

Abstract
In alkaptonuria, deficiency of homogentisate 1,2-dioxygenase leads to the accumulation of homogentisic acid (HGA) and its metabolites in the body, resulting in ochronosis. Reports of patients with alkaptonuria who have decreased kidney function are rare, but this seems to play an important role in the natural history of the disease.
We describe a 68-year-old female with chronic kidney disease (CKD) of unknown etiology who started peritoneal dialysis (PD) after 5 years of follow-up and who was diagnosed with alkaptonuria at this time. Progressive exacerbation of ochronotic manifestations had been noted during these last few years, as kidney function worsened. After PD initiation, the disease continued to progress, and death occurred after one year and a half, due to severe aortic stenosis-related complications. Her 70-year-old sister was evaluated and also diagnosed with alkaptonuria. She had no renal dysfunction. Higher HGA excretion and significantly milder ochronosis than that of her sister were found.
We present two alkaptonuric sisters with similar comorbidities except for the presence of CKD, who turned out to have totally different evolutions of their disease. This report confirms that kidney dysfunction may be an important factor in determining the natural history of alkaptonuria.
Keywords: Alkaptonuria, chronic kidney disease, ochronosis
Background
Alkaptonuria is an autosomal recessive disorder due to the deficiency of homogentisate 1,2-dioxygenase (HGO), an enzyme in the tyrosine degradation pathway [1] (Figure 1). This leads to the accumulation of homogentisic acid (HGA), which is oxidized to benzoquinones that polymerize, and leads to the formation of an ochronotic pigment that binds to connective tissue. As a result, ochronosis occurs, with darkening of cartilaginous tissues and bone, arthropathy (particularly, spine and large joints), cardiac valve and coronary artery involvement, and kidney and prostate stones [1, 2].
Fig. 1.

Biochemical pathway of the disease. Deficiency of homogentisate 1,2-dioxygenase (HGO) in the tyrosine degradation pathway leads to alkaptonuria.
The clinical landmark for early diagnosis of alkaptonuria is darkening of urine upon standing, due to excretion of large quantities of HGA. Definitive diagnosis relies on its gram quantities measurement in a 24-h collection [1, 3, 4]. Molecular diagnosis (search for mutation in the HGO gene) is not mandatory for clinical management. Treatment is mainly supportive for now, but a long-term study to test the efficacy of nitisinone (predicted to block the accumulation of HGA) in the improvement of the disease morbidity has been recently completed, and the results awaited [5].
Kidney clearance of HGA is an effective mechanism to prevent its large quantity body accumulation in patients with alkaptonuria. Active renal tubular secretion plays an important role in this clearance [4]. Reports of patients with this disorder who have concomitant chronic renal failure are rare, but they show that kidney function might play an important role in the natural history of the disease [4, 6, 7]. Here, we report the case of two sisters with alkaptonuria with markedly different evolutions of the disease, in which the presence of chronic kidney disease (CKD) could have been one important discriminating factor.
Case reports
Patient 1
A 68-year-old female was referred to our peritoneal dialysis (PD) program due to CKD stage V. She had had a record of kidney dysfunction which began 10 years earlier, and had been followed up in nephrology consultation during the previous 5 years.
At her first nephrology consultation, markers of baseline kidney function were: serum creatinine 141.4 µmol/L (1.6 mg/dL), blood urea nitrogen 9.5 mmol/L (26.6 mg/dL) and creatinine clearance 0.70 mL/s/1.73 m2 (41.8 mL/min/1.73 m2). Urinary sediment had no alterations. Daily proteinuria was 1.2 6 g/24 h. Kidney disease etiology related to immunological disorder, plasma cell dyscrasia, viral disease and nephrolithiasis was excluded. She had a history of hypertension controlled by monotherapy, and no history of diabetes mellitus. Diagnosis of spinal disk herniation and surgical correction had been performed 13 years before, and she had a record of chronic use of non-steroidal anti-inflammatory drugs (NSAIDs) due to chronic back pain. No biopsy was performed due to its technical difficulty (maintenance of the kidney's inferior pole in subcostal position during inspiration). Mild dark pigmentation of skin and sclera was noted at this time. Echocardiogram showed a mild aortic stenosis.
During the 5-year follow-up period in nephrology consultation, bilateral hip replacement due to arthrosis was performed at different times. Due to dark pigmentation, articular cartilage was sent for histological diagnosis and the pigment was erroneously classified as hemosiderin. Elevated liver enzymes and cholestasis led to diagnostic investigation of hemochromatosis and this was excluded (Table 1).
Table 1.
Patient #1–Laboratory findings at the time of nephrology referral
| Blood chemistry | |
|---|---|
| Albumin | 32 g/L (3.2 g/dL) |
| Calcium | 2.8 mmol/L (11.2 mg/dL) |
| Phosphorus | 1.1 mmol/L (3.4 mg/dL) |
| PTH | 370 ng/L |
| ASTa | 36 IU/L |
| ALTa | 41 IU/L |
| Alkaline phosphatase | 551 IU/L |
| Y-glutamyltransferase | 169 IU/L |
| Serum iron | 0.015 µmol/L (86 ng/mL) |
| Ferritin | 238 pmol/L (106 ng/mL) |
| TSAT | 27% |
aNormal value: 4–43 IU/L.
Kidney function progressively worsened, and the patient eventually started renal replacement therapy by PD. Progressive exacerbation of dark pigmentation of sclera, ear helices, skin of face, trunk and limbs, as well as urine, that had been noted in the last few years, continued after starting PD (Figure 2). Also, PD effluent became dark some minutes after complete drainage. Based on these signs, alkaptonuria diagnosis was suspected, and it was confirmed by the measurement of elevated levels of urine HGA (Table 2). Genetic testing for HGO mutation was not performed. No history of consanguinity was found in the family. Articular radiology showed typical findings of ochronosis, including calcification of the intervertebral disk, with vacuum phenomenon and disk space narrowing, and pubic symphysis involvement. HLA B27 was negative. Deterioration of the patient's general condition was noted during the following year, with exacerbation of generalized pain, extreme darkening of the skin and progression to severe symptomatic aortic stenosis. The patient initially refused aortic surgery and eventually died one year and a half after beginning PD due to cardiac complications.
Fig. 2.
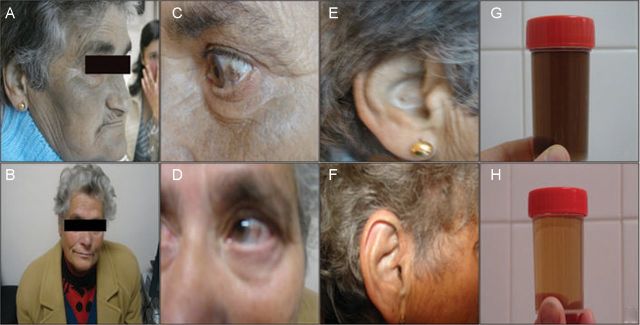
Ochronotic pigmentation of skin, sclera, ear cartilage and urine in Patient 1 (A, C, E and G, respectively) and Patient 2 (B, D, F and H, respectively). Note enhanced pigmentation in Patient 1.
Table 2.
Renal function and HGA values at alkaptonuria diagnosis
| Parameter | Patient #1 | Patient #2 |
|---|---|---|
| Cr Cl (mL/s/1.73 m2)[(mL/min/1.73 m2)] | 0.14 (8.96) | 1.21 (72.5) |
| Urine HGA (g/day)a | 0.9138 | 4.406 |
aNormal value: 0.01–0.1 g/day (found in patients without alkaptonuria, including those with CKD).
Patient 2
Her 70-year-old female sister was evaluated and the report showed dark pigmentation of sclera, ear cartilage and dark urine upon standing, but with much less intensity than her sister. She had no history of joint pathology nor pain complaints, although the radiological study confirmed the presence of ochronotic arthropathy (Figure 3). Alkaptonuria was confirmed by the measurement of urine HGA (Table 2). Mild aortic stenosis was diagnosed by echocardiogram, with no related symptoms. Kidney disease diagnosis work-up showed serum creatinine of 70.7 µmol/L (0.8 mg/dL), normal urinary sediment, proteinuria of 0.170 g/24 h and absence of echographic changes (no signs of lithiasis).
Fig. 3.
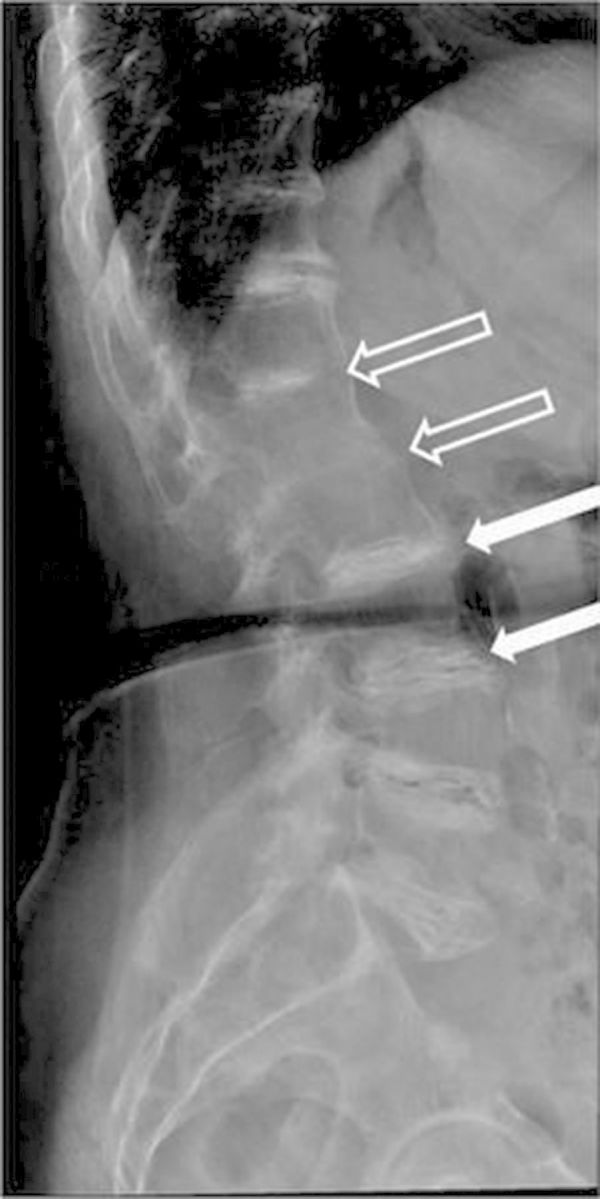
Lumbar spinal radiograph of Patient 2. Linear calcifications at every intervertebral disk space, with vacuum phenomena and space narrowing (white arrows), are common findings in ochronotic arthritis. Also visible D12-L1-L2 ankylosis (open arrows).
Discussion
Kidney disease related to alkaptonuria is rare. Case reports have pointed out nephrolithiasis as the main etiology [1]. Chemically, calculi have been defined as containing ochronotic pigment and calcium salts [8]. Renal biopsies, showing both pigment deposition [6, 7] and chronic interstitial nephritis features, point to its possible causal relationship, leading to the term ochronotic nephropathy [7]. Unfortunately in this case (Patient 1), we could not have a histological documentation of CKD etiology due to technical reasons, and its association with alkaptonuria was unrevealed. Clinically, a causal relationship between alkaptonuria and kidney disease seemed unlikely due to the absence of lithiasis history; also, there were no significant signs of glomerular or vascular etiology. Therefore, chronic use of NSAIDs (not present in her sister's history) was considered the most probable cause of patient 1's CKD, and this different nephrotoxic exposure could explain the heterogeneity of the kidney manifestations between the two siblings.
Therefore, this report does not stress the causal relationship between alkaptonuria and CKD, but the potential impact that the latter can have on the natural history of the former. Although tyrosine catabolism and HGA production occur mainly in the liver, with just a minor contribution from the kidneys [9], almost all HGA produced in the body is eliminated by the kidney. Glomerular filtration is not sufficient to achieve this task, and tubular secretion is responsible for the majority of it [4].
In alkaptonuric patients, end-stage renal disease (ESRD) as a result of CKD progression has been rarely reported. Since 1983, four cases (3 males, with ages 24, 50 and 61 years, and 1 female aged 19 years) have been published [7, 10, 11, 12]. Even if alkaptonuria is a rare condition, affecting 1 in 250 000 to 1 in 1 000 000 people worldwide [2], and probably there are a number of undiagnosed or unreported ESRD cases, its prevalence in alkaptonuria can be estimated as very low.
In Patient 1 (sibling with ESRD), kidney HGA excretion was ∼20% of her sister's, leading to a greater accumulation of HGA in the body and severity of the disease. As ochronotic pigment deposition manifestations rapidly progressed after reaching ESRD, aortic stenosis also progressed and ultimately led to the patient's death. Relationship of pigment deposition and aortic stenosis is well documented [2], and the clinical evolution supports their relationship in this patient.
Although HGA peritoneal clearance was observed, with PD effluent becoming dark after drainage, this proved to be inefficient as a way to remove HGA, as signs and symptoms of the disease continued to worsen after initiation of PD. Peritoneal membrane failure was not observed, but the relatively short time of PD treatment precludes definitive conclusions about the potential effect of ochronosis on the membrane function. Elimination of HGA by renal replacement therapy has already been reported by Heng et al. [12], when dark haemofiltration waste fluid was observed, but as in this case, intensive renal support failed to improve outcome.
Treatment with ascorbic acid has shown to be inefficient for the treatment of this disease [1, 13], and both of our patients were not put on this medication. Also, dietary therapy restricting phenylalanine and tyrosine is difficult to maintain and has had no demonstrable efficacy in improving the symptoms of alkaptonuria [14]. Nitisinone inhibits 4-hydroxyphenylpyruvate dioxygenase, the enzyme that produces HGA, and it has been shown to be effective in reducing the urinary HGA levels and improving symptoms [1, 14]. Exclusion criteria for nitisinone therapy used in these studies and particularly in the NIH clinical trial [5], such as elevated serum creatinine, bilateral hip joint replacement and anaemia, were present in our patient, so its hypothetical use was also excluded.
Kidney specific HGO activity has been emphasized by the report of Introne et al. [4], when a kidney transplant was performed in a patient with alkaptonuria and CKD, and a reduction of urinary HGA levels and normalization of plasma HGA levels were observed. Kidney transplantation can thus have beneficial effects on the natural history of patients with alkaptonuria and CKD, both by restoring normal HGA excretion and also by providing HGO activity.
Here, we present two sisters affected with alkaptonuria with totally different evolutions of their disease, one with a fairly benign course, with radiological but no symptomatic articular involvement and mild cardiac involvement. She had normal renal function for her age, allowing much higher HGA excretion than her sister, who had kidney disease and whose joint, skin and cardiac alkaptonuria manifestations rapidly progressed to severe as renal function worsened. This report confirms that kidney function may be an important determinant of the natural history of alkaptonuria.
Conflict of interest statement
None declared.
Acknowledgements
The authors would like to acknowledge Dr. Carla Lima, Dr. Jesus Garrido, Dr. Sérgio Lemos and Dr. Edgar Lorga for their valuable contributions to the manuscript.
References
- 1.Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347:2111–2121. doi: 10.1056/NEJMoa021736. doi:10.1056/NEJMoa021736. [DOI] [PubMed] [Google Scholar]
- 2.Fisher AA, Davis MW. Alkaptonuric ochronosis with aortic valve and joint replacements and femoral fracture: a case report and literature review. Clin Med Res. 2004;2:209–215. doi: 10.3121/cmr.2.4.209. doi:10.3121/cmr.2.4.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watts RW, Watts RA. Alkaptonuria: A 60-yr Follow-up. Rheumatology (Oxford) 2007;46:358–359. doi: 10.1093/rheumatology/kel345. doi:10.1093/rheumatology/kel345. [DOI] [PubMed] [Google Scholar]
- 4.Introne WJ, Phornphutkul C, Bernardini I, McLaughlin K, Fitzpatrick D, Gahl WA. Exacerbation of the ochronosis of alkaptonuria due to renal insufficiency and improvement after renal transplantation. Mol Genet Metab. 2002;77:136–142. doi: 10.1016/s1096-7192(02)00121-x. doi:10.1016/S1096-7192(02)00121-X. [DOI] [PubMed] [Google Scholar]
- 5.National Human Genome Research Institute (NHGRI) ClinicalTrials.gov. Bethesda, MD: National Library of Medicine (US); 2000–2005. Long-term study of nitisinone to treat alkaptonuria. http://clinicaltrials.gov/ct2/show/NCT00107783. NLM Identifier: NCT00107783. [Google Scholar]
- 6.Kazancioglu R, Taylan I, Aksak F, et al. Alkaptonuria and renal failure: a case report. J Nephrol. 2004;17:441–445. [PubMed] [Google Scholar]
- 7.Venkataseshan VS, Chandra B, Graziano V, et al. Alkaptonuria and renal failure: a case report and review of the literature. Mod Pathol. 1992;5:464–471. [PubMed] [Google Scholar]
- 8.Sutor DJ, Wolley SE, Krizek V. The composition of calculi from patients with alkaptonuria. Br J Urol. 1970;42:386–388. doi: 10.1111/j.1464-410x.1970.tb04470.x. doi:10.1111/j.1464-410X.1970.tb04470.x. [DOI] [PubMed] [Google Scholar]
- 9.La Du BN. Alkaptonuria. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic & Molecular Bases of Inherited Disease. 8th edn. Vol. 2. New York: McGraw-Hill; 2001. pp. 2109–2123. [Google Scholar]
- 10.Abreo K, Abreo F, Zimmerman SW, et al. A fifty-year-old man with skin pigmentation, arthritis, chronic renal failure and methemoglobinemia. Am J Med Genet. 1983;14:97–114. doi: 10.1002/ajmg.1320140115. doi:10.1002/ajmg.1320140115. [DOI] [PubMed] [Google Scholar]
- 11.Ladjouze-Rezig A, Rodriguez de Cordoba S, Aquaron R. Ochronotic rheumatism in Algeria: clinical, radiological, biological and molecular studies–a case study of 14 patients in 11 families. Joint Bone Spine. 2006;73:284–292. doi: 10.1016/j.jbspin.2005.03.010. doi:10.1016/j.jbspin.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 12.Heng AE, Courbebaisse M, Kemeny JL, et al. Hemolysis in a patient with alkaptonuria and chronic kidney failure. Am J Kidney Dis. 2010;56:e1–e4. doi: 10.1053/j.ajkd.2009.11.023. doi:10.1053/j.ajkd.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 13.Forslind K, Wollheim FA, Akesson B. Alkaptonuria and ochronosis in three siblings. Ascorbic acid treatment monitored by urinary HGA excretion. Clin Exp Rheumatol. 1988;6:289–292. [PubMed] [Google Scholar]
- 14.Suwannarat P, O'Brien K, Perry MB, et al. Use of nitisinone in patients with alkaptonuria. Metabolism. 2005;54:719–728. doi: 10.1016/j.metabol.2004.12.017. doi:10.1016/j.metabol.2004.12.017. [DOI] [PubMed] [Google Scholar]