerous previous studies (59–65) have also noted difficulties in obtaining 16S rRNA gene sequences for members of the Beggiatoaceae from samples visibly dominated by large sulfur-oxidizing bacteria. Some differences among rRNA gene and transcript libraries are expected, given that different organisms maintain different ribosome numbers and that microbial populations can be inactive (66–68), but the discrepancy between the rRNA gene and transcript gene abundance of “Ca. Thiopilula” in this case was striking and was not observed for other populations in the sample (Fig. 4). Biases against members of the Beggiatoaceae with standard primer sets are well known (1, 69), and indeed, we were unable to amplify a “Ca. Thiopilula” 16S rRNA gene from single cells until primers were modified based on information from the metatranscriptome. However, the primer sets used for 16S rRNA gene amplicon sequencing (V5-V6 and V3) have no mismatches with the Beggiatoaceae sequences in Fig. 3, including all three Barbados “Ca. Thiopilula” sequences. “Ca. Thiopilula” spp. and other members of the Beggiatoaceae are known to have introns in their rRNA genes (63), which theoretically could have produced long sequences that would have been removed with size selection by gel extraction or the Caliper LabChip XT instrument during library preparation. However, no intron sequences were present in the partial 16S rRNA gene sequence from the picked-cell extract. Other possibilities include bias during the DNA extraction step (although we used multiple bead beatings in an attempt to offset this bias) or a relatively small amount of “Ca. Thiopilula” DNA relative to its rRNA content and to that of other bacteria. Perhaps, the small cytoplasmic volume relative to the cell size of large sulfur-oxidizing bacteria represents a special case in which, compared to other microorganisms, more rRNA than DNA is produced, although many large bacteria are also known to exhibit extreme polyploidy and thus also have more DNA per cell (exceeding 10,000 genome copies/cell in some cases) (70, 71).
FIG 4.

Composition of the biofilm and sediment communities based on 16S rRNA gene libraries (A and B), 16S rRNA transcripts from the metatranscriptome (C), and expressed universal marker genes (44) in the metatranscriptome (D). The 16S rRNA gene libraries were generated from the white biofilm only, with bacterium-specific primers (A), and postincubation biofilm and sediment, with universal primers (B). The two “Ca. Thiopilula” transcripts in Fig. 2 represent 3.3% of the community in panel C.
Diversity and structure of the cold seep microbial community.
The Barbados “Ca. Thiopilula” organisms were associated with many smaller microorganisms, both aggregated in the white biofilm material and in the shallow sediments (Fig. 2). rRNA gene libraries were generated from both the white biofilm material (127.5k sequences following quality screening and assembly, 16S rRNA gene V3 region) as well as the sediment and biofilm samples that were used in the incubation experiment (51k sequences following quality screening and assembly, V5-V6 region, collected following incubation). Both libraries were dominated by diverse Epsilon-, Delta-, and Gammaproteobacteria and included bacteria from the Bacteriodetes and the OD1 and BD1-5 clades (Fig. 4). The incubation material included both the white biofilm and underlying sediment and had relatively fewer Epsilonproteobacteria and more Gammaproteobacteria than the white biofilm alone (Fig. 4A and B). The vast majority of archaea in the incubation library (Fig. 4B) were affiliated with anaerobic oxidation of methane group II (ANME-II) in the Methanosarcinales (see Fig. S2 in the supplemental material).
rRNA transcripts and universal marker genes (44) expressed during the incubation experiment (Fig. 4C and D; see also Table S1 in the supplemental material) showed a taxonomic distribution generally similar to that of the incubation rRNA gene library but with different proportions of the major groups. rRNA transcripts had a larger contribution from Epsilonproteobacteria and ANME-II archaea than did the rRNA gene libraries. Universal marker genes in the metatranscriptome included a large subset of sequences assigned to the Firmicutes (Fig. 4D). “Ca. Thiopilula” sequences were present in the metatranscriptome (3.3% of rRNA transcripts) (Fig. 3) but were exceedingly rare in the rRNA gene libraries. The 16S rRNA gene library from the white biofilm (V3 primers) contained only 14 total sequences associated with “Ca. Thiopilula” (0.01% of the library), and the incubation rRNA gene library did not contain any “Ca. Thiopilula” sequences.
The most abundant rRNA gene and transcript sequences identified as likely sulfur-oxidizing microorganisms in the community were representatives of the Epsilonproteobacteria (Fig. 4). Most epsilonproteobacteria from the rRNA gene data sets were relatives of Sulfurovum, with a smaller number of Sulfurimonas-like sequences (Fig. 4). Based on the frequent occurrence of environmental sequences from these two clades in sulfidic habitats, and from the study of the isolates that have been characterized to date, Sulfurovum spp. and Sulfurimonas spp. are likely microaerophilic sulfur oxidizers (72). The Barbados cold seep community also contains numerous presumed organoheterotrophic taxa, including abundant Vibrionales sequences in the Gammaproteobacteria (primarily Photobacterium spp.). Other abundant taxa include anaerobic and aerobic methanotrophs (ANME-II archaea and Methylococcales bacteria) and presumed sulfate reducers in the Desulfobulbaceae and Desulfobacteraceae families within the Deltaproteobacteria.
“Ca. Thiopilula” gene expression under anoxic, sulfidic conditions.
Presumably, vacuolated sulfur-oxidizing bacteria are especially adept at life in episodically anoxic sediments, because they can store energy in the form of elemental S and nitrate as well as in other storage compounds such as polyphosphate and glycogen (11). In order to explore the ecophysiology of the Barbados “Ca. Thiopilula” organisms when exposed to anoxia, we used metatranscriptomics to characterize gene expression by the “Ca. Thiopilula” mat community when incubated under anoxic, sulfidic conditions. The metatranscriptome generated from the incubation sediment contained 26.5 million reads and 4.8 Gb of sequence data following quality filtering, of which 7.8% were mRNA transcripts. De novo assembly produced 15.5k contigs between 200 and 7,323 bp, with average and N50 lengths of 482 and 505 bp, respectively.
FISH analyses of sediment and biofilm before and after incubation confirmed that the large sulfur-oxidizing bacteria were alive throughout the incubation (Fig. 2E). Using a custom PhymmBL database supplemented with all other available Beggiatoaceae genome sequences, 34 total contigs were assigned to the Beggiatoaceae family, of which 24 sequences were annotated to RefSeq by MG-RAST and 21 sequences were assigned to clusters of orthologous groups (COG) and nonsupervised orthologous groups (NOG) by either MG-RAST or RAMMCAP (Table 1). The Beggiatoaceae contigs ranged from 200 to 1,002 bp in length, with coverage of between 1.59× and 25.8×. Expressed Beggiatoaceae sequences include multiple ribosomal proteins and pilus assembly proteins and several sequences involved in sulfur and nitrogen metabolism. Because of their abundance in the metatranscriptome, and because only a single large sulfur-oxidizing bacterium morphotype was observed in the microbial mat, we assume that all Beggiatoaceae contigs originated from the “Ca. Thiopilula” population.
TABLE 1.
Summary of Beggiatoaceae contigs in the cold seep metatranscriptomed
| Contig | Length (bp) | Coverage (fold) | PhymmBL scorea | RefSeq annotation(s)b | COG/NOG identification(s)c | COG/NOG description | Top BLASTX match(es) (to nr database) |
|---|---|---|---|---|---|---|---|
| 976 | 1,002 | 7.2 | 0.98 | Hypothetical protein | NOG85955 | Hydroxylamine protein | Hydroxylamine oxidoreductase |
| 1182 | 937 | 7.6 | 0.98 | Hypothetical protein | NOG85955 | Hydroxylamine protein | Hydroxylamine oxidoreductase |
| 1909 | 781 | 11.4 | 0.98 | Adenylylsulfate reductase subunit alpha | COG1053 | Succinate dehydrogenase/fumarate reductase, flavoprotein subunit | AprA |
| 3197 | 635 | 10.9 | 0.98 | 50S ribosomal protein L3 | COG0087 | Ribosomal protein L3 | 50S ribosomal protein L3 |
| 4195 | 565 | 20.2 | 0.65 | Hsp20/alpha crystallin family protein; Hsp20 family protein; small heat shock protein | COG0071 | Molecular chaperone (small heat shock protein) | Heat shock protein Hsp20 |
| 5444 | 501 | 25.8 | 0.93 | 50S ribosomal protein L9; 30S ribosomal protein S18 | COG0071; COG0359 | Ribosomal protein S18; ribosomal protein L9 | 50S ribosomal protein L9 |
| 5731 | 490 | 6.6 | 0.78 | Translation elongation factor G | COG0480 | Translation elongation factors (GTPases) | Elongation factor G |
| 5916 | 483 | 6.8 | 0.97 | Sulfite reductase, dissimilatory-type alpha subunit | COG2221 | Dissimilatory sulfite reductase (desulfoviridin), alpha and beta subunits | Reverse-type dissimilatory siroheme sulfite reductase subunit A |
| 6495 | 462 | 6.6 | 0.95 | Nitrate transporter | Nitrate transporter | ||
| 6503 | 461 | 3.6 | 0.65 | Hypothetical protein | NOG126924 | No NOG description | Hypothetical protein |
| 7097 | 443 | 31.3 | 0.88 | Tfp pilus assembly protein | COG4969 | Tfp pilus assembly protein PilE | PilE/pilin |
| 8024 | 417 | 3.8 | 0.66 | Putative pentachlorophenol-4-monooxygenase | COG0654 | 2-Polyprenyl-6-methoxyphenol hydroxylase and related FAD-dependent oxidoreductases | Hypothetical protein; 2-polyprenyl-6-methoxyphenol hydroxylase-like oxidoreductase monooxygenase |
| 8452 | 407 | 2.6 | 0.88 | Phosphoribosylaminoimidazole synthetase; phosphoribosylformyl-glycinamidine cycloligase | COG0150 | Phosphoribosylaminoimidazole (AIR) synthetase | Phosphoribosylaminoimidazole synthetase |
| 9001 | 394 | 5.6 | 0.68 | Protein of unknown function, DUF938 | Hypothetical protein | ||
| 9545 | 382 | 4.5 | 0.69 | COG4726 | Tfp pilus assembly protein PilX | Hypothetical protein | |
| 12436 | 331 | 8.6 | 0.97 | Truncated adenosine-5′-phosphosulfate reductase alpha subunit | Adenosine-5′-phosphosulfate reductase | ||
| 12633 | 328 | 4.6 | 0.68 | LSU ribosomal protein l6p, 50S ribosomal protein L6 | COG0097 | Ribosomal protein L6P/L9E | 50S ribosomal protein L6 |
| 13086 | 320 | 3.8 | 0.67 | Signal recognition particle protein Srp54 | COG0541 | Signal recognition particle GTPase | GTP-binding signal recognition particle Srp54 G-domain-containing protein |
| 13206 | 319 | 2.2 | 0.68 | Radical SAM domain-containing protein | COG0641 | Arylsulfatase regulator (Fe-S oxidoreductase) | Radical SAM domain-containing protein; anaerobic sulfatase-maturating protein |
| 13888 | 307 | 4.4 | 0.93 | l-Threonine O-3-phosphate decarboxylase | COG0079 | Histidinol-phosphate/aromatic aminotransferase and cobyric acid decarboxylase | l-Threonine O-3-phosphate decarboxylase |
| 14042 | 305 | 6.8 | 0.70 | RNase HI | NOG69418 | RNase H | RNase HI |
| 14149 | 303 | 9.1 | 0.86 | Fimbrial protein precursor | COG4969 | Tfp pilus assembly protein, major pilin PilA | Fimbrial protein |
| 15353 | 282 | 3.3 | 0.69 | Dihydrolipoyl dehydrogenase | COG1249 | Pyruvate/2-oxoglutarate dehydrogenase complex, dihydrolipoamide dehydrogenase (E3) component, and related enzymes | Dihydrolipoyl dehydrogenase |
| 16386 | 265 | 3.2 | 0.66 | Transposase IS116/IS110/IS902 family protein | COG3547 | Transposase and inactivated derivatives | Transposase |
PhymmBL score for the family Beggiatoaceae (see Fig. S1 in the supplemental material for more information regarding PhymmBL scores).
Based on MG-RAST annotation.
Based on either MG-RAST or RAMMCAP annotation.
Only sequences with either an MG-RAST or an RAMMCAP annotation are shown. LSU, large subunit; SAM, S-adenosylmethionine.
During incubation, the Barbados “Ca. Thiopilula” population expressed two genes involved in sulfur oxidation, reverse-type dissimilatory sulfite reductase subunit A (dsrA) and adenosine-5′-phosphosulfate (APS) reductase subunit A (aprA). Both of these genes encode enzymes for the oxidation of intracellular sulfur globules (43, 73–75), and the expression of these genes is consistent with observations of intracellular S0 in “Ca. Thiopilula” cells. Reverse-type dissimilatory sulfite reductase (Dsr) catalyzes the oxidation of S0 to sulfite, and APS reductase (Apr) converts the sulfite to APS. APS is then dephosphorylated to sulfate, which is transported out of the cell (43). Phylogenetic analysis of the expressed DsrA sequence shows that it groups with Beggiatoa and Thiomargarita sequences in a large group with other reverse-type DsrA sequences from sulfur-oxidizing bacteria (Fig. 5A). Reverse DsrA phylogenies are generally congruent with 16S rRNA gene trees (50), so the pattern shown in Fig. 5A is unlikely to result from horizontal gene transfer.
FIG 5.

Maximum likelihood analysis of reverse DsrA (A) and hydroxylamine oxidoreductase-like (B) sequences. DsrA sequences from Moorella thermoacetica (GenBank accession number NC_007644) and Pyrobaculum islandicum (GenBank accession number U75249) were used as outgroups in panel A, and the tree in panel B is midpoint rooted. Numbers at nodes indicate support based on 100 fast bootstrap replicates (only values of >50 are shown). GenBank accession numbers are shown after the organism name.
The “Ca. Thiopilula” population also expressed multiple genes that are likely involved in nitrate reduction. The two longest contigs in the Beggiatoaceae bin are most similar to a recently characterized octaheme cytochrome that was proposed to be involved in the reduction of nitrogenous compounds in vacuolated marine Beggiatoa (“Ca. Maribeggiatoa”) (41, 42). This Beggiatoa protein, BOGUAY_0691, was found to have nitrite reductase, hydroxylamine oxidase, and hydrazine oxidase properties in vitro (42). Although its physiological role remains uncertain, MacGregor et al. (41, 42) argued that the latter two functions are unlikely and that BOGUAY_0691 probably serves as a nitrite reductase in vivo. Indeed, its expression under anoxic, sulfidic conditions in our incubation experiments is further evidence for its involvement in reductive rather than oxidative dissimilatory N metabolism. Phylogenetic analysis of the Barbados “Ca. Thiopilula” sequence places it in a clade with the BOGUAY_0691 Beggiatoa sequence, a “Ca. Omnitrophus fodinae” sequence classified as a hydroxylamine oxidoreductase, and the sequences of several other hypothetical proteins from sulfide-oxidizing symbionts (Fig. 5B). This clade is only distantly related to well-characterized hydroxylamine oxidoreductases from Nitrosomonas spp. and other aerobic ammonium oxidizers (Fig. 5B) (76). Similar octaheme cytochromes have been proposed to function in a novel dissimilatory reduction of nitrate to ammonia (DNRA) pathway (77–79). DNRA has been shown for Thioploca and is suggested to occur in other members of the Beggiatoaceae (80). However, further characterization of these octaheme cytochromes will be required to identify their metabolic role in the Beggiatoaceae and determine whether or not they are involved in DNRA. The Barbados “Ca. Thiopilula” population also expressed a nitrate transporter in the major facilitator superfamily (Table 1). Expression of this transporter under anoxic conditions, combined with that of the BOGUAY_0691-like proteins, suggests that the Barbados “Ca. Thiopilula” organisms were reducing nitrate during the anoxic incubation.
The expression of genes for both sulfur oxidation and nitrate reduction is consistent with the presence of intracellular sulfur and a central vacuole in the Barbados “Ca. Thiopilula” organisms. It therefore appears that, like other vacuolated members of the Beggiatoaceae, Barbados “Ca. Thiopilula” is capable of anaerobic sulfur oxidation via nitrate respiration. Because Dsr is thought to be essential for the oxidation of stored elemental sulfur, it is likely that internal elemental sulfur was serving as the electron donor (Fig. 6). The nitrate transporter could be used for nitrate transport across either the cytoplasmic membrane or the vacuolar membrane, where respiration of nitrate is thought to occur in vacuolated Beggiatoa (81) (Fig. 6). Nitrate (30 to 50 μM) was present in both the core top water and the incubation supernatant (Table 2), so we cannot rule out the possibility that the organism was responding to nitrate in the sediments rather than respiring nitrate stored in its vacuole.
FIG 6.
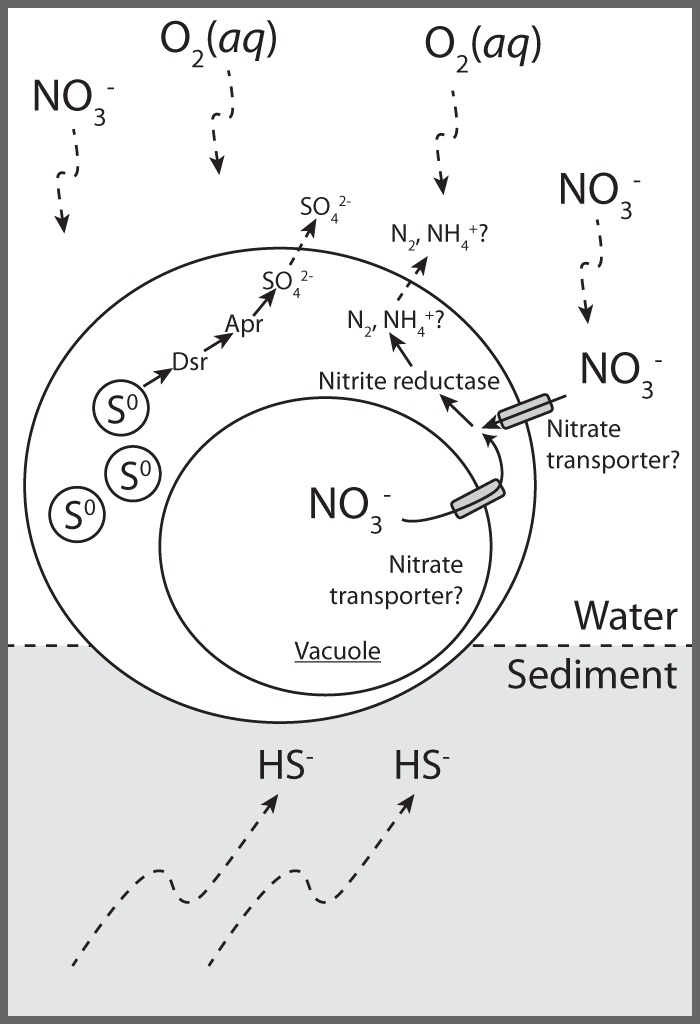
Conceptual biogeochemical schematic of “Ca. Thiopilula” cells in the cold seep setting, based on metatranscriptomic analysis of the incubation sediment. The proposed roles for the expressed nitrogen and sulfur metabolism genes are indicated.
TABLE 2.
Geochemistry of core top water and incubation supernatantsa
| Element/compound | Concn in: |
||
|---|---|---|---|
| Core top water | Incubation supernatant |
||
| 0 h | 6 h | ||
| Ba (μmol/kg) | 0.5 | 0.5 | 0.7 |
| Ca (mmol/kg) | 10.6 | 8.9 | 7.7 |
| Fe (μmol/kg) | 4.8 | 4.6 | 5.4 |
| K (mmol/kg) | 12.0 | 10.3 | 9.4 |
| Li (μmol/kg) | 37.4 | 56.3 | 48.2 |
| Mg (mmol/kg) | 55.6 | 40.4 | 36.4 |
| Mn (μmol/kg) | 1.5 | 1.6 | 1.6 |
| Na (mmol/kg) | 469.1 | 400.8 | 381.6 |
| Sr (μmol/kg) | 89.2 | 82.6 | 71.2 |
| Cl− (mmol/kg) | ND | 495.9 | 472.4 |
| Br− (μmol/kg) | 1.0 | 0.9 | 0.8 |
| SO42− (mmol/kg) | 30.4 | 18.5 | 16.4 |
| NO3− (μmol/kg) | 31.3 | 34.3 | 48.3 |
ND, no data (sample was acidified with HCl). Samples were collected at the start and at the end (6 h) of incubation.
Because no reference genome is available for “Ca. Thiopilula,” identification of “Ca. Thiopilula” transcripts was dependent on sequence similarity to other members of the Beggiatoaceae. Metatranscriptome contigs are too short to be binned based on tetranucleotide signatures or similar methods (82, 83). Therefore, classification of contigs is subject to “database bias,” in which unequal representation of taxonomic groups in genome databases can cause sequences to be incorrectly assigned to the “next best” taxon (e.g., see reference 84). Horizontal gene transfer, which has occurred between the Beggiatoaceae and other groups (85), would further result in inaccurate transcript assignment. Simulated PhymmBL classification of genomes that were not present in the database showed that while >85% and >70% of fragments were accurately classified at the class and family levels, respectively, only 10 to 50% of the total sequences could be assigned (see Fig. S1 in the supplemental material). Therefore, we expect that additional transcripts from the Barbados “Ca. Thiopilula” population are present in the data set but are either unclassified or assigned to other taxa. “Ca. Thiopilula” sequences account for 3.3% of 16S rRNA transcripts, but only 0.14% of the mRNA sequences could be assigned to the Beggiatoaceae. This likely explains the failure to detect certain transcripts for genes in pathways for sulfur and nitrogen metabolism (e.g., the presence of dsrA but the absence of dsrB and dsrC) (Table 1). Other sequences may also be expressed at levels that were below the limit of detection in our data set.
Nonetheless, de novo metatranscriptome assembly remains a suitable strategy for gene expression analysis of environmental microbial communities (e.g., see reference 86), and our findings here represent the first transcriptomic information for “Ca. Thiopilula”-like organisms. Cultivation-independent techniques have proven essential for an understanding of the ecophysiology of the Beggiatoaceae (e.g., see reference 80), and our approach here represents another avenue for the study of large sulfur-oxidizing bacteria that are currently not in culture (see also reference 11). While isotope labeling approaches indicate sulfide oxidation linked to DNRA in related organisms such as Thioploca spp. (80) and the aerobic oxidation of sulfide by Thiomargarita was measured by using microsensors (87), the anaerobic metabolism of Thiomargarita-like bacteria has not been previously documented. Our results link specific genes associated with anaerobic nitrate and sulfur metabolism in other organisms to metabolic pathways that have previously only been suspected to be active in Thiomargarita and “Ca. Thiopilula” (Fig. 6). Future analyses combining gene expression analyses with other cultivation-independent techniques will be essential to further define other physiological attributes of “Ca. Thiopilula”-like organisms from diverse environments.
Metatranscriptomic analysis of microorganisms co-occurring with “Ca. Thiopilula” in the Barbados cold seep microbial community.
Abyssal cold seeps like those described in the present study receive negligible inputs of energy and organic carbon from the surface ocean. Instead, primary productivity at the sediment surface is supported largely by sulfide production from AOM, which is, in turn, fueled by advection of methane from deep-seated sources. Based on their abundance in the DNA and RNA libraries, the microbes very likely responsible for the majority of AOM at this site are the abundant ANME-II archaea and Deltaproteobacteria. Indeed, some of the most highly expressed transcripts in the metatranscriptome overall were the methyl coenzyme M reductase and associated genes (mcrABCD) by the Methanomicrobia (Fig. 7). The most abundant rRNA sequences identified as archaea in the metatranscriptome are an ANME-IIa phylotype and an ANME-IIc phylotype, which represent 4% and 2.5% of the total SSU rRNA transcripts, respectively (see Fig. S2a in the supplemental material). The most abundant rRNA sequences identified as Deltaproteobacteria are two phylotypes in the SEEP-SRB1 group (family Desulfobacteraceae), which represent 1.8% and 1.7% of the total SSU rRNA transcripts, and two phylotypes in the Desulfobulbaceae family, which represent 1.7% and 1.5% of the total SSU rRNA transcripts (see Fig. S2b in the supplemental material). Members of the SEEP-SRB1 clade are generally thought to be the common syntrophic partners of ANME-II organisms (88, 89). In contrast, in a recent study of deep cold seeps in the Japan Trench (5,346-m water depth), SEEP-SRB1 bacteria were rare, and the most abundant Deltaproteobacteria were instead members of the SEEP-SRB3 and SEEP-SRB4 clades in the Desulfobulbaceae (90). However, the abundance of SEEP-SRB1 sequences in the Barbados sediments indicates that the ANME-II/SEEP-SRB1 association may indeed occur at abyssal depths, although the abundance of Desulfobulbaceae group Deltaproteobacteria suggests that other syntrophic associations are possible (see also reference 91).
FIG 7.
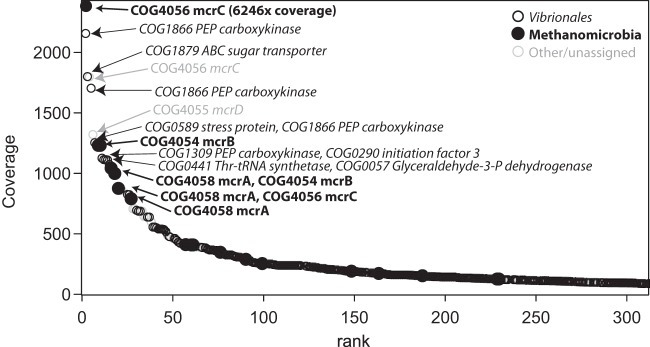
Rank abundance of sequences assigned to COG categories in the metatranscriptome, based on coverage information. Sequences assigned to the order Vibrionales (primarily Photobacterium spp.) and the class Methanomicrobia are especially abundant in the data set. The identity and COG description of the most highly expressed sequences are indicated.
The sulfide generated in the cold seep sediments supports abundant sulfide-oxidizing bacteria. In addition to “Ca. Thiopilula,” sulfur-oxidizing Epsilonproteobacteria (primarily Sulfurovum spp.) were abundant and active in the mat community (Fig. 4). Sulfurovum spp. have been previously reported at other abyssal methane seeps (90) and may represent important free-living chemolithoautotrophic primary producers at deep cold seeps. The metatranscriptome includes nitrate reductase sequences affiliated with the Epsilonproteobacteria (see Fig. S3 in the supplemental material), which is consistent with a facultative anaerobic life-style of the isolated “Sulfurovumales” strains. Aerobic methanotrophs are also present in the data set (Methylococcales) (Fig. 4), and methanotrophy is another potential source of biomass production at the sediment-water interface.
Many of the most highly expressed genes in the data set are affiliated with Photobacterium spp. in the Vibrionales clade (Gammaproteobacteria) (Fig. 7). Highly expressed Vibrionales transcripts include some that code for carbohydrate metabolism and transport functions, which is consistent with an organoheterotrophic physiology. Alternatively, these highly expressed genes may represent a stress response by this otherwise depth-adapted bacterium. Two abundant transcripts encode enzymes in the gluconeogenesis pathway (phosphoenolpyruvate [PEP] carboxykinase and glyceraldehyde-3-phosphate dehydrogenase) (Fig. 7), and Vezzi et al. (92) reported the upregulation of gluconeogenesis as a stress response in Photobacterium profundum. Photobacterium spp. appear to be active and important organoheterotrophic members of the Barbados community under anoxic conditions, and carbohydrates in the EPS produced by other members of the mat community, potentially including “Ca. Thiopilula,” may act to enrich for these piezophilic heterotrophs.
Overall, the inferred composition of the cold seep microbial community at Barbados is consistent with shallow oxygen penetration and anoxic conditions directly below the sediment-water interface. The presence of both obligate anaerobic and aerobic organisms within immediate proximity of the sediment surface indicates that the white biofilm is poised at a sharp redox interface. ANME-II organisms are thought to be less sensitive to oxygen than ANME-I archaea (93), and their dominance in shallow sediments at this site is consistent with a sharp but unstable redox interface (Fig. 2A). Microorganisms that can oxidize sulfide using both nitrate and presumably oxygen, like “Sulfurovumales” and “Ca. Thiopilula” (see Fig. S3 in the supplemental material), are well suited to this setting. Indeed, as a vacuolated sulfur oxidizer that is able to store both metabolic electron donors and acceptors, “Ca. Thiopilula” may be especially well adapted to methane seep conditions that are known to fluctuate with changes in fluid flow.
Summary and implications.
Although Thiomargarita-like bacteria have been previously reported to compose microbial mats at cold seeps, this study is the first to report gene expression data from such mat communities. The mats described here contained a population of vacuolated sulfur-oxidizing bacteria identified as representatives of the genus “Ca. Thiopilula.” The Barbados organisms were morphologically similar to “Ca. Thiopilula aggregata” bacteria, which are known to occur as aggregates of multiple cells living in close proximity to other vacuolated sulfur-oxidizing bacteria, but were found here at abyssal depths as isolated cells. The expression of genes associated with sulfur oxidation and nitrogen reduction suggests that “Ca. Thiopilula” oxidizes reduced inorganic sulfur compounds using nitrate in a manner similar to that of some related vacuolated bacteria. Gene expression data further suggest that the canonical relationship in which sulfate reduction and AOM produce sulfide that fuels sulfide oxidation is also occurring in Barbados cold seep sediments at abyssal depths, with a diminutive Thiomargarita-like bacterium occupying the niche filled by its larger brethren at shallower methane seeps.
Supplementary Material
ACKNOWLEDGMENTS
We thank the anonymous reviewers for comments that improved the manuscript. We acknowledge C. Nguyen for computing support and the Minnesota Supercomputing Institute for the use of facilities. R. Knurr in the UMN Department of Earth Sciences Analytical Geochemistry Laboratory performed the IC and ICP-AES analyses. We give special thanks to A. Becker and K. Beckman at the UMGC for insightful discussion and advice concerning metatranscriptome sequencing and to A. Hague and D. Gohl at the UMGC for assistance with rRNA gene amplicon sequencing. We thank V. Edgecomb for insightful discussion and advice on metatranscriptomics and C. Van Dover and the crew of the R/V Atlantis and Jason (AT 21-02) for assisting with sample collection in Barbados.
This work was supported by a grant-in-aid from the University of Minnesota and by National Science Foundation grants EAR-1057119 to J.V.B. and OCE-1031050 to C. Van Dover and C. Cunningham at Duke University, the latter of which funded cruise AT 21-02. D.S.J. was supported by a generous postdoctoral fellowship from the Agouron Institute.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00039-15.
REFERENCES
- 1.Salman V, Amann R, Girnth AC, Polerecky L, Bailey JV, Høgslund S, Jessen G, Pantoja S, Schulz-Vogt HN. 2011. A single-cell sequencing approach to the classification of large, vacuolated sulfur bacteria. Syst Appl Microbiol 34:243–259. doi: 10.1016/j.syapm.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Schulz H, Brinkhoff T, Ferdelman T, Mariné MH, Teske A, Jørgensen B. 1999. Dense populations of a giant sulfur bacterium in Namibian shelf sediments. Science 284:493–495. doi: 10.1126/science.284.5413.493. [DOI] [PubMed] [Google Scholar]
- 3.Fossing H, Gallardo VA, Jørgensen BB, Hüttel M, Nielsen LP, Schulz H, Canfield DE, Forster S, Glud RN, Gundersen JK. 1995. Concentration and transport of nitrate by the mat-forming sulphur bacterium Thioploca. Nature 374:713–715. doi: 10.1038/374713a0. [DOI] [Google Scholar]
- 4.McHatton SC, Barry JP, Jannasch HW, Nelson DC. 1996. High nitrate concentrations in vacuolate, autotrophic marine Beggiatoa spp. Appl Environ Microbiol 62:954–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teske A, Nelson DC. 2006. The genera Beggiatoa and Thioploca, p 784–810. In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (ed), The prokaryotes. Springer, New York, NY. [Google Scholar]
- 6.Salman V, Bailey JV, Teske A. 2013. Phylogenetic and morphologic complexity of giant sulphur bacteria. Antonie Van Leeuwenhoek 104:169–186. doi: 10.1007/s10482-013-9952-y. [DOI] [PubMed] [Google Scholar]
- 7.Bailey JV, Orphan VJ, Joye SB, Corsetti FA. 2009. Chemotrophic microbial mats and their potential for preservation in the rock record. Astrobiology 9:843–859. doi: 10.1089/ast.2008.0314. [DOI] [PubMed] [Google Scholar]
- 8.Jørgensen BB, Kasten S. 2006. Sulfur cycling and methane oxidation, p 271–309. In Schulz H, Zabel M (ed), Marine geochemistry, 2nd ed Springer, Berlin, Germany. [Google Scholar]
- 9.Fossing H, Jørgensen BB. 1990. Oxidation and reduction of radiolabeled inorganic sulfur compounds in an estuarine sediment, Kysing Fjord, Denmark. Geochim Cosmochim Acta 54:2731–2742. doi: 10.1016/0016-7037(90)90008-9. [DOI] [Google Scholar]
- 10.Brock J, Schulz-Vogt HN. 2011. Sulfide induces phosphate release from polyphosphate in cultures of a marine Beggiatoa strain. ISME J 5:497–506. doi: 10.1038/ismej.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulz HN, Schulz HD. 2005. Large sulfur bacteria and the formation of phosphorite. Science 307:416–418. doi: 10.1126/science.1103096. [DOI] [PubMed] [Google Scholar]
- 12.Sievert S, Kiene R, Schulz-Vogt H. 2007. The sulfur cycle. Oceanography 20:117–123. doi: 10.5670/oceanog.2007.55. [DOI] [Google Scholar]
- 13.Bailey J, Corsetti F, Greene S, Crosby C, Liu P, Orphan V. 2013. Filamentous sulfur bacteria preserved in modern and ancient phosphatic sediments: implications for the role of oxygen and bacteria in phosphogenesis. Geobiology 11:397–405. doi: 10.1111/gbi.12046. [DOI] [PubMed] [Google Scholar]
- 14.Bailey JV, Joye SB, Kalanetra KM, Flood BE, Corsetti FA. 2007. Evidence of giant sulphur bacteria in Neoproterozoic phosphorites. Nature 445:198–201. doi: 10.1038/nature05457. [DOI] [PubMed] [Google Scholar]
- 15.Williams LA, Reimers C. 1983. Role of bacterial mats in oxygen-deficient marine basins and coastal upwelling regimes: preliminary report. Geology 11:267–269. [Google Scholar]
- 16.Krajewski K, Cappellen PV, Trichet J, Kuhn O, Lucas J, Martín-Algarra A, Prévôt L, Tewari V, Gaspar L, Knight R. 1994. Biological processes and apatite formation in sedimentary environments. Eclogae Geol Helv 87:701–746. [Google Scholar]
- 17.Lepland A, Joosu L, Kirsimäe K, Prave AR, Romashkin AE, Črne AE, Martin AP, Fallick AE, Somelar P, Üpraus K, Mand K, Roberts NMW, van Zuilen MA, Wirth R, Schreiber A. 2014. Potential influence of sulphur bacteria on Palaeoproterozoic phosphogenesis. Nat Geosci 7:20–24. doi: 10.1038/ngeo2005. [DOI] [Google Scholar]
- 18.Grünke S, Lichtschlag A, de Beer D, Felden J, Salman V, Ramette A, Schulz-Vogt H, Boetius A. 2012. Mats of psychrophilic thiotrophic bacteria associated with cold seeps of the Barents Sea. Biogeosci Discuss 9:3917–3948. doi: 10.5194/bgd-9-3917-2012. [DOI] [Google Scholar]
- 19.Olu K, Lance S, Sibuet M, Henry P, Fiala-Médioni A, Dinet A. 1997. Cold seep communities as indicators of fluid expulsion patterns through mud volcanoes seaward of the Barbados accretionary prism. Deep Sea Res Part 1 Oceanogr Res Pap 44:811–841. doi: 10.1016/S0967-0637(96)00123-9. [DOI] [Google Scholar]
- 20.Olu K, Sibuet M, Harmegnies F, Foucher J-P, Fiala-Médioni A. 1996. Spatial distribution of diverse cold seep communities living on various diapiric structures of the southern Barbados prism. Prog Oceanogr 38:347–376. doi: 10.1016/S0079-6611(97)00006-2. [DOI] [Google Scholar]
- 21.Bartram AK, Lynch MDJ, Stearns JC, Moreno-Hagelsieb G, Neufeld JD. 2011. Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl Environ Microbiol 77:3846–3852. doi: 10.1128/AEM.02772-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newton RJ, Huse SM, Morrison HG, Peake CS, Sogin ML, McLellan SL. 2013. Shifts in the microbial community composition of gulf coast beaches following beach oiling. PLoS One 8:e74265. doi: 10.1371/journal.pone.0074265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R. 2007. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res 35:e120. doi: 10.1093/nar/gkm541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones D, Tobler D, Schaperdoth I, Mainiero M, Macalady J. 2010. Community structure of subsurface biofilms in the thermal sulfidic caves of Acquasanta Terme, Italy. Appl Environ Microbiol 76:5902–5910. doi: 10.1128/AEM.00647-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daims H, Brühl A, Amann R, Schleifer K-H, Wagner M. 1999. The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst Appl Microbiol 22:434–444. doi: 10.1016/S0723-2020(99)80053-8. [DOI] [PubMed] [Google Scholar]
- 26.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Kobert K, Flouri T, Stamatakis A. 2014. PEAR: a fast and accurate Illumina paired-end read merger. Bioinformatics 30:614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, Buchner A, Lai T, Steppi S, Jobb G, Förster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, König A, Liss T, Lüßmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer K-H. 2004. ARB: a software environment for sequence data. Nucleic Acids Res 32:1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmieder R, Lim YW, Edwards R. 2012. Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics 28:433–435. doi: 10.1093/bioinformatics/btr669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng Y, Leung HC, Yiu S, Chin FY. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 35.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A. 2008. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li W. 2009. Analysis and comparison of very large metagenomes with fast clustering and functional annotation. BMC Bioinformatics 10:359. doi: 10.1186/1471-2105-10-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun S, Chen J, Li W, Altintas I, Lin A, Peltier S, Stocks K, Allen EE, Ellisman M, Grethe J, Wooley J. 2011. Community cyberinfrastructure for advanced microbial ecology research and analysis: the CAMERA resource. Nucleic Acids Res 39:D546–D551. doi: 10.1093/nar/gkq1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brady A, Salzberg SL. 2009. Phymm and PhymmBL: metagenomic phylogenetic classification with interpolated Markov models. Nat Methods 6:673–676. doi: 10.1038/nmeth.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacGregor BJ, Biddle JF, Harbort C, Matthysse AG, Teske A. 2013. Sulfide oxidation, nitrate respiration, carbon acquisition, and electron transport pathways suggested by the draft genome of a single orange Guaymas Basin Beggiatoa (Cand. Maribeggiatoa) sp. filament. Mar Genomics 11:53–65. doi: 10.1016/j.margen.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 42.MacGregor BJ, Biddle JF, Siebert JR, Staunton E, Hegg EL, Matthysse AG, Teske A. 2013. Why orange Guaymas Basin Beggiatoa spp. are orange: single-filament-genome-enabled identification of an abundant octaheme cytochrome with hydroxylamine oxidase, hydrazine oxidase, and nitrite reductase activities. Appl Environ Microbiol 79:1183–1190. doi: 10.1128/AEM.02538-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mußmann M, Hu FZ, Richter M, de Beer D, Preisler A, Jørgensen BB, Huntemann M, Glöckner FO, Amann R, Koopman WJ. 2007. Insights into the genome of large sulfur bacteria revealed by analysis of single filaments. PLoS Biol 5:e230. doi: 10.1371/journal.pbio.0050230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ciccarelli FD, Doerks T, Von Mering C, Creevey CJ, Snel B, Bork P. 2006. Toward automatic reconstruction of a highly resolved tree of life. Science 311:1283–1287. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- 45.Miller CS, Baker BJ, Thomas BC, Singer SW, Banfield JF. 2011. EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol 12:R44. doi: 10.1186/gb-2011-12-5-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pruesse E, Peplies J, Glöckner FO. 2012. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28:1823–1829. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 48.Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Armougom F, Moretti S, Poirot O, Audic S, Dumas P, Schaeli B, Keduas V, Notredame C. 2006. Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res 34:W604–W608. doi: 10.1093/nar/gkl092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loy A, Duller S, Baranyi C, Mußmann M, Ott J, Sharon I, Béjà O, Le Paslier D, Dahl C, Wagner M. 2009. Reverse dissimilatory sulfite reductase as phylogenetic marker for a subgroup of sulfur-oxidizing prokaryotes. Environ Microbiol 11:289–299. doi: 10.1111/j.1462-2920.2008.01760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Le SQ, Gascuel O. 2008. An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 53.Abascal F, Zardoya R, Posada D. 2005. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 54.Bailey JV, Salman V, Rouse GW, Schulz-Vogt HN, Levin LA, Orphan VJ. 2011. Dimorphism in methane seep-dwelling ecotypes of the largest known bacteria. ISME J 5:1926–1935. doi: 10.1038/ismej.2011.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grünke S, Felden J, Lichtschlag A, Girnth AC, de Beer D, Wenzhöfer F, Boetius A. 2011. Niche differentiation among mat-forming, sulfide-oxidizing bacteria at cold seeps of the Nile Deep Sea Fan (Eastern Mediterranean Sea). Geobiology 9:330–348. doi: 10.1111/j.1472-4669.2011.00281.x. [DOI] [PubMed] [Google Scholar]
- 56.Guezennec J, Fiala-Medioni A. 1996. Bacterial abundance and diversity in the Barbados Trench determined by phospholipid analysis. FEMS Microbiol Ecol 19:83–93. doi: 10.1111/j.1574-6941.1996.tb00201.x. [DOI] [Google Scholar]
- 57.Lance S, Henry P, Le Pichon X, Lallemant S, Chamley H, Rostek F, Faugères J-C, Gonthier E, Olu K. 1998. Submersible study of mud volcanoes seaward of the Barbados accretionary wedge: sedimentology, structure and rheology. Mar Geol 145:255–292. doi: 10.1016/S0025-3227(97)00117-5. [DOI] [Google Scholar]
- 58.Nielsen PH, De Muro MA, Nielsen JL. 2000. Studies on the in situ physiology of Thiothrix spp. present in activated sludge. Environ Microbiol 2:389–398. doi: 10.1046/j.1462-2920.2000.00120.x. [DOI] [PubMed] [Google Scholar]
- 59.Angert ER, Northup DE, Reysenbach A-L, Peek AS, Goebel BM, Pace NR. 1998. Molecular phylogenetic analysis of a bacterial community in Sulphur River, Parker Cave, Kentucky. Am Miner 83:1583–1592. [Google Scholar]
- 60.Edgcomb VP, Kysela DT, Teske A, de Vera Gomez A, Sogin ML. 2002. Benthic eukaryotic diversity in the Guaymas Basin hydrothermal vent environment. Proc Natl Acad Sci U S A 99:7658–7662. doi: 10.1073/pnas.062186399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gillan DC, Speksnijder AG, Zwart G, De Ridder C. 1998. Genetic diversity of the biofilm covering Montacuta ferruginosa (Mollusca, Bivalvia) as evaluated by denaturing gradient gel electrophoresis analysis and cloning of PCR-amplified gene fragments coding for 16S rRNA. Appl Environ Microbiol 64:3464–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.López-García P, Duperron S, Philippot P, Foriel J, Susini J, Moreira D. 2003. Bacterial diversity in hydrothermal sediment and epsilonproteobacterial dominance in experimental microcolonizers at the Mid-Atlantic Ridge. Environ Microbiol 5:961–976. doi: 10.1046/j.1462-2920.2003.00495.x. [DOI] [PubMed] [Google Scholar]
- 63.Salman V, Amann R, Shub DA, Schulz-Vogt HN. 2012. Multiple self-splicing introns in the 16S rRNA genes of giant sulfur bacteria. Proc Natl Acad Sci U S A 109:4203–4208. doi: 10.1073/pnas.1120192109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sekar R, Mills DK, Remily ER, Voss JD, Richardson LL. 2006. Microbial communities in the surface mucopolysaccharide layer and the black band microbial mat of black band-diseased Siderastrea siderea. Appl Environ Microbiol 72:5963–5973. doi: 10.1128/AEM.00843-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stevens H, Ulloa O. 2008. Bacterial diversity in the oxygen minimum zone of the eastern tropical South Pacific. Environ Microbiol 10:1244–1259. doi: 10.1111/j.1462-2920.2007.01539.x. [DOI] [PubMed] [Google Scholar]
- 66.Orsi W, Biddle JF, Edgcomb V. 2013. Deep sequencing of subseafloor eukaryotic rRNA reveals active fungi across marine subsurface provinces. PLoS One 8:e56335. doi: 10.1371/journal.pone.0056335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Orsi WD, Edgcomb VP, Christman GD, Biddle JF. 2013. Gene expression in the deep biosphere. Nature 499:205–208. doi: 10.1038/nature12230. [DOI] [PubMed] [Google Scholar]
- 68.Teske A, Wawer C, Muyzer G, Ramsing NB. 1996. Distribution of sulfate-reducing bacteria in a stratified fjord (Mariager Fjord, Denmark) as evaluated by most-probable-number counts and denaturing gradient gel electrophoresis of PCR-amplified ribosomal DNA fragments. Appl Environ Microbiol 62:1405–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kalanetra KM, Joye SB, Sunseri NR, Nelson DC. 2005. Novel vacuolate sulfur bacteria from the Gulf of Mexico reproduce by reductive division in three dimensions. Environ Microbiol 7:1451–1460. doi: 10.1111/j.1462-2920.2005.00832.x. [DOI] [PubMed] [Google Scholar]
- 70.Lane N, Martin W. 2010. The energetics of genome complexity. Nature 467:929–934. doi: 10.1038/nature09486. [DOI] [PubMed] [Google Scholar]
- 71.Mendell JE, Clements KD, Choat JH, Angert ER. 2008. Extreme polyploidy in a large bacterium. Proc Natl Acad Sci U S A 105:6730–6734. doi: 10.1073/pnas.0707522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Campbell BJ, Engel AS, Porter ML, Takai K. 2006. The versatile ε-proteobacteria: key players in sulphidic habitats. Nat Rev Microbiol 4:458–468. doi: 10.1038/nrmicro1414. [DOI] [PubMed] [Google Scholar]
- 73.Dahl C, Engels S, Pott-Sperling AS, Schulte A, Sander J, Lübbe Y, Deuster O, Brune DC. 2005. Novel genes of the dsr gene cluster and evidence for close interaction of Dsr proteins during sulfur oxidation in the phototrophic sulfur bacterium Allochromatium vinosum. J Bacteriol 187:1392–1404. doi: 10.1128/JB.187.4.1392-1404.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dahl C, Prange A. 2006. Bacterial sulfur globules: occurrence, structure and metabolism, p 21–51. In Shivley JM. (ed), Inclusions in prokaryotes. Springer, Berlin, Germany. [Google Scholar]
- 75.Hagen KD, Nelson DC. 1997. Use of reduced sulfur compounds by Beggiatoa spp.: enzymology and physiology of marine and freshwater strains in homogeneous and gradient cultures. Appl Environ Microbiol 63:3957–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bergmann DJ, Hooper AB, Klotz MG. 2005. Structure and sequence conservation of hao cluster genes of autotrophic ammonia-oxidizing bacteria: evidence for their evolutionary history. Appl Environ Microbiol 71:5371–5382. doi: 10.1128/AEM.71.9.5371-5382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Campbell BJ, Smith JL, Hanson TE, Klotz MG, Stein LY, Lee CK, Wu D, Robinson JM, Khouri HM, Eisen JA. 2009. Adaptations to submarine hydrothermal environments exemplified by the genome of Nautilia profundicola. PLoS Genet 5:e1000362. doi: 10.1371/journal.pgen.1000362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hanson TE, Campbell BJ, Kalis KM, Campbell MA, Klotz MG. 2013. Nitrate ammonification by Nautilia profundicola AmH: experimental evidence consistent with a free hydroxylamine intermediate. Front Microbiol 4:180. doi: 10.3389/fmicb.2013.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kraft B, Tegetmeyer HE, Sharma R, Klotz MG, Ferdelman TG, Hettich RL, Geelhoed JS, Strous M. 2014. The environmental controls that govern the end product of bacterial nitrate respiration. Science 345:676–679. doi: 10.1126/science.1254070. [DOI] [PubMed] [Google Scholar]
- 80.Otte S, Kuenen JG, Nielsen LP, Paerl HW, Zopfi J, Schulz HN, Teske A, Strotmann B, Gallardo VA, Jørgensen BB. 1999. Nitrogen, carbon, and sulfur metabolism in natural Thioploca samples. Appl Environ Microbiol 65:3148–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Beutler M, Milucka J, Hinck S, Schreiber F, Brock J, Mußmann M, Schulz-Vogt HN, de Beer D. 2012. Vacuolar respiration of nitrate coupled to energy conservation in filamentous Beggiatoaceae. Environ Microbiol 14:2911–2919. doi: 10.1111/j.1462-2920.2012.02851.x. [DOI] [PubMed] [Google Scholar]
- 82.Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP, Banfield JF. 2009. Community-wide analysis of microbial genome sequence signatures. Genome Biol 10:R85. doi: 10.1186/gb-2009-10-8-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Teeling H, Meyerdierks A, Bauer M, Amann R, Glöckner FO. 2004. Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ Microbiol 6:938–947. doi: 10.1111/j.1462-2920.2004.00624.x. [DOI] [PubMed] [Google Scholar]
- 84.Fuchsman CA, Rocap G. 2006. Whole-genome reciprocal BLAST analysis reveals that Planctomycetes do not share an unusually large number of genes with Eukarya and Archaea. Appl Environ Microbiol 72:6841–6844. doi: 10.1128/AEM.00429-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Flood B, Bailey J, Biddle J. 2014. Horizontal gene transfer and the rock record: comparative genomics of phylogenetically distant bacteria that induce wrinkle structure formation in modern sediments. Geobiology 12:119–132. doi: 10.1111/gbi.12072. [DOI] [PubMed] [Google Scholar]
- 86.Baker BJ, Sheik CS, Taylor CA, Jain S, Bhasi A, Cavalcoli JD, Dick GJ. 2013. Community transcriptomic assembly reveals microbes that contribute to deep-sea carbon and nitrogen cycling. ISME J 7:1962–1973. doi: 10.1038/ismej.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schulz HN, de Beer D. 2002. Uptake rates of oxygen and sulfide measured with individual Thiomargarita namibiensis cells by using microelectrodes. Appl Environ Microbiol 68:5746–5749. doi: 10.1128/AEM.68.11.5746-5749.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Knittel K, Boetius A, Lemke A, Eilers H, Lochte K, Pfannkuche O, Linke P, Amann R. 2003. Activity, distribution, and diversity of sulfate reducers and other bacteria in sediments above gas hydrate (Cascadia Margin, Oregon). Geomicrobiol J 20:269–294. doi: 10.1080/01490450303896. [DOI] [Google Scholar]
- 89.Schreiber L, Holler T, Knittel K, Meyerdierks A, Amann R. 2010. Identification of the dominant sulfate-reducing bacterial partner of anaerobic methanotrophs of the ANME-2 clade. Environ Microbiol 12:2327–2340. doi: 10.1111/j.1462-2920.2010.02275.x. [DOI] [PubMed] [Google Scholar]
- 90.Felden J, Ruff S, Ertefai T, Inagaki F, Hinrichs KU, Wenzhöfer F. 2014. Anaerobic methanotrophic community of a 5346-m-deep vesicomyid clam colony in the Japan Trench. Geobiology 12:183–199. doi: 10.1111/gbi.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pernthaler A, Dekas AE, Brown CT, Goffredi SK, Embaye T, Orphan VJ. 2008. Diverse syntrophic partnerships from deep-sea methane vents revealed by direct cell capture and metagenomics. Proc Natl Acad Sci U S A 105:7052–7057. doi: 10.1073/pnas.0711303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vezzi A, Campanaro S, D'Angelo M, Simonato F, Vitulo N, Lauro F, Cestaro A, Malacrida G, Simionati B, Cannata N. 2005. Life at depth: Photobacterium profundum genome sequence and expression analysis. Science 307:1459–1461. doi: 10.1126/science.1103341. [DOI] [PubMed] [Google Scholar]
- 93.Knittel K, Lösekann T, Boetius A, Kort R, Amann R. 2005. Diversity and distribution of methanotrophic archaea at cold seeps. Appl Environ Microbiol 71:467–479. doi: 10.1128/AEM.71.1.467-479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–175. In Stackebrandt E, Goodfellow M (ed) Nucleic acid techniques in bacterial systematics. Wiley, New York, NY. [Google Scholar]
- 95.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.