

Abstract
Metastasis is the leading cause of cancer-related deaths, but it is unclear how cancer cells escape their primary sites in epithelia and disseminate to other sites in the body. One emerging possibility is that transformed epithelial cells could invade the underlying tissue by a process called cell extrusion, which epithelia use to remove cells without disrupting their barrier function. Typically, during normal cell turnover, live cells extrude apically from the epithelium into the lumen and later die by anoikis; however, several oncogenic mutations shift cell extrusion basally, towards the tissue that the epithelium encases. Tumour cells with high levels of survival and motility signals could use basal extrusion to escape from the tissue and migrate to other sites within the body.
A crucial primary step for cancer metastasis is invasion, but we know very little about the mechanisms that govern it. As metastasis is the main reason that patients succumb to cancer, understanding the mechanisms that initiate metastasis will be crucial for targeting aggressive tumours. Because it has been difficult to directly follow tumour cell invasion from the epithelia, where most human cancers arise, we do not yet have a clear picture of the mechanisms that drive this process. In considering how tumour cells invade, it is helpful to understand how normal epithelia function and behave. Epithelia form a selective and protective barrier for all of the tissues that they encase. The polarized epithelium contains an apical surface that faces the lumen (external environment) and a basal surface that faces the basement membrane. Epithelia are the first line of defence against pathogens and toxins and, therefore, the cells that constitute epithelia are exposed to potential damage. As a result, many epithelia constantly turn over by cell division and death. We found that to maintain homeostatic epithelial cell numbers, when epithelia become too crowded owing to cell division elsewhere in the layer, some cells extrude and later die1. By extruding, cells that are destined for death are seamlessly ejected from the monolayer by concerted contraction of the cells that surround them2. Typically, because these cells extrude apically, they detach from the matrix and its associated survival signals, and die by anoikis. However, because metastatic tumour cells can, in some cases, override anoikis by upregulating survival signalling3,4, we propose that extrusion could enable them to escape the epithelium. Normally, epithelia extrude cells apically into the lumen, which would function to remove any transformed cells, thereby essentially suppressing tumorigenesis. Intriguingly, we have found that oncogenic signalling can alter normal apical extrusion and cause cells to instead extrude basally under the epithelium. In this way, basal extrusion could enable transformed cells that are refractory to cell death to invade the underlying stroma. In this Opinion article, we discuss how misregulation of extrusion and normal epithelial survival mechanisms could enable tumours to initiate metastasis by subverting a process that normally triggers epithelial cell death.
Mechanisms of epithelial cell extrusion
Dying cells could pose a threat to the tight barrier that epithelia form, but they do not. Instead, epithelial cells that are destined to die are extruded by contraction of an actin and myosin ring in the surrounding cells, which squeeze cells out of the epithelium while closing the potential gap that could have formed from the exit of the cells (FIG. 1). All of the epithelia that have been observed, across animals from Drosophila melanogaster, zebrafish, mice and humans, extrude epithelial cells through what seems to be a highly conserved mechanism1,2,5–8. For a cell to extrude, it produces and secretes the bioactive lipid sphingosine-1-phosphate (S1P), which then binds to S1P receptor 2 (S1P2; also known as S1PR2; a G protein-coupled receptor) in the surrounding cells9 to contract an intercellular actomyosin band that squeezes the cell out of the epithelium2. Additionally, we and others have observed that the cell being extruded also contracts, thereby aiding in its removal10. Epithelia extrude either live cells during homeostasis or dying cells in response to apoptotic stimuli1,2,6,11. We have found that epithelia maintain cell number homeostasis by extruding live cells once cells at a given site become up to 1.6-fold to 1.8-fold more crowded1. Crowding-induced extrusion occurs through activation of the mechanosensitive ion channel, PIEZO1, which presumably triggers calcium currents12 to activate S1P-dependent extrusion of live cells, which later die by anoikis. Apoptotic stimuli, such as chemotherapies, toxins or pathogens that trigger cell death can also activate cell extrusion through the S1P–S1P2 pathway9, possibly in response to caspase activation11. Thus, both during natural cell turnover and following the induction of apoptosis, the S1P–S1P2 pathway activates cells to extrude in a manner that ensures no gaps form in the epithelia as cells are expelled.
Figure 1. The direction in which a cell extrudes has important consequences for its fate.

Extrusion removes either live or dying epithelial cells in response to crowding during homeostasis or apoptotic stimuli, respectively. Typically, live cells extrude apically from the epithelium into the lumen by contracting an intercellular actomyosin band basolaterally to squeeze the cell out (left). Apically extruded cells generally die in the lumen by the loss of survival signals from the matrix — a process called anoikis (top). Tumour cells with upregulated survival signalling could still be eliminated through the lumen by apical extrusion, which could function like a tumour-suppressor mechanism (top). Less frequently, cells extrude basally by apical contraction (right), back into the tissue that the epithelium encases. Several oncogenic mutations disrupt apical extrusion, thereby driving cells to extrude basally instead. Because basally extruded cells with upregulated survival signals can bypass anoikis, this may provide a novel mechanism to enable oncogenic cells with upregulated survival signalling to initiate invasion (bottom).
Apical versus basal extrusion
Normally, epithelia extrude cells apically into the lumen, but in some situations, cells can also extrude basally into the underlying tissue. The direction in which a cell extrudes depends on where the actomyosin band contracts in neighbouring cells13. To extrude a cell apically, contraction occurs towards the base of the cell, whereas to extrude it basally, contraction occurs at the apex (FIG. 1). The direction in which a cell extrudes can have important consequences for the fate of the cell, especially when live, transformed cells are extruded. Apical extrusion eliminates cells with upregulated survival signalling through the lumen (FIG. 1). For example, in the intestine, apical extrusion would remove putative tumour cells into the waste canal. Similarly, transient mosaic expression of oncogenic HRASV12 or v-src transforms cells and causes them to self-segregate away from the wild-type epithelium in a process that is similar to but different from extrusion, which essentially removes them14,15. In mammary or prostate glands, apical extrusion could lead to carcinoma in situ — a tumour type with good prognosis in which cells accumulate in the luminal space and are generally non-invasive16,17. However, basal extrusion preserves live cells within the organ (FIG. 1). During development, basal extrusion could enable cells to dedifferentiate from the epithelium and then differentiate into new cell types, as during neuroblast delamination in D. melanogaster18. For tumours, basal extrusion could enable transformed cells to invade the tissue that the epithelium encases to initiate metastasis. Intriguingly, oncogenic mutations can subvert the normal extrusion pathway, shifting the direction of extrusion from apical to basal, and this suggests a link between basal extrusion and invasiveness. A basally extruded cell could either divide and accumulate beneath the epithelium or invade, depending on its ability to cross the basement membrane by either degrading or invading the underlying matrix19. Many tumours express various matrix metallo proteinases, which suggests that matrix degradation may be intrinsic to transformation, enabling basally extruded transformed cells to transit through the basement membrane. However, in vivo studies have suggested that cancer cells can breach the basement membrane without degrading it, by extending invadopodia that squeeze through gaps in the matrix and push it apart20,21. Determining whether basally extruded cells can breach the basement membrane and how they do so will be important goals for future in vivo studies.
Apical extrusion seems to require at least two activities: S1P–S1P2 signalling and microtubule dynamics. Microtubules reorient to the basolateral interfaces of both the extruding and neighbouring cells to localize RHO guanine nucleotide exchange factor 1 (ARHGEF1; also known as p115RHOGEF) and thereby activate RHO-mediated actomyosin contraction under the extruding cell, driving it out apically13 (FIG. 2a). Disruption of microtubule dynamics shifts extrusion basally13. Although microtubules reorient in both the extruding cell and its neighbours, cell-autonomous knockdown of a crucial microtubule regulator, adenomatous polyposis coli (APC), suggests that the direction in which a cell extrudes requires dynamic microtubules only within the extruding cell22. Because the S1P–S1P2–RHO pathway controls only apical but not basal extrusion23, one possibility is that, in the extruding cell, microtubules target S1P to restrict contraction and membrane recycling basolaterally, where it is needed to drive apical extrusion (FIG. 2a). When any machinery that controls apical extrusion is aberrant, cell-autonomous contraction of cortical actin and myosin at existing apical junctions could enable a cell to extrude basally. Recent studies show that cell-autonomous apical contraction precedes the basolateral contraction in the neighbouring cells, and this suggests that loss of basolateral contraction would naturally lead to basal extrusion10. However, how complete apical contraction is controlled is still unknown, as are other signals and mechanisms that might collaborate to control apical extrusion.
Figure 2. Modes of diverting extrusion basally.

a | During wild-type apical extrusion, the cell that is destined for extrusion (dark beige), as well as its neighbouring cells (light beige), reorient their microtubules to the basolateral interface. Reorientation of microtubules in the cell destined for extrusion is required for apical extrusion and presumably restricts the biologically active lipid sphingosine-1-phosphate (S1P) to the basolateral surface, where it binds to S1P receptor 2 (S1P2) expressed in the neighbouring cells to trigger apical extrusion. Microtubule reorientation in the neighbouring cells might reinforce RHO- mediated actomyosin contraction (arrows) at the basolateral surface. b | Mutations in the tumour suppressor adenomatous polyposis coli (APC) that disrupt microtubule dynamics can function cell-autonomously in the extruding cell to drive extrusion basally. Cell-autonomous apical contraction of cortical actin and myosin (arrows) at apical epithelial cell junctions can extrude the cell basally into the basement membrane. c | Oncogenic KRAS disrupts apical extrusion by downregulating both S1P and S1P2. Autophagy degrades S1P, and both S1P puncta and apical extrusion can be rescued by disrupting autophagy. Without apical extrusion signalling, junctional apical actin and myosin contraction (arrows) results in basal extrusion.
Diverting extrusion basally
Oncogenic signalling
Although there is no direct proof that basal extrusion drives tumour cell invasion, we have found that oncogenic mutations can manipulate apical extrusion, a process that normally promotes cell death, into a process that could allow cells to escape into the stroma. Mutations that disrupt the tumour-suppressive function of APC or constitutively activate KRAS disrupt normal apical extrusion by disrupting either cytoskeletal dynamics or S1P signalling, respectively (FIG. 2b,c). APC is mutated or lost in >80% of colorectal cancers and is downregulated in some breast and prostate cancers24–26. We found that cells preferentially extruded basally when a truncated form of APC associated with tumour formation was expressed in either cell lines or in zebrafish epidermis22. The APC truncation mutant lacks the carboxyl terminus that binds to both microtubules and F-actin27–29. Because apical extrusion relies on coupling micro tubules to cortical actin to control where contraction occurs, loss of this domain disrupts this targeting, thereby driving extrusion basally. APC functions cell-autonomously to drive apical extrusion, as expression of the microtubule-binding domain of APC in the extruding cell alone is sufficient to rescue apical extrusion in colorectal cancer cell lines expressing the truncation mutant. This suggests that single cells accumulating sporadic APC mutations could extrude basally. This new function for APC in controlling the direction of extrusion could collaborate with its known function in driving uncontrolled proliferation through upregulated WNT signalling30,31, thereby enabling APC-mutant cells to both invade and proliferate.
Oncogenic KRAS mutations are crucial drivers of aggressive tumours, such as those of the pancreas, lung and colon32–34. When we expressed the oncogenic KRASV12G mutation in MDCK (canine kidney epithelial cell) monolayers, they predominantly extruded basally in a cell-autonomous manner23. When grown in three-dimensional cysts surrounded by matrix, the basally extruded oncogenic KRAS-mutant cells proliferated into smaller cysts or migrated away as single cells. Similarly, constitutive mosaic expression of oncogenic HRAS in MCF-10A mammary epithelial cell cysts caused cells to either basally extrude or lead collective cell migration of the neighbouring wild-type cells within the cyst35. Although three-dimensional cultures are more representative of in vivo epithelia than monolayers, it is not clear how well the Matrigel that surrounds these cysts mimics the underlying matrix and stroma in real tissue. Therefore, the compelling behaviour of basally extruded cells from cysts will ultimately need to be assessed in vivo to determine whether basally extruded transformed cells can also escape beneath the epithelium in real tissue. We have found that unlike mutated APC, which disrupts microtubules, cells that express oncogenic KRAS degrade S1P and partially down-regulate S1P2, both of which are required for apical extrusion. S1P is degraded owing to high levels of autophagy, specifically in extruding KRAS-transformed cells. Disruption of autophagy (either genetically or chemically) rescued S1P accumulation and apical extrusion23. Because cells expressing oncogenic KRAS rely on autophagy for their increased survival36,37, current clinical trials are using chloroquine (an autophagy inhibitor) to target these cells. This treatment could also prevent tumour invasion by promoting apical extrusion38. Chloroquine treatment has already shown promising results in overcoming chemotherapy resistance in human HER2 (also known as ERBB2)-positive breast cancers that are also addicted to autophagy39. However, mouse models of genetically engineered pancreatic cancers that lack p53 have found that chloroquine treatment can actually exacerbate tumour growth40, suggesting some human tumours driven by KRAS mutants might also be chloroquine-resistant.
Alternative mechanisms to divert extrusion basally
Apart from APC and KRAS, other factors that are typically associated with poor cancer prognosis could also shift the direction in which cells extrude from epithelia. Proteins controlling epithelial cell polarity, such as those in the scribble homologue (SCRIB)–lethal giant larvae homologue (LGL)–discs large homologue (DLG) and partitioning defective 6 homologue (PAR6)–PAR3–atypical protein kinase C (aPKC) complexes, are mutated in numerous carcinomas41 and have been linked to their progression and metastasis42. Disruption of epithelial polarity could randomize the direction in which a cell extrudes, thereby increasing the incidence of basal extrusion (FIG. 3a). Another possibility is that activation of basal extrusion alone could occur without invoking other signals that activate apical extrusion (FIG. 3b). In several studies, basal extrusion seems to occur as a default pathway when apical extrusion fails10,23. Although it is not clear what controls basal extrusion, one candidate could be hyperactivation of RHO, which is associated with tumour cell migration43,44,20. It is plausible that simply activating RHO would cause cells to contract at the apical cell–cell junctions, where most actin and myosin II exists. In addition, comparatively weaker cell–cell or cell–matrix adhesions could cause some cells to preferentially detach (FIG. 3c). Studies of nanomechanical forces of single cells, both in vitro and from human breast cancer biopsies, revealed that metastatic breast tumour cells are less stiff than the rest of the tumour or surrounding normal tissue45, and this can substantially enhance their motility in vitro46. Similarly, reduced cell–cell adhesion by decreased expression of E-cadherin that is found in some metastatic tumours47 could make these cells more susceptible to extrusion than others. Although reduced tension of a single cell within an epithelium under expansion forces could make that cell spread, under intrinsic crowding forces in regions of extrusion1, it would instead be more likely to extrude. Loss of E-cadherin and reduced cell stiffness have been linked to metastatic tumours, but it is unclear whether these factors drive cell extrusion or are a consequence of losing contact with the epithelium following extrusion. Therefore, further studies will need to identify what makes a cell susceptible to extrusion and whether factors that promote extrusion do so in a preferentially apical or basal manner.
Figure 3. Alternative mechanisms to divert extrusion basally.

a | Improper localization or disruption of proteins controlling apical–basal epithelial cell polarity could increase basal extrusion by randomizing the direction in which a cell extrudes and disrupting apical extrusion. For example, E-cadherin and partitioning defective (PAR) protein complexes, which are crucial for localizing actin and myosin at apical contacts, could organize actin elsewhere if polarity is disrupted. b | Cell-autonomous apical contraction (arrows) alone could result in basal extrusion. Although it is not clear what activates contraction during basal extrusion, one candidate could simply be activation of RHO, which would cause actin and myosin — which are typically concentrated apically at adherens junctions — to contract at the top of the cell. c | Stiffness of tumour cells has been shown to have an important role during cell invasion. Under compressive forces, Cells that are less stiff (green), would be more primed to extrude than neighbouring, stiffer cells. Similarly, reduced E-cadherin levels, which are also associated with metastatic cells, would result in reduced attachment to surrounding cells, and these cells with lower E-cadherin levels might be more likely to become detached by extrusion. Although either of the above mechanisms could enable cells to extrude apically or basally, cells may be more likely to extrude basally, which seems to be the default direction in the absence of canonical extrusion signalling. S1P, sphingosine-1-phosphate; S1P2, S1P receptor 2.
Basal extrusion and tumour invasion
We propose that tumour cells could exploit epithelial extrusion as a mechanism to initiate invasion into the underlying stroma. Current models for how cells escape the primary tumour to initiate metastasis fit into two broad categories: collective cell and single cell invasion21,48,49. The main difference between these modes of invasion is whether or not tumour cells maintain intracellular contacts as they migrate through the stroma and matrix (BOX 1). Histological sections showing streams of cells and single cells emanating from the tumour lend support to both types of motility21. In considering the hypothesis that tumour cells can invade using basal extrusion, it is important to establish the fate of transformed cells after they have extruded basally. When we transform MDCK cells with oncogenic KRAS, cells that extrude basally from cysts show two different behaviours: they either migrate away singly (FIG. 4a) or they proliferate, thereby forming a smaller cyst that is attached to the parent cyst23 (FIG. 4b). Therefore, basal extrusion of transformed cells could enable cells to potentially migrate to other sites using either mode of invasion. It may be that some cells are primed to lose E-cadherin after extrusion to become more mesenchymal-like or stem cell-like (FIG. 4a). Basal extrusion seems to drive cells to dedifferentiate during development, as is the case during neuroblast delamination from the neuroepithelium in D. melanogaster or blood stem cell budding from the endothelia of many vertebrates50,18. Similarly, in cancer, tumour-initiating cells may bud from the epithelium by basal extrusion, thereby increasing their ability to proliferate and survive in foreign sites. Alternatively, basally extruded cells may retain their E-cadherin, as suggested by the ability of extruded cells expressing oncogenic KRAS to form new intact cysts. As seen in MCF-10A cysts, an initial basally extruded cell with an HRAS mutation could lead the collective cell migration of other attached cells35. Cells that do not lose E-cadherin expression after basal extrusion may still be less differentiated, having more intrinsic ability to divide, but restricted to an epithelial rather than a mesenchymal or pluripotent cell fate. Given the right cues in vivo, these cells could undergo collective cell migration rather than simply divide (FIG. 4b). Although epithelia that are cultured in three dimensions behave more like epithelia in vivo, they lack the complex components of real tissue. Thus, future work will need to determine whether basal extrusion could drive either single or collective cell migration in an in vivo model system.
Box 1. Current models of carcinoma invasion.
EMT: invasion of single tumour cells from a mass
Epithelial-to-mesenchymal transition (EMT) might provide a mechanism by which some single cells disseminate from an epithelial tumour mass47,67,68 (see the figure, part a). During EMT, tumour cells downregulate E-cadherin (thereby weakening cell–cell adhesions) and migrate away as single cells. The loss of E-cadherin and activation of matrix-degrading proteases changes cell behaviour to be more mesenchymal, which allows these cells to adapt and survive in different parts of the body, independently of normal epithelial survival signals. EMT could promote the dedifferentiation of cells into stem cells in development and cancer67,69. Mesenchymal cells could then transdifferentiate to an epithelial phenotype in different sites within the body to promote metastatic outgrowth. Several types of carcinomas with poor prognosis express various inducers of EMT, such as snail homologue 1 (SNAI1) and SLUG (also known as SNAI2), TWIST, and zinc finger E-box-binding homeobox 1 (ZEB1) and ZEB2, which downregulate E-cadherin, and this supports the idea that tumours use EMT to initiate invasion.
Collective tumour cell invasion from a mass
Tumour cells have been seen emanating in streams from a tumour cell mass in what is termed collective cell migration21,48,70 (see the figure, part b). Cells can migrate as a continuous mass that disseminates from the primary tumour or as smaller discontinuous cohorts of nearby cells. Unlike single cells that invade by EMT, these cells maintain cell–cell adhesions and are cohesive as they migrate. Migration of a cell front requires degradation of the matrix and the secretion of matrix-degrading proteases. Owing to histological evidence and the fact that most metastases retain E-cadherin, most metastatic carcinomas are thought to migrate collectively. In addition, it is thought that metastatic cells may alter their ability to migrate singly or collectively, depending on the matrix and the tissue that they encounter.
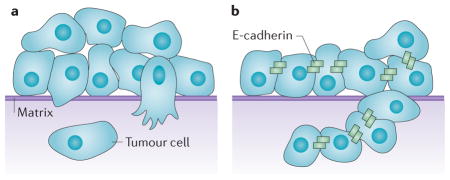
Figure 4. Model of single and collective cell-autonomous basal extrusion of tumour cells during tumour invasion.
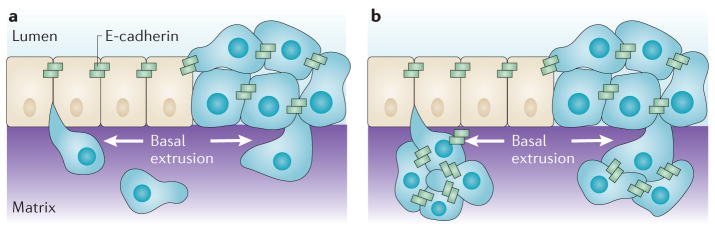
Transformed cells (blue) could invade by basal extrusion, either when surrounded by wild-type cells (beige) or from homogenously transformed neighbouring cells (blue). a | Basally extruded cells could downregulate E-cadherin once they lose contact with other epithelial cells and migrate away as single cells. b | Basally extruded cells could retain E-cadherin expression and proliferate and migrate together by collective cell migration.
An important point that distinguishes basal extrusion from the other models is that extrusion of cells can occur away from the main tumour mass (FIG. 4a,b). We have found that although oncogenic KRAS can drive cells to lose contact inhibition, the sites where cells basally extrude are not necessarily the same as the areas where masses form23. This could account for why pancreatic, colon and lung tumours that are driven by KRAS are typically metastatic with poor prognosis. The idea that tumours could metastasize independently of a primary tumour is alarming but not unheard of51,52. Indeed, molecular profiling of different tumour types suggests that some tumours develop with higher likelihoods of metastasizing than others53–56. Although transformed cells could invade at sites that are distinct from the primary mass using epithelial-to-mesenchymal transition (EMT) or collective cell migration, the invasive aspects in these models typically derive from general loss of epithelial organization and compressive forces that arise in surrounding tissue owing to growth of the primary tumour.
Concluding remarks
It is not clear why some tumours metastasize, whereas others do not. Our understanding of tumour invasion is mitigated by our ability to capture the natural formation and invasion of a tumour from an epithelium in vivo. Two-photon confocal intravital imaging of mammary mouse tumours has provided unprecedented resolution of migrating tumour cells in vivo57–62. One caveat to this approach, however, is the necessary introduction of a wound to provide a window for microscope access to the tumour, which could result in signalling and inflammation that is not present in naturally occurring tumours63. Future studies using zebrafish could provide an excellent animal model to directly visualize tumours invading directly from epithelia, as embryos are transparent and can be readily imaged without the invasive techniques that are intrinsic to current tumour models64. The zebrafish epidermis provides an excellent model for the epithelial bilayer that encases lungs and mammary glands. We are developing tools to label, knock down and express genes in single cells, where we can directly follow cells dividing, migrating and invading from the epithelium in situ (G.M.S., A. V. Gardner, G. Eisenhoffer and J.R., unpublished observations). Additionally, a new method to test the metastatic potential of human tumours has been developed in zebrafish embryos65,66. In this model, within only 48 hours after injecting cancer cells into the yolk sacs of 2-day-old zebrafish embryos, the metastatic potential was found to mimic mouse xenografts that were observed over the course of 2 months54, and this zebrafish model allows tumour cell dissemination to be filmed live.
Once a tumour cell has gained access to the circulation, the extravasation, survival and establishment of micrometastases in a distal organ are thought to be surprisingly efficient4. Therefore, identifying whether tumour cells use basal extrusion to initiate invasion into the underlying stroma before entering the circulation is of crucial importance to our understanding of metastasis. Understanding the molecular and genetic profiles of cells that can extrude basally and survive might help us to define the metastatic potential of some tumours. Furthermore, identifying whether basal extrusion might be enhanced with zinc finger E-box-binding homeobox 1 (ZEB1), snail homologue 1 (SNAI1) and SLUG (also known as SNAI2), which drive EMT, or with matrix metalloproteinases, which are typically upregulated with collective cell migration, will be important to determine the relationship of extrusion to these previously defined invasion modes. New imaging methods that allow us to follow metastases from their initial local invasion to their colonization in distant organs will also be important for determining how cells invade and migrate and whether basal extrusion is a crucial step in this process. A better understanding of how different tumours invade will be essential for preventing their spread.
Acknowledgments
The authors thank the US National Institutes of Health (NIH) for an Innovator Award DP2OD002056-01 and an RO1 1R01GM102169-01 to J.R., and for an NIH Developmental Biology Training Grant 5T32 HDO7491 to G.M.S.
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Eisenhoffer GT, et al. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature. 2012;484:546–549. doi: 10.1038/nature10999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenblatt J, Raff MC, Cramer LP. An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr Biol. 2001;11:1847–1857. doi: 10.1016/s0960-9822(01)00587-5. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiehart DP, Galbraith CG, Edwards KA, Rickoll WL, Montague RA. Multiple forces contribute to cell sheet morphogenesis for dorsal closure in Drosophila. J Cell Biol. 2000;149:471–490. doi: 10.1083/jcb.149.2.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marinari E, et al. Live-cell delamination counterbalances epithelial growth to limit tissue overcrowding. Nature. 2012;484:542–545. doi: 10.1038/nature10984. [DOI] [PubMed] [Google Scholar]
- 7.Madara JL. Maintenance of the macromolecular barrier at cell extrusion sites in intestinal epithelium: physiological rearrangement of tight junctions. J Membr Biol. 1990;116:177–184. doi: 10.1007/BF01868675. [DOI] [PubMed] [Google Scholar]
- 8.Guan Y, et al. Redistribution of the tight junction protein ZO-1 during physiological shedding of mouse intestinal epithelial cells. Am J Physiol Cell Physiol. 2011;300:C1404–C1414. doi: 10.1152/ajpcell.00270.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu Y, Forostyan T, Sabbadini R, Rosenblatt J. Epithelial cell extrusion requires the sphingosine-1-phosphate receptor 2 pathway. J Cell Biol. 2011;193:667–676. doi: 10.1083/jcb.201010075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuipers D, et al. Epithelial repair is a two-stage process driven first by dying cells and then by their neighbours. J Cell Sci. 2014;127:1229–1241. doi: 10.1242/jcs.138289. [DOI] [PubMed] [Google Scholar]
- 11.Andrade D, Rosenblatt J. Apoptotic regulation of epithelial cellular extrusion. Apoptosis. 2011;16:491–501. doi: 10.1007/s10495-011-0587-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coste B, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330:55–60. doi: 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slattum G, McGee KM, Rosenblatt J. P115 RhoGEF and microtubules decide the direction apoptotic cells extrude from an epithelium. J Cell Biol. 2009;186:693–702. doi: 10.1083/jcb.200903079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hogan C, et al. Characterization of the interface between normal and transformed epithelial cells. Nature Cell Biol. 2009;11:460–467. doi: 10.1038/ncb1853. [DOI] [PubMed] [Google Scholar]
- 15.Kajita M, et al. Interaction with surrounding normal epithelial cells influences signalling pathways and behaviour of Src-transformed cells. J Cell Sci. 2010;123:171–180. doi: 10.1242/jcs.057976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donker M, et al. Breast-conserving treatment with or without radiotherapy in ductal carcinoma in situ: 15-year recurrence rates and outcome after a recurrence, from the EORTC 10853 randomized Phase III trial. J Clin Oncol. 2013;31:4054–4059. doi: 10.1200/JCO.2013.49.5077. [DOI] [PubMed] [Google Scholar]
- 17.Bleyer A, Welch HG. Effect of three decades of screening mammography on breast-cancer incidence. N Engl J Med. 2012;367:1998–2005. doi: 10.1056/NEJMoa1206809. [DOI] [PubMed] [Google Scholar]
- 18.Hartenstein V, Younossi-Hartenstein A, Lekven A. Delamination and division in the Drosophila neurectoderm: spatiotemporal pattern, cytoskeletal dynamics, and common control by neurogenic and segment polarity genes. Dev Biol. 1994;165:480–499. doi: 10.1006/dbio.1994.1269. [DOI] [PubMed] [Google Scholar]
- 19.Kelley LC, Lohmer LL, Hagedorn EJ, Sherwood DR. Traversing the basement membrane in vivo: a diversity of strategies. J Cell Biol. 2014;204:291–302. doi: 10.1083/jcb.201311112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wyckoff JB, Pinner SE, Gschmeissner S, Condeelis JS, Sahai E. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr Biol. 2006;16:1515–1523. doi: 10.1016/j.cub.2006.05.065. [DOI] [PubMed] [Google Scholar]
- 21.Friedl P, Locker J, Sahai E, Segall JE. Classifying collective cancer cell invasion. Nature Cell Biol. 2012;14:777–783. doi: 10.1038/ncb2548. [DOI] [PubMed] [Google Scholar]
- 22.Marshall TW, Lloyd IE, Delalande JM, Nathke I, Rosenblatt J. The tumor suppressor adenomatous polyposis coli controls the direction in which a cell extrudes from an epithelium. Mol Biol Cell. 2011;22:3962–3970. doi: 10.1091/mbc.E11-05-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slattum G, Gu Y, Sabbadini R, Rosenblatt J. Autophagy in oncogenic K-Ras promotes basal extrusion of epithelial cells by degrading S1P. Curr Biol. 2014;24:19–28. doi: 10.1016/j.cub.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clevers H. Wnt breakers in colon cancer. Cancer Cell. 2004;5:5–6. doi: 10.1016/s1535-6108(03)00339-8. [DOI] [PubMed] [Google Scholar]
- 25.Oving IM, Clevers HC. Molecular causes of colon cancer. Eur J Clin Invest. 2002;32:448–457. doi: 10.1046/j.1365-2362.2002.01004.x. [DOI] [PubMed] [Google Scholar]
- 26.van Es JH, Giles RH, Clevers HC. The many faces of the tumor suppressor gene APC. Exp Cell Res. 2001;264:126–134. doi: 10.1006/excr.2000.5142. [DOI] [PubMed] [Google Scholar]
- 27.Kita K, Wittmann T, Nathke IS, Waterman-Storer CM. Adenomatous polyposis coli on microtubule plus ends in cell extensions can promote microtubule net growth with or without EB1. Mol Biol Cell. 2006;17:2331–2345. doi: 10.1091/mbc.E05-06-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mogensen MM, Tucker JB, Mackie JB, Prescott AR, Nathke IS. The adenomatous polyposis coli protein unambiguously localizes to microtubule plus ends and is involved in establishing parallel arrays of microtubule bundles in highly polarized epithelial cells. J Cell Biol. 2002;157:1041–1048. doi: 10.1083/jcb.200203001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zumbrunn J, Kinoshita K, Hyman AA, Nathke IS. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 β phosphorylation. Curr Biol. 2001;11:44–49. doi: 10.1016/s0960-9822(01)00002-1. [DOI] [PubMed] [Google Scholar]
- 30.Minde DP, Anvarian Z, Rudiger SG, Maurice MM. Messing up disorder: how do missense mutations in the tumor suppressor protein APC lead to cancer? Mol Cancer. 2011;10:101. doi: 10.1186/1476-4598-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schepers A, Clevers H. Wnt signaling, stem cells, and cancer of the gastrointestinal tract. Cold Spring Harb Perspect Biol. 2012;4:a007989. doi: 10.1101/cshperspect.a007989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neuzillet C, Hammel P, Tijeras-Raballand A, Couvelard A, Raymond E. Targeting the Ras-ERK pathway in pancreatic adenocarcinoma. Cancer Metastasis Rev. 2013;32:147–162. doi: 10.1007/s10555-012-9396-2. [DOI] [PubMed] [Google Scholar]
- 33.Aviel-Ronen S, Blackhall FH, Shepherd FA, Tsao MS. K-ras mutations in non-small-cell lung carcinoma: a review. Clin Lung Cancer. 2006;8:30–38. doi: 10.3816/CLC.2006.n.030. [DOI] [PubMed] [Google Scholar]
- 34.Jiang Y, Kimchi ET, Staveley-O’Carroll KF, Cheng H, Ajani JA. Assessment of K-ras mutation: a step toward personalized medicine for patients with colorectal cancer. Cancer. 2009;115:3609–3617. doi: 10.1002/cncr.24434. [DOI] [PubMed] [Google Scholar]
- 35.Liu JS, Farlow JT, Paulson AK, Labarge MA, Gartner ZJ. Programmed cell-to-cell variability in Ras activity triggers emergent behaviors during mammary epithelial morphogenesis. Cell Rep. 2012;2:1461–1470. doi: 10.1016/j.celrep.2012.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.White E. Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 2013;27:2065–2071. doi: 10.1101/gad.228122.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mathew R, White E. Autophagy, stress, and cancer metabolism: what doesn’t kill you makes you stronger. Cold Spring Harb Symp Quant Biol. 2011;76:389–396. doi: 10.1101/sqb.2012.76.011015. [DOI] [PubMed] [Google Scholar]
- 38.Mancias JD, Kimmelman AC. Targeting autophagy addiction in cancer. Oncotarget. 2011;2:1302–1306. doi: 10.18632/oncotarget.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cufi S, et al. The anti-malarial chloroquine overcomes primary resistance and restores sensitivity to trastuzumab in HER2-positive breast cancer. Sci Rep. 2013;3:2469. doi: 10.1038/srep02469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenfeldt MT, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 41.Royer C, Lu X. Epithelial cell polarity: a major gatekeeper against cancer? Cell Death Differ. 2011;18:1470–1477. doi: 10.1038/cdd.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macara IG, McCaffrey L. Cell polarity in morphogenesis and metastasis. Phil Trans R Soc B. 2013;368:20130012. doi: 10.1098/rstb.2013.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gildea JJ, et al. RhoGDI2 is an invasion and metastasis suppressor gene in human cancer. Cancer Res. 2002;62:6418–6423. [PubMed] [Google Scholar]
- 44.Struckhoff AP, Rana MK, Worthylake RA. RhoA can lead the way in tumor cell invasion and metastasis. Front Biosci. 2011;16:1915–1926. doi: 10.2741/3830. [DOI] [PubMed] [Google Scholar]
- 45.Plodinec M, et al. The nanomechanical signature of breast cancer. Nature Nanotechnol. 2012;7:757–765. doi: 10.1038/nnano.2012.167. [DOI] [PubMed] [Google Scholar]
- 46.Lee MH, et al. Mismatch in mechanical and adhesive properties induces pulsating cancer cell migration in epithelial monolayer. Biophys J. 2012;102:2731–2741. doi: 10.1016/j.bpj.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 48.Sahai E. Mechanisms of cancer cell invasion. Curr Opin Genet Dev. 2005;15:87–96. doi: 10.1016/j.gde.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 49.Yilmaz M, Christofori G, Lehembre F. Distinct mechanisms of tumor invasion and metastasis. Trends Mol Med. 2007;13:535–541. doi: 10.1016/j.molmed.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 50.Kissa K, Herbomel P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature. 2010;464:112–115. doi: 10.1038/nature08761. [DOI] [PubMed] [Google Scholar]
- 51.Rhim AD, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chambers KF, et al. Stroma regulates increased epithelial lateral cell adhesion in 3D culture: a role for actin/cadherin dynamics. PLoS ONE. 2011;6:e18796. doi: 10.1371/journal.pone.0018796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sorlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perez-Enciso M, Tenenhaus M. Prediction of clinical outcome with microarray data: a partial least squares discriminant analysis (PLS-DA) approach. Hum Genet. 2003;112:581–592. doi: 10.1007/s00439-003-0921-9. [DOI] [PubMed] [Google Scholar]
- 55.Livasy CA, et al. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod Pathol. 2006;19:264–271. doi: 10.1038/modpathol.3800528. [DOI] [PubMed] [Google Scholar]
- 56.Prat A, Ellis MJ, Perou CM. Practical implications of gene-expression-based assays for breast oncologists. Nature Rev Clin Oncol. 2012;9:48–57. doi: 10.1038/nrclinonc.2011.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zomer A, et al. Intravital imaging of cancer stem cell plasticity in mammary tumors. Stem Cells. 2013;31:602–606. doi: 10.1002/stem.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang W, et al. Coordinated regulation of pathways for enhanced cell motility and chemotaxis is conserved in rat and mouse mammary tumors. Cancer Res. 2007;67:3505–3511. doi: 10.1158/0008-5472.CAN-06-3714. [DOI] [PubMed] [Google Scholar]
- 59.Nakasone ES, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Condeelis J, Weissleder R. In vivo imaging in cancer. Cold Spring Harb Perspect Biol. 2010;2:a003848. doi: 10.1101/cshperspect.a003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roussos ET, et al. Mena deficiency delays tumor progression and decreases metastasis in polyoma middle-T transgenic mouse mammary tumors. Breast Cancer Res. 2010;12:R101. doi: 10.1186/bcr2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boimel PJ, et al. Contribution of CXCL12 secretion to invasion of breast cancer cells. Breast Cancer Res. 2012;14:R23. doi: 10.1186/bcr3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elinav E, et al. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13:759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- 64.White R, Rose K, Zon L. Zebrafish cancer: the state of the art and the path forward. Nature Rev Cancer. 2013;13:624–636. doi: 10.1038/nrc3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jung DW, et al. A novel zebrafish human tumor xenograft model validated for anti-cancer drug screening. Mol BioSyst. 2012;8:1930–1939. doi: 10.1039/c2mb05501e. [DOI] [PubMed] [Google Scholar]
- 66.Konantz M, et al. Zebrafish xenografts as a tool for in vivo studies on human cancer. Ann NY Acad Sci. 2012;1266:124–137. doi: 10.1111/j.1749-6632.2012.06575.x. [DOI] [PubMed] [Google Scholar]
- 67.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 68.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nieto MA. The early steps of neural crest development. Mech Dev. 2001;105:27–35. doi: 10.1016/s0925-4773(01)00394-x. [DOI] [PubMed] [Google Scholar]
- 70.Wang W, et al. Tumor cells caught in the act of invading: their strategy for enhanced cell motility. Trends Cell Biol. 2005;15:138–145. doi: 10.1016/j.tcb.2005.01.003. [DOI] [PubMed] [Google Scholar]