

Abstract
Rationale: The growing recognition of the importance of splicing, together with rapidly accumulating RNA-sequencing data, demand robust high-throughput approaches, which efficiently analyze experimentally derived whole-transcriptome splice profiles.
Results: We have developed a computational approach, called SNPlice, for identifying cis-acting, splice-modulating variants from RNA-seq datasets. SNPlice mines RNA-seq datasets to find reads that span single-nucleotide variant (SNV) loci and nearby splice junctions, assessing the co-occurrence of variants and molecules that remain unspliced at nearby exon–intron boundaries. Hence, SNPlice highlights variants preferentially occurring on intron-containing molecules, possibly resulting from altered splicing. To illustrate co-occurrence of variant nucleotide and exon–intron boundary, allele-specific sequencing was used. SNPlice results are generally consistent with splice-prediction tools, but also indicate splice-modulating elements missed by other algorithms. SNPlice can be applied to identify variants that correlate with unexpected splicing events, and to measure the splice-modulating potential of canonical splice-site SNVs.
Availability and implementation: SNPlice is freely available for download from https://code.google.com/p/snplice/ as a self-contained binary package for 64-bit Linux computers and as python source-code.
Contact: pmudvari@gwu.edu or horvatha@gwu.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Alternative splicing is well known as an essential mechanism of gene regulation which may affect protein function (Braunschweig et al., 2013; Singh and Cooper, 2012). To date, ∼95% of the mammalian genes are estimated to be subjected to alternative splicing; current knowledge explains only a portion of the underlying molecular events (Barash et al., 2010; Bernstein et al., 2012; Moore and Silver, 2008; Pan et al., 2008). Most of the knowledge on the splicing was obtained through alignment of DNA and RNA sequences and systematic search for functional elements, facilitated by the recent advances of the sequencing technologies (Bernstein et al., 2012; Barbosa-Morais et al., 2012; Clark and Thanaraj, 2002; Liao et al., 2005; Merkin et al., 2012). Among the major mechanisms affecting the splicing process are nucleotide changes that disrupt or create binding sites for splicing, transcriptional and other regulatory factors (Barash et al., 2010; Bernstein et al., 2012; Barbosa-Morais et al., 2012; McManus and Graveley, 2011; Merkin et al., 2012; Moore and Silver, 2008; Pan et al., 2008; Wang and Burge, 2008). In addition to the four consensus sequences critical for the spliceosome assembly—5′ splice site (5′SS), 3′SS, the branch sequence, and the polypyrimidine tract (PPT)—changes in the exonic and intronic splicing enhancers and silencers (ESE, ISE, ESS and ISS) and other, less patterned sequences, are increasingly acknowledged as splicing modulators (De Conti et al., 2013; Woolfe et al., 2010). The emerging recognition of the importance of splicing has stimulated efforts for modeling the splice-modulating potential of genetic variants through probabilistic predictions based on junction nucleotide composition, dependencies among neighboring bases, local optimality in the context of the gene structure, homology alignments, interaction with splicing factors and comparison with experimentally verified splice-modulating motifs (Brunak et al., 1991; Brendel and Kleffe, 1998; Dogan et al., 2007; Faber et al., 2011; Kamath et al., 2012; Pertea et al., 2001; Piva et al., 2012; Riva et al., 2012; Woolfe et al., 2010; Yeo and Burge, 2004). These approaches helped annotate numerous previously unknown splice-modulating variants; however, being mostly based on pre-existing knowledge, they may miss variants acting through unknown mechanisms.
Several recent analyses have highlighted the importance of systematic, genome-wide evaluation of sequence-specific splicing events (Barash et al., 2010; Han et al., 2013; Merkin et al., 2012; Sterne-Weiler and Sanford, 2014). A survey of the splicing patterns on relatively small genome portion—250 exons in HapMap-genotyped individuals—has suggested that common splice-modulating SNVs can be frequent in the genome (Cheung et al., 2005; Coulombe-Huntington et al., 2009; Hull et al, 2007). An analysis of splice-sensitive microarrays demonstrated high frequency of alternative splicing events within the usually unexplored areas of the genome, and, at significance levels much below standard multiple-testing thresholds, implying that the extent of cis-regulated differential splicing between individuals may be far greater than estimated (ElSharawy et al., 2009). Concurrent with the above are findings from another study, which employs software—SNPSplicer—to compare matching genomic DNA and complementary DNA (cDNA) from individuals with different genotypes (ElSharawy et al., 2006).
We developed a computational approach, SNPlice, which identi-fies potential splice-modulating variants from RNA-seq data generated through massively parallel sequencing. SNPlice assesses sequencing reads for co-occurrence of variant base and a nearby exon-intron boundary. Given that canonical splicing is expected to retain predominantly exon–exon junctions in the transcriptome, we based our strategy on the assumption that transcriptome sequencing reads harboring an exon–intron boundary indicate a biological process of junction alteration and, possibly, altered splicing.
2 Methods
2.1 Samples
SNPlice performance was tested on 65 human RNA-seq datasets (Supplementary Table S1). Five in-house primary cell lines derived from Retinal Pigment Epithelia (RPE) of healthy organ donors' eyes (Maminiskis et al., 2006), were used for allele-specific sequencing of selected SNPlice highlighted variants. Sixty matching normal and tumor datasets from the Cancer Genomic Hub (https://cghub.ucsc.edu) were used to assess the between-samples variability of SNPlice findings.
2.2 RNA libraries preparation and sequencing
Total RNA from the RPE primary cell lines was isolated using standard protocols (TRIzol Reagent, Life Technologies, Foster City, CA). The libraries were prepared using TruSeq RNA sample preparation kit (Illumina, Inc.) that includes poly-A selection, according to the manufacturer recommendations. Paired-end sequencing (50nt read length) was performed on Illumina HiSeq 2000 platform.
2.3 Read alignment and variants call and annotation
The generated and downloaded sequencing datasets were processed as follows. The paired end raw reads were aligned against hg19 using TopHat2 (Kim et al., 2013), version 2.0.8, with default settings and allowing two mismatches in the alignment. Identical read sequences mapping to the same loci were counted once per individual to reduce the potential impact of PCR duplicates on the counts. Variants' calls were obtained using the mpileup utility of SAMTools (http://samtools.sourceforge.net/mpileup-.shtml) (Li et al., 2009). Base Alignment Quality was used to score the variant calls and consensus calling was done using BCF tools. Maximum depth call was set at 8000. All variants were subjected to SNPlice. Cufflinks was used to assemble the transcripts without reference junction annotation; transcript abundance was quantified in fragments per kilobase per million (FPKM) fragments mapped, as previously described (Trapnell et al., 2013). The variants were annotated using SeattleSeq Annotation Tools version 8.01, dbSNP build 138 (http://snp.gs.washington.edu/SeattleSeq-Annotation138/).
2.4 Computational algorithm
We developed a Python program, SNPlice, using the pysam Python module to find reads spanning SNV loci and exon–intron boundaries or exon–exon junctions (defined as spanning reads), and quantify the extent of co-occurrence of variant alleles in unspliced reads. The program identifies reads that span a SNV locus and an exon boundary within the length of the sequencing read, and classifies them according to the nucleotide observed at the SNV site (Supplementary Fig. S1). Reads are further required to extend past the exon–intron boundary by at least 5 bp to ensure the alignment can be confidently placed in the adjacent intron or in the following exon (Supplementary Fig. S2). The length of the alignment of each read to the reference is then checked—reads whose alignment length on the reference genome is similar (±2 bp) to the length of the read are classified as intronic, while those whose alignment length on the reference genome is similar (± 2 bp) to the length of the read plus the length of the intron are classified as exonic. Reads whose nucleotide at the SNV position do not match either the reference or the alternative base, or whose alignment length does not match these intronic nor exonic definitions are disregarded, not because they are uninteresting, but because we must ensure the read counts are not inflated by false-positive or other types of poor alignments. For each SNV locus and exon-intron boundary, then, we compute four read counts (Fig. 1): NVARee represents number of reads bearing the variant nucleotide and mapped across the exon–exon junction, NVARei represents the number of reads bearing the variant nucleotide and mapped across the exon–intron boundary, NREFee represents the number of reads bearing the reference nucleotide and mapped across the exon–exon junction, and NREFei represents the number of reads bearing the reference nucleotide and mapped across the exon-intron boundary. We evaluate these counts in a 2 × 2 contingency table to assess the spliced versus unspliced status of variant versus reference allele reads. We compute the traditional log odds-ratio using pseudocounts of 0.5 to avoid numerical issues with zeros (Gart and Zweifel, 1967; Haldane, 1955; Parzen et al., 2002). The log odds-ratio increases as intronic reads become more enriched in variant allele reads. The statistical significance of the counts is determined using Fisher’s exact test, which assesses the lack of independence in the observation of variant and intronic reads. Fisher’s exact test P values were corrected for multiple trials by estimating the False Discovery Rate (FDR, Benjamini and Hochberg, 1995). SNVs of interest were visually examined through the Integrative Genome Viewer IGV (Thorvaldsdottir et al., 2013).
Fig. 1.
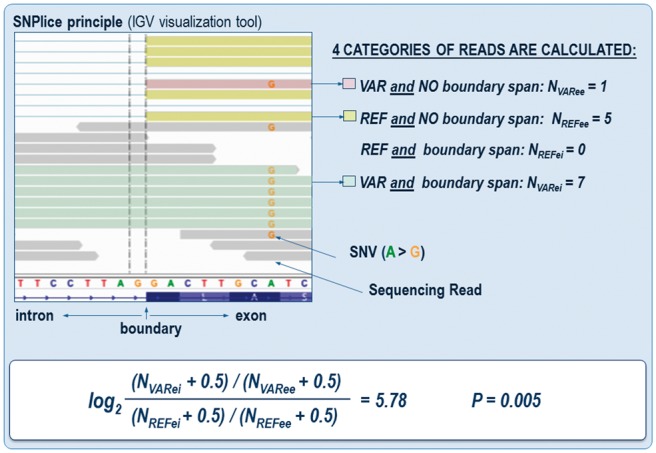
SNPlice principle. Alignment of reads spanning a potential splice modulating SNV and an exon-intron boundary, illustrating the SNPlice analytical strategy. Sequencing reads with variant and reference nucleotide at the SNV position in the proximity of the boundary are shown (color coded). Variant-harboring reads often continue in the intron (P = 0.005), indicating association with potential junction alteration. The double vertical line indicates the first position of the intron
SNPlice is freely available for download from https://code.google.com/p-/snplice/ as a self-contained binary package and as python source code. SNPlice version 1.7.2 was used for the analyses described in this paper. The workflow is illustrated on Figure 2. Following analysis of raw reads using Tophat, and Samtools (mpileup utility), the resulting aligned reads (BAM format), junctions (BED format) and variants (VCF format) are read directly by SNPlice. Users can choose to include only annotated junctions through aligning the RNA-seq reads to the reference transcriptome. Prior to SNPlice, the variants can be annotated using SeattleSeq (VCF to VCF format)—all SNV annotations are retained in the SNPlice output. The analysis can be performed on reads from individual samples where observed heterozygous variants provide the basis for SNPlice analysis, or on pooled reads from many samples, making it possible to study the effect of the variants across heterozygous and homozygous samples.
Fig. 2.
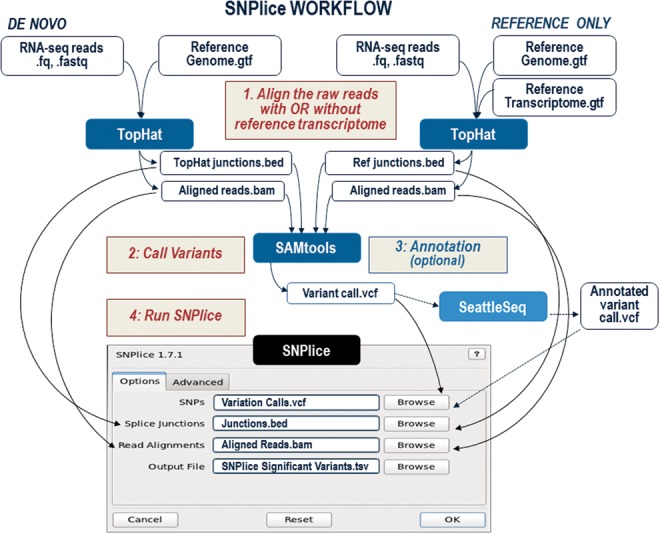
SNPlice workflow. To analyze annotated junctions only, RNAseq reads can be aligned to the reference transcriptome
2.5 Allele-specific Sanger sequencing
We designed allele-specific PCR, followed by Sanger sequencing, to illustrate the co-occurrence of variant nucleotide and exon–intron boundary for two of SNPlice highlighted loci. For each allele-specific PCR, three primers were designed: a common forward exonic primer to amplify the SNV locus, and two reverse primers hybridizing in the downstream exon or intron, respectively (Supplementary Table S2). First-strand cDNA was synthesized with SuperScript III reverse transcriptase (Invitrogen, Inc) using 1 μg of total RNA and mixture of oligo dT primer and random hexamers. Two separate reactions, amplifying the region containing the SNV, and either the exon-exon junction, or exon–intron boundary, were performed in parallel using high fidelity LA Taq DNA polymerase (Takara), according to the manufacturer recommendations. The products were gel-purified and subjected to bi-directional Sanger sequencing (ACGT Inc.), with the forward and the reverse primers used for the amplification.
3 Results
SNPlice processing of 65 human transcriptomes identified 141 unique variants significantly (FDR < 0.05) associated with intronic reads, 36 of which were observed in two or more unrelated samples (the 20 top-scored variants are shown in Supplementary Table S3). Between 0 and 18 variants were found per individual sample; this number positively correlated with the sequencing depth (Supplementary Fig. S3A). Comparison between the matched datasets revealed that 55% of the variants were called significant by SNPlice in both normal and tumor dataset; in all cases when the variant was called in only one of the dataset, this was the set with higher number of informative reads. We then compared the SNPlice significant calls across all samples heterozygous for the variant position (Supplementary Fig. S3B). Approximately one-third of the variants were called significant in all heterozygote samples in our datasets.
To assess SNPlice performance on pools of samples, we combined the read counts across the five RPE RNA-seq datasets and analyzed the summed counts using the same procedure as in SNPlice. This analysis identified 33 SNPlice significant variants, as compared to the 18 found through the individual analysis of the 5 datasets.
Of the 141 variants identified as significant by SNPlice in individual samples, 122 were positioned three and more nucleotides from the boundary of the exon. No correlation was seen between the distance of the variant to the splice site and the strength of the association. The most frequently observed nucleotide substitution (in regards to the sense orientation of the open reading frame) was C > T; in contrast, very few G > T and T > A substitutions were observed (Supplementary Fig. S3C). In regards to gene structure, 32% of the exonic SNVs were positioned in an in-frame exon, 14% in the first exon of the gene, 10% were in the last and 3% were within the 50 bp upstream of the last exon-exon junction (for at least one isoform). One SNV was a nonsense variant resulting in a premature stop codon, 57 were missense substitutions and the rest were synonymous.
3.1 Assessment of splice-modulating potential of SNVs highlighted through SNPlice
Illustrative examples of SNPlice highlighted variants are listed in Table 1. Rs10749291 is a synonymous substitution in exon 4 of the gene SFXN4 (c.258A > G, p.Q86Q, isoform ENST00000355697). IGV examination of the heterozygote sample confirmed the variant nucleotide only within reads encompassing the closest exon–intron junction, which was an acceptor site located six nucleotides from the SNV (Fig. 3A). Interestingly, the closest donor site (located 21 nucleotides from the SNV), was also encompassed almost exclusively by reads containing the variant nucleotide, leading to high proportion of molecules bearing the exon–intron junction on both sides of the exon (P < 0.001, Fisher’s exact test). No reads bearing the reference nucleotide were mapped to the intron on either side of the exon. Next, we examined the homozygote samples for the reference and variant allele. Consistent with the lack of effect on the junction, the homozygous reference samples did not show any indication for alternatively spliced molecules (Fig. 3B). On the contrary, the homozygous variant samples showed a higher proportion (P = 0.001) of nonspliced reads than observed in the heterozygote sample (Fig. 3C). Thus, SNPlice identified a variant that confidently associates with junction alteration, in a manner consistent with allele-quantitative effect.
Table 1.
SNVs located outside canonical splice-site sequences and assessed significant through SNPlice for association with intron retention. Comparative splice-altering potential is estimated through SplicePort (threshold -1, FDR<0.1), SpliceAid2, and Skippy. DTB – Distance to Boundary, A – acceptor; D-donor.
| Chr:location (hg19) | REF/VAR | Rs# | Gene/exon | DTB (nt) | Ex size (nt) | P value | q (FDR) | SplicePort (A/D) |
SpliceAid2 |
Skippy |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| REF | VAR | REF |
VAR |
ESE loss | ESE gain | ESS gain | ||||||||||
| motif | factor | motif | factor | |||||||||||||
| 10:120920588 | T/C | 10749291 |
|
|
27 |
|
|
|
|
|
|
|
|
0 | 0 | 1 |
| 2:198283305 | T/C | 788023 |
|
7 (A) | 80 | 0.0001 | 0.03 |
|
- | gttttc |
|
tcttc |
|
0 | 4 | 0 |
| 22:38877461 | T/G | 12004 |
|
8 (A) | 253 | 0.001 | 0.05 |
|
|
- | - | tccccat | hnRNPK | 0 | 0 | 0 |
| 4:1720346 | A/G | 11552262 |
|
7 (A) | 475 | 0.001 | 0.05 |
|
|
- | - | - | - | 1 | 0 | 5 |
| 18:21119777 | C/T | 1140458 |
|
3 (D) | 116 | 0.008 | 1.00 |
|
- | acaac | YB-1 | - | - | 1 | 0 | 0 |
| 12:113357209 | G/A | 1131476 |
|
16 (A) | 165 | 0.027 | 1.00 |
|
- | - | - | - | - | 0 | 0 | 0 |
Fig. 3.

Read alignment and assembly in the region of rs10749291. (A) Heterozygote sample: exclusive residing of the variant nucleotide within reads encompassing the exon-intron boundaries on both sides (located 6 and 21 nucleotides from the SNV), is seen. Read abundance shows relatively high proportion of intronic coverage on both sites of the boundary (arrows). (B) Homozygote reference sample: no reads are encompassing either one of the two boundaries, as also indicated by the complete absence of intronic coverage. (C) Homozygote variant sample: higher proportion of non-spliced reads, as compared to the heterozygote sample, supportive for allele-quantitative effect. (D) IGV visualization of the Cufflinks assembly (.gtf) showing the presence of an isoform retaining the introns on both sides of exon 4 of SFXN4 (bottom track); the reference assembly is on the top. The gene orientation is indicated by arrows along the introns
To assess the potential alternative transcript linked to rs10749291, we analyzed the assembly of the SFXN4 gene generated by Cufflinks (Trapnell et al., 2010). Consistent with IGV examination, the assembly showed presence of an isoform retaining the introns and the further exons on both sides of exon 4 (Fig. 3D). The predicted open reading frame expands 50 codons downstream of exon 3 into intron 3, until it reaches an in-frame stop codon TAA (Supplementary Fig. S4). Despite being a predicted target of nonsense mediated mRNA decay (NMD), such an isoform has been isolated from multiple human tissues: (http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/av.cgi?db=human&l=SFXN4.jAug10). It is predicted to encode a protein of 134 amino acids that contains no characteristic SFXN4 domain or trans-membrane motifs. Rs10749291 is a common variant (estimated minor allele frequency, MAF ∼0.49) that has not been previously reported to associate with splicing alterations. Our data strongly suggest that rs10749291 is implicated in the generation of the above-described alternative SFXN4 transcript. We next assessed the modulating potential of rs10749291 through splice-predictive tools. SplicePort estimated that the variant diminishes the acceptor strength, SpliceAid2 modeled that the change switches the binding site preference from the splice-regulator TIA-1 to PPT-binding protein PTB (Cavaloc et al., 1999; Caputi and Zahler, 2002; Jurica et al., 2002; Supplementary Fig. 5), and Skippy predicted gain of one new ESS (See Table 1).
Three more exonic variants scored by SNPlice as significant through individual sample analysis are presented in Table 1. Rs788023 is a C > T substitution located 7nt from the acceptor of exon 5 of SF3B1 (Supplementary Fig. S6A). Analysis of the transcript assembly of SF3B1 showed the presence of an isoform retaining the intron on the acceptor side of SNV-harboring exon, as well as the downstream positioned exon–intron–exon structure. SF3B1 isoforms expressing intron 4 as a part of an alternative 5′-UTR are described, and predicted to encode a partial protein of 345 AA (http://srv00.ibbe.cnr.it/ASPicDB/newresults.php?organism=-human&job=list4830/job7596). The second variant, rs12004, is situated 8 nt from the acceptor inside exon 4 of KDELR3 (Supplementary Fig. 6B); the variant nucleotide is modeled to create a new binding site for the spliceosome component hnRNPK (Caputi and Zahler, 2002). Reads assembly from the same sample showed presence of an alternative known last exon of KDELR3, present in major protein-coding KDELR3 isoforms (i.e. CCDS46705.1). The third SNV, rs11552262, is located 7 nt from the acceptor inside exon 2 of TMEM129 (Supplementary Fig. S6C). This variant was predicted to switch the splice site from acceptor to donor, and to destroy one existing and create three new ESEs. Reads assembly in the region of rs11552262 showed very low expression (FPKM < 0.2) of an isoform with partial intron retention on the acceptor site of exon 2, that has not been described before.
The remaining two variants in Table 1, rs1140458 in NPC1 and rs1131476 in OAS1 reached statistical significance only after pooling the reads from the individual samples in the RPE dataset. Both variants were predicted through SplicePort to destroy existing canonical splice sites—donor for NPC1, and acceptor for OAS1. In addition, rs1140458 in NPC1 was predicted to lead to loss of a binding site for the splice factor YB-1 (Ray et al., 2009), and loss of an exonic enhancer. We decided to further analyze these two variants through allele-specific Sanger sequencing.
3.2 Allele-specific Sanger sequencing of variant-boundary sites highlighted by SNPlice
To assess for the SNPlice suggested co-occurrence of variant nucleotide and exon–intron boundary, an AS-RT-PCR was designed to amplify in parallel the exon–exon junction, and the exon–intron boundary containing molecules (Supplementary Fig. S7A), for each of the two selected SNVs (rs1140458 in NPC1 and rs1131476 in OAS1). The resulting Sanger sequencing chromatograms are shown on Supplementary Figures S7B and S7C, respectively. The chromatograms were examined for relative signal (peak) of the variant versus the reference peak in the exon–exon and the exon–intron amplicons. Ideally, to confirm association of variant nucleotide with intron-containing reads, we anticipate predominant presence of the variant signal (versus reference) in the chromatograms from the exon–intron product, while the reference would be retained in the canonically spliced exon–exon junction containing amplicons.
As seen on Supplementary Figures S7B and S7C, the sequencing confirmed the allele-specific junction alteration that was identified by SNPlice. The top chromatogram on Supplementary Figure 7B shows canonically spliced NPC1 fragment (as indicated by the presence of an exon–exon junction between exons 18 and 19 of NPC1), and rs1140458 in heterozygous state, as indicated by equally strong signal from the reference and the variant base. In contrast, amplicons harboring the exon-intron boundary (second from the top chromatogram on Supplementary Fig. S7B) contained almost exclusively the variant nucleotide peak. The results were consistent with the reverse primer (Supplementary Fig. S7B, bottom chromatograms). Retaining of intron 19 donor sequence into the NPC1 open reading frame is predicted to result in a stop codon generation immediately after the last codon of exon 18. A protein coding NPC1 isoform terminated after exon 18 has been described (uc010xba, 766 AA, UCSC genes); our results suggest potential involvement of rs1140458 in the uc010xba formation.
Furthermore, even stronger association with junction alteration was detected for rs1131476 in OAS1. While the canonical splicing apparently tolerated molecules harboring the rs1140458 variant in NPC1 (supported by presence of both reference and variant peak in the exon–exon amplicons on Supplementary Fig. S7B), OAS1 alleles harboring rs1131476 variant nucleotide, were completely absent in the canonically spliced RNA product (Supplementary Fig. S7C, top chromatogram). Similarly to the rs1140458 variant in NPC1, exon–intron harboring molecules were characterized by sole presence of the variant peak (Supplementary Fig. S7C, second from the top chromatogram).
The results were consistent also with the reverse primer (Supplementary Fig. S7C, bottom chromatograms). Visual examination of the proximal to rs1131476 sequences of OAS1 identified the presence of closely positioned coallelic intronic variant, rs10774671, located in the canonical acceptor site. From the two in-phase variants, rs10774671 is more likely to be implicated in the junction alteration, due to its splice-site location. Without necessarily being involved in the underlying biological process, the coallelic rs1131476 is indicative of alternatively spliced OAS1 alleles, and can potentially serve as a marker in association analyses. Rs1131476 and rs10774671 are located at the boundary between intron 5 and exon 6 of OAS1 (ENST00000202917). An isoform expressing intron 5 as part of a 3’-UTR downstream of an alternative earlier OAS1 last exon is described (http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/av.cgi?db=human&l=OAS1.c-Aug10). Whether the detected in our experiment intron-retaining RNA molecules are part of the described, or of a novel OAS1 isoform, is subject of future investigations. Nevertheless, our data strongly suggest involvement of the rs1131476 and/or rs10774671 in the generation of an intron retaining expressed OAS1 isoform. Rs1131476 and rs10774671 illustrate an important example where SNPlice highlights SNVs physically linked (in-phase) to other splice-altering features (such as SNVs positioned in splice-sites, See Supplementary Fig. S7C).
4 Discussion
Until recently, direct assessment of allelic phase for DNA and RNA features of diploid genomes has been possible mostly through molecular cloning. Next-generation sequencing technologies are based on the production of a nearly ideal copy of a single DNA or RNA molecular segment, commonly termed ‘sequencing read’. Due to the uninterrupted process of template-based sequencing read generation, the features of the original molecule are preserved in the copy. As a consequence, molecular characteristics belonging to the same molecule, such as variant nucleotide and exon–intron boundary, can be evaluated in their allelic corelationship, and tools utilizing this information have began to emerge (Viner et al., 2014). SNPlice utilizes the allele-specific signal available in the massively parallel sequencing platforms to assess co-occurrence between variant nucleotide and nearby exon–intron boundaries. Hence, SNPlice highlights variants preferentially occurring on intron-containing molecules, possibly resulting from altered splicing. In addition, SNPlice considers paired-end sequencing reads, making it possible to assess longer distance variant-effected splicing. IGV examination of highlighted loci showed consistent with SNPlice co-occurrence of variants and junctions, which was further illustrated through allele-specific Sanger sequencing. Thus, SNPlice provides useful means for screening of transcriptome reads to identify novel splice altering variants, and to assess the splice-altering potential of splice-site positioned substitutions. Furthermore, SNPlice analysis of RNA-seq datasets from diseased tissues can identify rare pathogenic variants that are currently considered neutral due to lack of evidence for effect on the protein function. Finally, comparing SNPlice findings between groups of patients and controls could identify disease-implicated splicing deregulation.
Applying SNPlice, we were able to identify variants that selectively reside in alternatively spliced RNA molecules; the harboring motifs are to be submitted to SpliceAid-F (Giulietti et al., 2013). Cufflinks assembly of the reads in the SNV region suggested expression of alternative isoforms, in some cases, expressed at protein level. Most of these isoforms have been previously annotated through independent experiments, thus supporting the expression of the intron-retaining RNA molecules identified in our study. However, the described SNVs have never been directly linked to the expression of these particular isoforms. In this regard, SNPlice offers an experimentally based tool to identify novel relationships between encoded and expressed genomic layers.
Relatively few splice-site positioned variants (i.e. within the two nucleotides immediate to the boundary) were scored significant through SNPlice. This is not surprising, as many variants with strong effect on the splicing are expected to be eliminated through the evolutionary selection. One additional explanation is that, if the junction alteration creates a frame-shift, molecules bearing the alternative junction and the variant nucleotide may be degraded through nonsense-mediated mRNA decay (NMD). While this is expected to eliminate or reduce the number of variant-bearing reads, such an effect is difficult to assess using RNA data alone. However, splice-modulating variants that lead to NMD can be detected through SNPlice assessment of cell cultures under experimentally inhibited NMD.
Currently, the genome-scale splice-modulating potential of SNVs is estimated mostly through modeling tools built on knowledge derived from gene- or variant-focused experiments. Comparative analysis of selected significant variants from SNPlice results through SplicePort, SpliceAid2 and Skippy, were concordant with at least one of the three tools. Some variants, however, did not seem to affect known donors or acceptors, or to alter known binding motif or ESE. Because SNPlice assesses empirically derived data, it may highlight variants acting through unknown mechanisms, which are not currently implemented in the predictive pipelines.
Individual SNPlice assessment of the transcriptomes in our dataset identified between 0 and 18 heterozygous variants per sample associated with junction alteration at false discovery threshold 0.05. Because the statistical analyses are based on transcriptome sequencing read counts, the computed values are linked to the coverage (resp. expression levels) at the particular locus. Therefore, for many loci, strong statistical significance is unlikely to be reached due to relatively low number of reads, which can also explain the incomplete consistency between the matching normal and tumor dataset. For example, the two wet-lab validated SNVs reached significance only through pooled analysis, and not in any individual sample. An approach to address low read number is to combine the read counts from multiple samples to be processed through SNPlice as a whole. As illustrated, the combined approach increased the number of significant SNVs, at the same time retaining most of the variants identified through the individual analysis. An additional inherent advantage of the combined approach is that it considers the reads from homozygote samples, thus increasing the count of informative reads. This is especially applicable to intronic variants, which, if exerting a strong effect on splicing, naturally tend to appear as mono-allelic expression in the transcriptome (see Supplementary Fig. S7C). However, pooling reads from different samples needs to be applied with caution, as it assumes identical splicing regulation, and eliminates the uniqueness of splice factors and conditions that may be directly linked to the involvement of the particular variant in the splice alteration.
An inherent advantage of SNPlice is its immediate link to the empirical data. As the perspective on splicing turns into highly dynamic, condition-specific mechanism, SNPlice is poised to support cell- and tissue-specific analyses through individual transcriptome assessment, as well as to study dynamic, pathogenic and developmental splicing patterns. In addition, since cells typically coordinate numerous changes in ‘splicing programs’ (Braunschweig et al., 2013) analyzing the whole transcriptome can capture splice changes in their mutual dependencies. Finally, being based on read counts, SNPlice provides means for quantitative estimations of the splice changes across multiple samples. Application of the pipeline to large-scale datasets is expected to reveal multiple new splice modulating SNVs, which in turn, may highlight novel splice regulatory mechanisms or disease implicated genetic changes. In addition, SNPlice provides an innovative strategy to re-visit the splice-modulating potential of SNVs located in canonical sequences that are traditionally considered critical for splicing regulation.
Funding
This work is supported by MGPC, GWU; NIH National Center for Advancing Translational Sciences [UL1TR000075]; Clinical and Translational Science Institute at Children’s National Medical Center [CTSI-CN to A.H.]; and the Georgetown University Dean's Pilot [GX4002-753 to N.G.].
Supplementary Material
References
- Barash Y., et al. (2010) Deciphering the splicing code. Nature, 465, 53–59. [DOI] [PubMed] [Google Scholar]
- Barbosa-Morais N.L., et al. (2012) The evolutionary landscape of alternative splicing in vertebrate species. Science, 338, 1587–1593. [DOI] [PubMed] [Google Scholar]
- Benjamini Y.H., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B, 57, 289–300. [Google Scholar]
- Bernstein B.E., et al. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U., et al. (2013) Dynamic integration of splicing within gene regulatory pathways. Cell, 152, 1252–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel V., Kleffe J. (1998) Prediction of locally optimal splice sites in plant pre-mRNA with applications to gene identification in Arabidopsis thaliana genomic DNA. Nucleic Acids Res., 26, 4748–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunak S., et al. (1991) Prediction of human mRNA donor and acceptor sites from the DNA sequence. J. Mol. Biol., 220, 49–65. [DOI] [PubMed] [Google Scholar]
- Caputi M., Zahler A.M. (2002) SR proteins and hnRNP H regulate the splicing of the HIV-1 tev-specific exon 6D. EMBO J., 21, 845–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaloc Y., et al. (1999) The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA, 5, 468–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung V.G., et al. (2005) Mapping determinants of human gene expression by regional and genome-wide association. Nature, 437, 1365–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark F., Thanaraj T.A. (2002) Categorization and characterization of transcript-confirmed constitutively and alternatively spliced introns and exons from human. Hum. Mol. Genet., 11, 451–464. [DOI] [PubMed] [Google Scholar]
- Coulombe-Huntington J., et al. (2009) Fine-scale variation and genetic determinants of alternative splicing across individuals. PLoS Genet., 5, e1000766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Conti L., et al. (2013) Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip Rev RNA, 4, 49–60. [DOI] [PubMed] [Google Scholar]
- Dogan R.I., et al. (2007) SplicePort–an interactive splice-site analysis tool. Nucleic Acids Res., 35, W285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElSharawy A., et al. (2006) SNPSplicer: systematic analysis of SNP-dependent splicing in genotyped cDNAs. Hum. Mutat., 27, 1129–1134. [DOI] [PubMed] [Google Scholar]
- ElSharawy A., et al. (2009) Systematic evaluation of the effect of common SNPs on pre-mRNA splicing. Hum. Mutat., 30, 625–632. [DOI] [PubMed] [Google Scholar]
- Faber K., et al. (2011) Genome-wide prediction of splice-modifying SNPs in human genes using a new analysis pipeline called AASsites. BMC Bioinformatics, 12 (Suppl. 4), S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gart J.J., Zweifel R. J. (1967) On the bias of various estimators of the logit and its variance with application to quantal bioassay. Biometrika, 54, 181–187. [PubMed] [Google Scholar]
- Giulietti M., et al. (2013) SpliceAid-F: a database of human splicing factors and their RNA-binding sites. Nucleic Acids Res., 41, D125–D131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane J.B.S. (1955) The estimation and significance of the logarithm of a ratio frequencies. Ann. Hum. Genet., 20, 309–311. [DOI] [PubMed] [Google Scholar]
- Han H., et al. (2013) MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature, 498, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull J., et al. (2007) Identification of common genetic variation that modulates alternative splicing. PLoS Genet., 3, e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurica M.S., et al. (2002) Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. Rna, 8, 426–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath U., et al. (2012) An evolutionary algorithm approach for feature generation from sequence data and its application to DNA splice site prediction. IEEE/ACM Trans. Comput. Biol. Bioinform., 9, 1387–1398. [DOI] [PubMed] [Google Scholar]
- Kim D.P., et al. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol., 14,R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao P.Y., et al. (2005) Splicing for alternative structures of Cav1.2 Ca2+ channels in cardiac and smooth muscles. Cardiovasc. Res., 68, 197–203. [DOI] [PubMed] [Google Scholar]
- Maminishkis A., et al. (2006) Confluent monolayers of cultured human fetal retinal pigment epithelium exhibit morphology and physiology of native tissue. Invest Ophthalmol. Vis. Sci., 47, 3612–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus C.J., Graveley B.R. (2011) RNA structure and the mechanisms of alternative splicing. Curr. Opin. Genet. Dev., 21, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin J., et al. (2012) Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science, 338, 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M.J., Silver P.A. (2008) Global analysis of mRNA splicing. RNA, 14, 197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q., et al. (2008) Nat. Genet., 40, 1413–1415. [DOI] [PubMed] [Google Scholar]
- Parzen M., et al. (2002) An estimate of the Odds Ratio that always exists. J. Comput. Graphical Stat., 11, 420–436. [Google Scholar]
- Pertea M., et al. (2001) GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res., 29, 1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piva F., et al. (2012) SpliceAid 2: a database of human splicing factors expression data and RNA target motifs. Hum. Mutat., 33, 81–85. [DOI] [PubMed] [Google Scholar]
- Ray D., et al. (2009) Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat. Biotechnol. 27, 667–670. [DOI] [PubMed] [Google Scholar]
- Riva A. (2012) Large-scale computational identification of regulatory SNPs with rSNP-MAPPER. BMC Genomics, 13 (Suppl 4), S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.K., Cooper T.A. (2012) Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med., 18, 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterne-Weiler T., Sanford J.R. (2014) Exon identity crisis: disease-causing mutations that disrupt the splicing code. Genome Biol., 15, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H., et al. (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform., 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., et al. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol., 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viner C., et al. (2014) Validation of predicted mRNA splicing mutations using high-throughput transcriptome data. Version 2. F1000Res. eCollection (http://f1000r.es/378). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Burge C.B. (2008) Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA, 14, 802–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfe A., et al. (2010) Genomic features defining exonic variants that modulate splicing. Genome Biol., 11, R20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G., Burge C.B. (2004) Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol., 11, 377–394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.