

Abstract
Clinical gene therapy has been increasingly successful, due both to an enhanced molecular understanding of human disease and to progressively improving gene delivery technologies. Among the latter, delivery vectors based on adeno-associated virus (AAV) have emerged as safe and effective – in one recent case leading to regulatory approval. Although shortcomings in viral vector properties will render extension of such successes to many other human diseases challenging, new approaches to engineer and improve AAV vectors and their genetic cargo are increasingly helping to overcome these barriers.
Introduction
The vast majority of the approximately 7,000 monogenic disorders – which collectively afflict millions worldwide, with often debilitating personal and societal consequences – have no treatment options. Sequencing efforts to date have identified the genes responsible for approximately 50% of these disorders, and with the rapidly progressing advances in next-generation sequencing technologies the remainder will likely be identified within a decade1. In parallel, the field of gene therapy has surmounted numerous hurdles for safe and efficient gene delivery, which has led to unprecedented treatments for some monogenic disorders. Furthermore, gene therapy is showing signs of success in several complex disorders, for example, chronic conditions such as heart disease, neurodegenerative disorders, stroke and diabetes mellitus. The prospect of single-administration treatments for monogenic and complex human diseases — developed by integrating knowledge of disease genetics and pathology with effective gene therapy — has the potential to be paradigm shifting for healthcare.
Therapeutic success to date has been enabled by the identification of several viruses that can be engineered into effective gene delivery vectors, including the non-pathogenic parvovirus adeno-associated virus (AAV; Figure 1), among others. In particular, an increasing number of phase I–III clinical trials using AAV vectors have yielded promising results (for an overview of published clinical trials using AAV, their achievements, and associated limitations, see Supplementary information S1 (table)). For instance, in trials for familial lipoprotein lipase (LPL) deficiency, an AAV1-based vector encoding the gain-of-function variant LPLS447X resulted in persistent gene expression and protein activity, which led to sustained decreases in the incidence of pancreatitis2–4. Based on these outcomes and its safety profile, this product — Glybera (alipogene tiparvovec) — received market approval in the European Union in October 2012, albeit under “exceptional circumstances” (see EMEA website [http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002145/WC500135472.pdf]), representing the first approved gene therapy in Western nations. Other monogenic disorders in which AAV vectors have demonstrated safety and efficacy include Leber’s congenital amaurosis type 25–10, choroideremia11, and hemophilia B12, among others (Supplementary information S1 (table)). In parallel to successes with monogenic disorders, AAV has been applied to idiopathic diseases. For example, administration of an AAV1 vector encoding the SERCA2a gene resulted in improvements of various key outcomes in patients with advanced heart failure13,14. Gene therapy with AAV is thus showing increasing promise for both Mendelian inherited and complex diseases.
Figure 1. Adeno-associated virus (AAV) biology and variant generation.

(a) Schematic of the 4.7 kb single-stranded DNA genome. The AAV genome, which is packaged within a nonenveloped, icosahedral capsid, contains three open reading frames (ORFs) flanked by inverted terminal repeats (ITRs), which form T-shaped hairpin ends. The rep ORF encodes four nonstructural proteins (Rep 40, Rep 52, Rep 68, and Rep 78) that are essential for viral replication, transcriptional regulation, genome integration, and virion assembly66. The cap ORF encodes three structural proteins (VP1-3), that form the 60-mer viral capsid66 with the aid of the assembly-activating protein (AAP; gray arrow)67,68, which is encoded in an alternate ORF located within cap. Hypervariable regions are denoted by colored arrows. Surface-exposed amino acids are indicated on the capsid protein (black lines). (b) Crystal structure of the AAV capsid, with VP3 hypervariable regions colored to match the corresponding genetic regions.69 (c) To generate recombinant versions of AAV, a gene of interest is inserted between the ITRs, replacing rep and cap, which are provided in trans, on so-called ‘packaging constructs’, along with adenoviral helper genes needed for replication70. The viral capsid governs the resulting vector’s ability to transduce cells, from initial cell surface receptor binding to nuclear entry and genome release, which can lead to stable transgene expression in post-mitotic tissue71. Eleven naturally occurring serotypes and over 100 variants of AAV exist, which differ in their amino acid sequence and thus in their gene delivery properties72,73.
That said, the effective delivery of genetic material has been and will continue to be a major challenge in the field (Box 1), since in many cases the naturally evolved infectious properties of viral vehicles are mismatched with the delivery needs of many therapeutic indications. A number of novel approaches have been used to overcome some of these barriers. For example, progressive improvements in knowledge of AAV capsid structure15,16 are facilitating rational design of AAV capsids, and considerable progress in AAV capsid library development17,18 and screening methodology19,20 are facilitating directed evolution of AAV capsids. Furthermore, although gene therapy to date has primarily been successful in gene replacement therapies for recessive disorders, advances with therapeutic payloads may soon enable treatment of genetically dominant diseases.
Box 1. Challenges of AAV gene delivery and efficacy.
Immune interactions
The immune system is highly effective at preventing the delivery of foreign nucleic acids, thereby posing many challenges to therapeutic gene delivery. Widespread natural exposure to AAV has resulted in a large fraction of the population harboring neutralizing anti-capsid antibodies in blood and other bodily fluids. Furthermore, following cellular transduction, AAV capsid epitopes can become cross-presented on MHC I complexes, leading to the elimination of transduced cells by capsid-specific cytotoxic T lymphocytes and corresponding loss of gene expression, as evident in the decline in Factor IX expression observed in an early clinical trial for hemophilia B24. Many human CD4+ and CD8+ T cell epitopes have been identified for AAV224,46,47 and AAV848, and MHC loci are among the most polymorphic in the human genome, making it difficult to engineer an AAV capsid that could evade recognition by all possible MHC combinations. However, it may be possible to engineer capsids that are not as readily processed by proteasomes or Transporter associated with Antigen Processing (TAP) proteins.
Transport to and tropism for target cells
For systemically administered viruses, the liver is often the default destination, which can represent a barrier when other organs are the intended targets. In addition, endothelial cell layers, especially within the blood-brain barrier, pose a physical barrier for entry into a tissue. A vector that gains access to an organ, or is directly administered to that organ, can then encounter numerous transport barriers to efficient transduction of the often large tissue volumes involved in disease, including cell bodies and intervening extracellular matrix to which many AAV variants bind49 (for example, heparan sulfate50).
Cellular barriers
Once vectors have arrived at the surface of a target cell, it may lack the primary and/or secondary receptor necessary for vector binding and internalization. Furthermore, endosomal escape, proteasomal escape, nuclear entry, and vector unpackaging all represent barriers to transduction.
Packaging capacity
Natural adeno-associated viruses have a single-stranded 4.7 kb DNA genome. Gene delivery vectors based on AAV have been shown to be capable of packaging genomes of up to approximately 5 kb at near wild-type titers and infectivity51,52, beyond which packaging efficiency significantly decreases and genomes with 5’ truncations become encapsidated51,52.
This Progress article focuses on recent innovations in vector engineering, specifically, the rational design and directed evolution of AAV variants, as well as novel approaches for modifying genetic cargo. Successful applications of several vector engineering strategies that have created novel AAV variants to overcome some of the current challenges facing AAV-mediated gene delivery are discussed, particularly recent developments in directed evolution approaches for capsid engineering.
Engineering delivery systems
Challenges with gene delivery using AAV (Box 1) arise from the simple consideration that the properties that constitute success for natural viral infections are distinct from those needed for most medical applications, and viruses did not evolve for the latter. However, vector engineering can release viruses from the constraints of natural evolution and thereby enable them to acquire novel and biomedically valuable phenotypes. Such vector engineering advances can be grouped into rational design and directed evolution efforts.
Rational design of AAV variants
In some cases, knowledge of delivery mechanisms, coupled with AAV structural analyses15,16, can aid vector improvement. For example, the basic discovery that phosphorylation of capsid tyrosine residues results in ubiquitination and promotes proteasomal degradation of AAV virions21 led to the development of vehicles in which tyrosines were mutated to phenylalanines via site-directed mutagenesis21,22. In one such study, these vectors were capable of 10-fold higher transgene expression in vitro and up to 30-fold higher transgene expression in vivo21. Recently, this approach was used to engineer a novel AAV2(Tyr-Phe) mutant capsid with a potentially reduced risk of cytotoxic T lymphocyte immune responses, which is a key limitation of clinical AAV-mediated gene therapy (Box 1)23. Specifically, MHC class I presentation of AAV capsid epitopes is believed to underlie cytotoxic T lymphocyte reactions against AAV-transduced hepatocytes in clinical trials for hemophilia B12,24. Since the tyrosine mutations affect proteasomal processing of capsids, they also offer the potential to reduce MHC class I presentation of capsid antigens, a process that in general begins with through proteasomal degradation of cytosolic proteins.
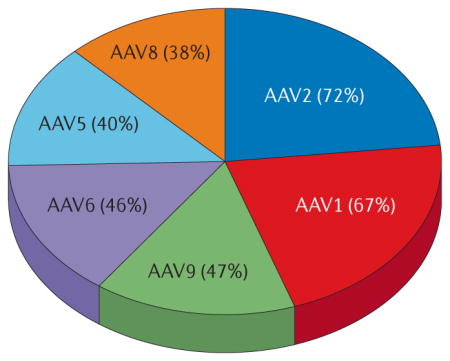
Rational design approaches have also been pursued to address challenges with pre-existing neutralizing antibodies (Box 1). The majority of the human population has been naturally exposed to AAV, and natural AAV variants and serotypes exhibit considerable sequence identity20. As a result, a substantial percentage of the human population harbors neutralizing antibodies that markedly limit gene delivery by many natural vectors (AAV2: 72%, AAV1: 67%, AAV9: 47%, AAV6: 46%, AAV5: 40% and AAV8: 38%)25. This issue has been circumvented in most clinical studies by excluding patients with neutralizing antibodies, but improvements will have to be developed to broaden the patient pool that can benefit from a therapy. Several strategies have been utilized to discover and mutate the epitopes responsible for anti-capsid antibody binding. Linear and conformational epitopes responsible for neutralizing antibody binding to the AAV capsid have been mapped for several antibodies26,27. Lochrie et al. subsequently used an in silico structural analysis of potential docking sites for a murine IgG2a antibody with the AAV2 surface to determine sterically accessible, candidate positions, which were then subjected to extensive site-directed mutagenesis to develop variants with reduced neutralization by mouse and human antibodies in vitro28. A more recent, alternate approach by Mingozzi et al. involved the generation of empty AAV2-based capsid particles with mutations that ablate primary cell receptor binding29. When mixed with recombinant vectors carrying therapeutic transgenes, these empty capsids functioned as decoys to bind low to moderate levels of neutralizing antibodies and thereby enhance transduction of the co-administered vector in mice and non-human primates to levels equal to or greater than transduction in naïve animals29.
In another example of rational design, the incorporation of high-affinity ligands into the AAV capsid can confer binding to alternate cell surface receptors and thereby restrict or re-direct viral tropism. In a recent preclinical study, Münch et al. inserted designed ankyrin repeat proteins specific to the HER2 receptor at the N terminus of the VP2 region of the AAV2 capsid (Figure 1), thereby increasing the specificity of the vector to tumor cells overexpressing the HER2 receptor by ~30-fold in vitro and ~20-fold in vivo30. In addition, structural alignment and knowledge of regions involved in receptor binding can enable shifts in tropism. For instance, Shen et al. used site-directed mutagenesis to incorporate the amino acids responsible for AAV9 binding to galactose residues at the corresponding sites in the AAV2 capsid to design dual glycan-binding AAV vectors that could use both heparan sulfate and galactose to enter cells31. By virtue of this dual receptor binding, the vector showed significantly higher infectivity of the liver than AAV2 and greater specificity to the liver than AAV9.
In many situations, however, knowledge of the viral structure–function relationships underlying a given gene delivery problem is insufficient to enable rational design of AAV’s complex virion. A vector engineering approach that has emerged in recent years to address this dilemma is directed evolution, which emulates the process of natural evolution.
Directed evolution of AAV variants
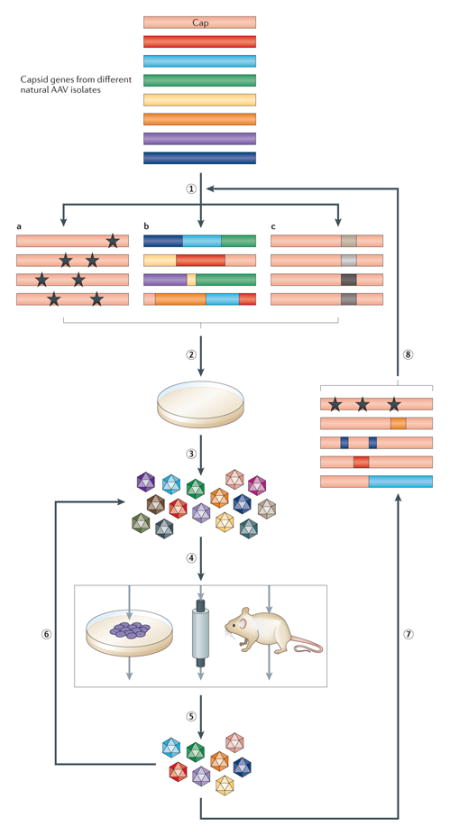
Directed evolution strategies harness genetic diversification and selection processes to enable the accumulation of beneficial mutations that progressively improve the function of a biomolecule (Box 2, Figure 2). In this process, wild-type AAV cap genes are diversified by several approaches to create large genetic libraries that are packaged to generate libraries of viral particles, and selective pressure is then applied to isolate novel variants that can overcome gene delivery barriers (Box 2)32,33. Importantly, the mechanistic basis underlying a gene delivery problem need not be known for directed evolution function, which can thus accelerate the development of enhanced vectors.
Box 2. Elements of directed evolution for capsid engineering.
Library generation strategies
Error-prone PCR
The most straightforward library generation approach is low fidelity polymerase chain reaction (PCR) — also known as error-prone PCR — which introduces random point mutations into the AAV cap ORF at a predetermined, modifiable rate34. This approach has been used to introduce mutations into either single32,34 or multiple17 AAV serotypes for subsequent selection. The introduction of point mutations usually results in relatively few changes to the capsid, but error-prone PCR can be combined with other mutagenesis strategies to further optimize variants.
Chimeric capsids
Using an in vivo viral recombination method53 or more commonly via DNA shuffling38,54–56, random chimeras of AAV cap genes can be generated, which yields chimeric cap gene libraries that are composed of multiple serotypes. These ‘bred’ capsids can combine their parental properties in novel ways. However, many of the mutated capsids may be incapable of packaging, which substantially reduces the diversity of the library.
Random peptide insertions
Random peptide sequences can be inserted into defined sites of the viral capsid, such as in the heparin binding domain of the AAV2 capsid57,58 and in AAV918, via ligation of degenerate oligonucleotides into the cap ORF. The insertion of random peptides into the capsid can potentially shift the binding of AAV vectors to a new cell surface receptor. Conversely, defined peptide-encoding sequences can be inserted into random locations of the AAV2 cap ORF via transposon mutagenesis59,60.
Randomization of surface loops
Diversity can also be concentrated onto multiple hypervariable regions of the AAV capsid, which lie on surface-exposed loops. Such ‘loop swap’ libraries were for example generated by replacing four surface loops of AAV2 with libraries of peptide sequences bioinformatically designed based on the level of conservation of each amino acid position among natural AAV serotypes and variants61. Similar to the random peptide insertion libraries, only a small area of the capsid is mutated, but this method can be paired with additional mutagenesis strategies to modify the full capsid.
Selection
Binding affinity columns
AAV variants with increased or decreased binding affinity for a cell surface protein can be selected using affinity columns32, where elution of different fractions yields variants with altered binding. While allowing for rapid selection of AAV variants with novel receptor binding affinity or specificity, this approach does not take into consideration other important aspects of the infection pathway such as extracellular or intracellular trafficking.
In vitro cell culture models
When cell culture models can emulate key aspects of anin vivo context, such as anti-capsid antibody binding or airway epithelial structure and polarization, in vitro selection strategies using primary cells isolated from tissue samples or immortal cell lines that mimic the behavior of cells in the human body can be effective in creating AAV variants with increased efficiency and/or specificity. In vitro selections have thus resulted in the development of improved AAV variants for a number of cell types, including human airway epithelia, human glia, and human embryonic stem cells 17,36,61. This selection strategy, however, does not accurately model a complete in vivo environment, such as immune interactions, organ biodistribution, tissue transport barriers, and the presence of other cell types within a tissue.
In vivo models
To more accurately capture a clinical gene therapy environment, selections can be directly performed in animal models19,20,38. AAV libraries can be administered through various dosing routes, including intravenous injection, intravitreal administration, or direct intracranial injection. The tissue or cell type of interest is then harvested to isolate AAV variants that have successfully infected that target. One challenge is that the resulting variants are capable of improved infectivity of the animal used for the selections, which does not necessarily translate to improved infectivity of human cells62,63. Human xenograft models can be employed to select for infection of grafted human cells,20 though this advantage is counter-balanced by absence of an intact immune system in the immunodeficient host and by unknown specificity for a given human target cell in the context of non-human primates or humans.
Genome recovery
Adenovirus-mediated replication
Providing successful viral genomes with the opportunity to increase their frequency in the gene pool is an inherent feature of natural selection. Adenovirus has been utilized as a helper virus to amplify AAV vectors that have reached the nucleus of the target cell in a number of in vitro selections17,36,37,55,58,61,64,65. Rescue by human adenovirus was also utilized in vivo to amplify vectors that have infected human hepatocytes, but not mouse hepatocytes20, an approach that can be effective when the target cells are accessible and permissive to adenovirus superinfection.
PCR amplification
While this approach risks the isolation of genomes that have localized to rather than productively infected a given cell or tissue, PCR has been highly successful in amplification and recovery of enhanced AAV variants in a number of in vivo selection studies19,38,39. This strategy is well-suited for situations where the cell of interest is not accessible or permissive to adenovirus infection19 or the use of replication competent libraries raises biosafety considerations.
Directed evolution was first applied to address the neutralizing antibody problem, and several promising studies reported successes, for example the generation of AAV2 variants that could withstand significantly higher levels of neutralizing antibodies in vitro34 and in vivo32 compared with wild-type AAV2. Recent work that involved multiple rounds of directed evolution using several different pools of human anti-AAV antibodies as selective pressures has also yielded new variants capable of enhanced antibody evasion in vitro and in vivo35. Specifically, AAV variants created through either saturation mutagenesis of several amino acids important for antibody binding or DNA shuffling required up to 20-fold higher in vitro concentrations of pooled human antibodies for neutralization vs. AAV1 (35-fold vs. AAV2). The antibody neutralization properties also translated to enhanced transduction in vivo, where variants were capable of significantly higher liver, heart, and muscle transduction than AAV2 in mice passively immunized with human antibodies.
In parallel, mutant AAV capsids have been evolved for more efficient and specific infection of previously non-permissive cell types. For example, vectors have been engineered for 100-fold increased transduction of human airway epithelia36, 50-fold higher transduction of neural stem cells37, and 3-fold higher transduction of human pluripotent stem cells17 in vitro. Furthermore, directed evolution has increasingly been implemented with in vivo models, particularly in situations where in vitro culture is an inadequate model, such as for systemic gene delivery or vector transport through complex tissues. Yang et al. conducted in vivo biopanning for more efficient infection of murine muscle, and a resulting chimeric variant capsid exhibited nearly equal cardiac infectivity, yet statistically significantly decreased liver localization, compared to AAV938. In addition, Gray et al. isolated an AAV8 variant for the ability to gain access to regions of the brain in which seizure had compromised the blood-brain barrier39. More recently, Lisowski et al. used a model involving immunodeficient mice carrying human hepatocyte xenografts to better simulate in vivo human hepatocyte infection20. Upon administering a chimeric AAV library, human adenovirus (which exhibits tropism for human cells) was added to induce replication of the desired AAV variants and thereby yield an AAV variant capable of efficient and selective human hepatocyte transduction20. Future work may extend these studies to large animal models, particularly non-human primates.
Tissue transport barriers to viral infection are also a key limitation for the clinical application of gene therapy (Box 1). For example, the cells most afflicted in retinal disease – photoreceptors and retinal pigment epithelium – lie behind hundreds of microns of dense tissue. Subretinal injections poses surgical risks compared to intravitreal injection and do not transduce the full retina. Hence, Klimczak et al. engineered an AAV variant capable of highly specific (94%) and efficient infection of Müller cells, which span across the full retina, upon intravitreal injection40. In a rat model of retinitis pigmentosa, transduction of these cells with the engineered AAV variant enabled the broad expression of a neuroprotective factor and slowed retinal degeneration41. In a recent study, Dalkara et al. used in vivo directed evolution to generate an AAV with the ability to transport genetic cargo through the retina and directly infect photoreceptors after intravitreal delivery19. The resulting variant was capable of substantially higher gene expression in mouse and non-human primate photoreceptors in vivo, and led to the rescue of murine models of X-linked retinoschisis and Leber’s congenital amaurosis type 219.
Through successful application to a variety of in vitro and in vivo systems, directed evolution has demonstrated the capacity to overcome a broad range of gene delivery challenges. Future work may increasingly integrate rational knowledge of capsid structure, as well as advances in DNA synthesis and sequencing, to further enhance this technology platform. In addition to the development of improved vectors, advances in their genetic payload promise to further extend the reach of gene therapy.
Engineering genetic payload
Two additional challenges of AAV-mediated gene therapy are the treatment of autosomal dominant genetic diseases – in which an allele must be removed rather than added – and in some cases the limited genomic carrying capacity of AAV (4.7 kb). These problems can potentially be addressed by modifying the genetic cargo rather than the capsid. In particular, the progressive emergence of sequence-specific endonucleases (reviewed in REF. 42) – including zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and RNA-guided engineered nucleases based on the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs)/Cas system – may offer innovative answers to such challenges. Endogenous gene repair has been a longstanding goal of genetic therapies, and sequence-specific endonucleases can increase the efficiency of homologous recombination between a defective allele and donor DNA.43 For example, in a murine hemophilia B model, Li et al. used an AAV8 vector encoding a ZFN targeted to the F9 gene (encoding Factor IX) to induce double-strand breaks in the genome and thereby facilitate homologous recombination with a co-delivered, promoterless Factor IX cDNA fragment44. The resulting gene correction was sufficient to improve blood clotting times44, raising the possibility that fragments of cDNAs that in their entirety are too large for AAV could be used to mediate the repair of focal mutations within large endogenous genes, such as dystrophin, CFTR, CEP290, ABCA4, MYO7A, USH2A, and F8 (encoding the Factor VIII blood clotting protein). In addition, RNA interference has been implemented for specific knockdown of pathogenic alleles45, but targeted DNA-binding proteins or nucleases delivered by AAV offer the promise for a more potent transcriptional knockdown or even complete therapeutic knockout of such genes. Although additional investigations must be conducted to elucidate the potential for off-target genotoxicity of this approach, pairing together innovative vehicles and payloads offers the opportunity for further extending the reach of gene therapy.
Conclusions
Clinical trials involving AAV delivery to accessible tissues have enabled successful treatment of several recessive monogenic disorders, which has provided strong momentum to the field. That said, considerable challenges with both delivery and payload remain. Fortunately, viruses, like many biomolecules, are highly plastic, and the engineering and evolution of ‘designer’ vectors with properties that are tailored to specific clinical needs may bring progressively more therapeutic targets within AAV’s reach. Moreover, the development of new cargoes – especially site-specific DNA endonucleases – raises the possibility of gene correction or even the treatment of dominant genetic disorders. Recent advances in human disease biology, AAV virology and engineering, and therapeutic payloads thus promise to extend clinical successes to additional monogenic and complex disorders.
Supplementary Material
Online summary.
Gene delivery vectors based on adeno-associated virus (AAV) have emerged as safe and effective for numerous clinical gene therapy applications.
AAV-based gene therapy must overcome challenges that arise because the properties that constitute success for natural viral infections can be distinct from those needed for most medical applications. As a result, vectors based on natural AAV variants have both positive qualities, and many shortcomings.
Vector engineering can release viruses from the constraints of natural evolution and thereby enable them to acquire novel and biomedically valuable phenotypes, which may otherwise not serve their interests in a natural setting.
Advances in AAV biology and structural analysis have led to the rational design of AAV variants capable of reduced proteasomal degradation, decreased neutralization by anti-AAV antibodies, and increased tropism for certain cell types.
Directed evolution – genetic diversification and selection for greatly improved function – has led to the development of novel AAV variants capable of enhanced receptor binding, decreased neutralization by anti-AAV antibodies, and increased (and in some cases targeted) tropism for many specific cell types in vitro and in vivo.
Recent advances in AAV vector development via rational design and directed evolution, as well as in novel genetic payloads, promise to extend clinical successes of AAV gene therapy.
Glossary
- Parvovirus
linear, non-segmented single-stranded DNA viruses whose genome size is typically in the range of 5000 nucleotides
- Leber’s congenital amaurosis type 2
rare, monogenic inherited eye disorder caused by mutations in the RPE65 gene that encodes a protein needed for the isomerohydrolase activity of the retinal pigment epithelium that results in loss of photoreceptor function
- Choroideremia
X-linked recessive disease caused by a mutation to the CHM gene and subsequent absence of the Rab escort protein-1 (REP1) that leads to progressive loss of vision due to degeneration of the retina and choroid
- Rational design
a capsid engineering approach that utilizes knowledge of AAV biology and structural analysis to guide capsid changes
- Directed evolution
a capsid engineering approach that emulates natural evolution through iterative rounds of genetic diversification and selection processes, thereby enabling the accumulation of beneficial mutations that progressively improve the function of a biomolecule
- Tropism
the cell or tissue type that can be infected by a virus or gene delivery vector
- Biopanning
in vivo method for selection of AAV variants from a library for more efficient infectivity of a cell or tissue type of interest
- Müller cells
glial cells that support neurons in the vertebrate retina
Biographies
Melissa A. Kotterman received her B.S. in Chemical Engineering and Biomedical Engineering from Carnegie Mellon University and her Ph.D. in Chemical and Biomolecular Engineering from University of California, Berkeley. Dr. Kotterman’s research goals focus on the development of gene delivery vectors as tools for use in both basic research and clinically therapeutic applications. Specifically, she has developed AAV vectors that improve gene delivery to human embryonic stem cells and adult neural stem cells and prevent neutralization by antibodies.
David V. Schaffer received his B.S. in Chemical Engineering from Stanford and his Ph.D. in Chemical Engineering from Massachusetts Institute of Technology. Dr. Schaffer also did a postdoctoral fellowship at the Salk Institute for Biological Studies. As Professor of Chemical and Biomolecular Engineering, Bioengineering, and Molecular and Cell Biology at the University of California, Berkeley, Dr. Schaffer applies engineering principles to problems in cell biology and bioengineering. His laboratory focuses on the related fields of stem cell biology and gene therapy. Dr. Schaffer is working on the development of specific viral vectors to improve gene delivery vehicle performance for therapeutic applications, including stem cell-based therapies.
References
- 1.Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013;14:681–91. doi: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- 2.Stroes ES, et al. Intramuscular administration of AAV1-lipoprotein lipase S447X lowers triglycerides in lipoprotein lipase-deficient patients. Arterioscler Thromb Vasc Biol. 2008;28:2303–4. doi: 10.1161/ATVBAHA.108.175620. [DOI] [PubMed] [Google Scholar]
- 3.Gaudet D, et al. Review of the clinical development of alipogene tiparvovec gene therapy for lipoprotein lipase deficiency. Atheroscler Suppl. 2010;11:55–60. doi: 10.1016/j.atherosclerosissup.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Carpentier AC, et al. Effect of alipogene tiparvovec (AAV1-LPL(S447X)) on postprandial chylomicron metabolism in lipoprotein lipase-deficient patients. J Clin Endocrinol Metab. 2012;97:1635–44. doi: 10.1210/jc.2011-3002. [DOI] [PubMed] [Google Scholar]
- 5.Hauswirth WW, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19:979–90. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bainbridge JWB, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- 7.Maguire A, Simonelli F. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maguire AM, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374:1597–605. doi: 10.1016/S0140-6736(09)61836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobson SG, et al. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130:9–24. doi: 10.1001/archophthalmol.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett J, et al. AAV2 gene therapy readministration in three adults with congenital blindness. Sci Transl Med. 2012;4:120ra15. doi: 10.1126/scitranslmed.3002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacLaren RE, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet. 2014;6736:2117–2120. doi: 10.1016/S0140-6736(13)62117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nathwani AC, et al. Adenovirus-Associated Virus Vector–Mediated Gene Transfer in Hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaski BE, et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009;15:171–81. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jessup M, et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124:304–13. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DiMattia MA, et al. Structural insight into the unique properties of adeno-associated virus serotype 9. J Virol. 2012;86:6947–58. doi: 10.1128/JVI.07232-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Govindasamy L, et al. Structural insights into adeno-associated virus serotype 5. J Virol. 2013;87:11187–99. doi: 10.1128/JVI.00867-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asuri P, et al. Directed evolution of adeno-associated virus for enhanced gene delivery and gene targeting in human pluripotent stem cells. Mol Ther. 2012;20:329–38. doi: 10.1038/mt.2011.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varadi K, et al. Novel random peptide libraries displayed on AAV serotype 9 for selection of endothelial cell-directed gene transfer vectors. Gene Ther. 2012;19:800–9. doi: 10.1038/gt.2011.143. [DOI] [PubMed] [Google Scholar]
- 19.Dalkara D, et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013;5:189ra76. doi: 10.1126/scitranslmed.3005708. [DOI] [PubMed] [Google Scholar]
- 20.Lisowski L, et al. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2013 doi: 10.1038/nature12875. advance on. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong L, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci U S A. 2008;105:7827–32. doi: 10.1073/pnas.0802866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong L, et al. Tyrosine-phosphorylation of AAV2 vectors and its consequences on viral intracellular trafficking and transgene expression. Virology. 2008;381:194–202. doi: 10.1016/j.virol.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martino AT, et al. Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. Blood. 2013;121:2224–33. doi: 10.1182/blood-2012-10-460733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manno CS, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–7. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 25.Boutin S, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21:704–12. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 26.Moskalenko M, et al. Epitope Mapping of Human Anti-Adeno-Associated Virus Type 2 Neutralizing Antibodies: Implications for Gene Therapy and Virus Structure. J Virol. 2000;74:1761–1766. doi: 10.1128/jvi.74.4.1761-1766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wobus CE, et al. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J Virol. 2000;74:9281–93. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lochrie MA, et al. Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J Virol. 2006;80:821–34. doi: 10.1128/JVI.80.2.821-834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mingozzi F, et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci Transl Med. 2013;5:194ra92. doi: 10.1126/scitranslmed.3005795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Münch RC, et al. Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol Ther. 2013;21:109–18. doi: 10.1038/mt.2012.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen S, et al. Engraftment of a galactose receptor footprint onto adeno-associated viral capsids improves transduction efficiency. J Biol Chem. 2013;288:28814–23. doi: 10.1074/jbc.M113.482380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol. 2006;24:198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- 33.Bartel Ma, Weinstein JR, Schaffer DV. Directed evolution of novel adeno-associated viruses for therapeutic gene delivery. Gene Ther. 2012;19:694–700. doi: 10.1038/gt.2012.20. [DOI] [PubMed] [Google Scholar]
- 34.Perabo L, et al. Combinatorial engineering of a gene therapy vector: directed evolution of adeno-associated virus. J Gene Med. 2006;8:155–62. doi: 10.1002/jgm.849. [DOI] [PubMed] [Google Scholar]
- 35.Bartel MA, et al. Directed Evolution of AAV for Enhanced Evasion of Human Neutralizing Antibodies. Am Soc Gene Cell Ther 15th Annu Meet. 2012;20:S140. [Google Scholar]
- 36.Excoffon KJD, et al. Directed evolution of adeno-associated virus to an infectious respiratory virus. Proc Natl Acad Sci U S A. 2009;106:3865–70. doi: 10.1073/pnas.0813365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jang JH, et al. An evolved adeno-associated viral variant enhances gene delivery and gene targeting in neural stem cells. Mol Ther. 2011;19:667–75. doi: 10.1038/mt.2010.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L, et al. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proc Natl Acad Sci U S A. 2009;106:3946–51. doi: 10.1073/pnas.0813207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gray SJ, et al. Directed evolution of a novel adeno-associated virus (AAV) vector that crosses the seizure-compromised blood-brain barrier (BBB) Mol Ther. 2010;18:570–8. doi: 10.1038/mt.2009.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klimczak RR, Koerber JT, Dalkara D, Flannery JG, Schaffer DV. A novel adeno-associated viral variant for efficient and selective intravitreal transduction of rat Müller cells. PLoS One. 2009;4:e7467. doi: 10.1371/journal.pone.0007467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dalkara D, et al. AAV Mediated GDNF Secretion From Retinal Glia Slows Down Retinal Degeneration in a Rat Model of Retinitis Pigmentosa. Mol Ther. 2011;19:1602–1608. doi: 10.1038/mt.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Russell DW, Hirata RK. Human gene targeting by viral vectors. Nat Genet. 1998;18:325–30. doi: 10.1038/ng0498-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–21. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McBride JL, et al. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol Ther. 2011;19:2152–62. doi: 10.1038/mt.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mingozzi F, et al. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat Med. 2007;13:419–22. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- 47.Madsen D, Cantwell ER, O’Brien T, Johnson PA, Mahon BP. Adeno-associated virus serotype 2 induces cell-mediated immune responses directed against multiple epitopes of the capsid protein VP1. J Gen Virol. 2009;90:2622–33. doi: 10.1099/vir.0.014175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu TL, et al. CD8+ T cell recognition of epitopes within the capsid of adeno-associated virus 8-based gene transfer vectors depends on vectors’ genome. Mol Ther. 2014;22:42–51. doi: 10.1038/mt.2013.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dalkara D, et al. Inner limiting membrane barriers to AAV-mediated retinal transduction from the vitreous. Mol Ther. 2009;17:2096–102. doi: 10.1038/mt.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Summerford C, Samulski RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. 1998;72:1438–45. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther. 2010;18:80–6. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghosh A, Duan D. Expanding adeno-associated viral vector capacity: a tale of two vectors. Biotechnol Genet Eng Rev. 2007;24:165–77. doi: 10.1080/02648725.2007.10648098. [DOI] [PubMed] [Google Scholar]
- 53.Bowles D, Rabinowitz J, Samulski R. Marker rescue of adeno-associated virus (AAV) capsid mutants: a novel approach for chimeric AAV production. J Virol. 2003;77:423–432. doi: 10.1128/JVI.77.1.423-432.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grimm D, et al. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol. 2008;82:5887–911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koerber JT, Jang JH, Schaffer DV. DNA shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol Ther. 2008;16:1703–9. doi: 10.1038/mt.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li W, et al. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol Ther. 2008;16:1252–60. doi: 10.1038/mt.2008.100. [DOI] [PubMed] [Google Scholar]
- 57.Müller OJ, et al. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat Biotechnol. 2003;21:1040–6. doi: 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- 58.Perabo L, et al. In vitro selection of viral vectors with modified tropism: the adeno-associated virus display. Mol Ther. 2003;8:151–157. doi: 10.1016/s1525-0016(03)00123-0. [DOI] [PubMed] [Google Scholar]
- 59.Koerber JT, Schaffer DV. Transposon-Based Mutagenesis Generates Diverse Adeno-Associated Viral Libraries with Novel Gene Delivery Properties. Methods Mol Biol. 2008;434:161–170. doi: 10.1007/978-1-60327-248-3_10. [DOI] [PubMed] [Google Scholar]
- 60.Koerber JT, Jang JH, Yu JH, Kane RS, Schaffer DV. Engineering adeno-associated virus for one-step purification via immobilized metal affinity chromatography. Hum Gene Ther. 2007;18:367–78. doi: 10.1089/hum.2006.139. [DOI] [PubMed] [Google Scholar]
- 61.Koerber JT, et al. Molecular evolution of adeno-associated virus for enhanced glial gene delivery. Mol Ther. 2009;17:2088–95. doi: 10.1038/mt.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao G, et al. Biology of AAV serotype vectors in liver-directed gene transfer to nonhuman primates. Mol Ther. 2006;13:77–87. doi: 10.1016/j.ymthe.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 63.Bell P, et al. Evaluation of adeno-associated viral vectors for liver-directed gene transfer in dogs. Hum Gene Ther. 2011;22:985–97. doi: 10.1089/hum.2010.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Waterkamp DA, Muller OJ, Ying Y, Trepel M, Kleinschmidt JA. Isolation of targeted AAV2 vectors from novel virus display libraries. J Gene Med. 2006;8:1307–1319. doi: 10.1002/jgm.967. [DOI] [PubMed] [Google Scholar]
- 65.Michelfelder S, et al. Vectors selected from adeno-associated viral display peptide libraries for leukemia cell-targeted cytotoxic gene therapy. Exp Hematol. 2007;35:1766–76. doi: 10.1016/j.exphem.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 66.Knipe DM, Howley PM. Fields’ Virology. Lippincott Williams & Wilkins; Philadelphia, PA, USA: 2007. p. 3177. [Google Scholar]
- 67.Sonntag F, Schmidt K, Kleinschmidt Ja. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc Natl Acad Sci U S A. 2010;107:10220–5. doi: 10.1073/pnas.1001673107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sonntag F, et al. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J Virol. 2011;85:12686–97. doi: 10.1128/JVI.05359-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xie Q, et al. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc Natl Acad Sci U S A. 2002;99:10405–10. doi: 10.1073/pnas.162250899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Flotte TR. Gene therapy progress and prospects: recombinant adeno-associated virus (rAAV) vectors. Gene Ther. 2004;11:805–10. doi: 10.1038/sj.gt.3302233. [DOI] [PubMed] [Google Scholar]
- 71.Flotte TR, et al. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proc Natl Acad Sci U S A. 1993;90:10613–7. doi: 10.1073/pnas.90.22.10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schaffer DV, Koerber JT, Lim K. Molecular engineering of viral gene delivery vehicles. Annu Rev Biomed Eng. 2008;10:169–94. doi: 10.1146/annurev.bioeng.10.061807.160514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu Z, Asokan A, Samulski RJ. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther. 2006;14:316–27. doi: 10.1016/j.ymthe.2006.05.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.