

Abstract
This article briefly reviews some of the mechanisms involved in the pathogenesis of neurological diseases, i.e. damage mechanisms (DM), and their interactions and overlap with protection and reparatory processes (i.e. endogenous defence activities). A relationship between DM and endogenous defence activity (EDA) regarding therapy principles will also be described. Currently, it is difficult to find the correct therapeutic approach for brain protection and recovery, especially because we do not fully understand all of the endogenous neurobiological processes, the complete nature of the pathophysiological mechanisms and the links between these two categories. Moreover, we continue to use a simplistic and reductionist approach in this respect. Endogenous neurobiological processes, such as neurotrophicity, neuroprotection, neuroplasticity and neurogenesis, are central to protection and recovery and represent the background of EDA. The biological reality of the nervous system is far more complex. In fact, there is an endogenous holistic process of neuroprotection and neurorecovery that should be approached therapeutically in an integrated way. The current tendency to exclusively frame drug activity in terms of single mechanisms and single focus effect might distract from other paradigms with greater explanatory power and hinder the development of more effective treatment strategies. A change of concept is required in pharmacological brain protection and recovery. Prospective considerations include an integrated pharmacological approach, focusing on drugs with multimodal activity and pleiotropic neuroprotective effect which are biological drugs, rather than single mechanism drugs, which usually are chemical drugs.
Keywords: damage mechanism, endogenous defence activity, neuroprotection, neurorecovery, multimodal drugs
Introduction
There are several limitations to the current way of thinking about brain protection and recovery.
The general reductionistic perception of the effects of brain lesions is that they are the linear sum of independent pathophysiological mechanisms, such as excitotoxicity, inflammation, apoptosis-like and oxidative stress that generate the pathways of pathological cascades. This vision has developed the subtractive suppressive strategy for neuroprotection therapies 1. The backbone of this therapeutic strategy is the presumption that if a certain pathophysiological mechanism is pharmacologically suppressed by a chemical drug, this will simply subtract that specific amount of damage from the total amount created by all pathophysiological mechanisms.
Then, it can be expected that remaining damage amount will be below the death threshold. Consequently, it can be assumed that the rest of the system remains unchanged. The expectation of discovering a key cell death pathway has affected the experimental design of neuroprotection studies. However, what is clear from clinical trials in neuroprotection is that the suppression subtractive strategy does not work.
The historical concept that neuroprotection means the suppression of pathophysiological processes (i.e. the idea that a single mechanism molecule might be effective in clinical practise) is obsolete today and represents the root cause of failure of clinical neuroprotection.
This failure is a measurement of the failure of the reductionist approach to the problem 2.
In fact, as explained below, when one acute pathophysiological process (e.g. excitotoxicity or inflammation) is pharmacologically suppressed, the long-term endogenous drivers of recovery (e.g. plasticity and trophicity) are disturbed.
The evaluations of the therapeutic effects of the suppressive single mechanism drugs in large randomized control trials (RCTs) of neuroprotection in stroke, traumatic brain injury (TBI) or degenerative disorders use endpoints of 90 or 180 days 3,4.
In most cases, we expect positive clinical results in neurorecovery, which is a long-term outcome, sustained by long-term processes (e.g. neuroplasticity) using molecules that often negatively interfere with plasticity.
Because endogenous post-lesional brain patterns are based on the early switch from neuroprotection to neuroplasticity, the inconsistency lays in the evaluation of therapeutic effect of drugs with short-term effect using long-term outcome.
It is not appropriate to approach neuroprotection and neurorecovery separately. This dichotomy might mislead the basic and clinical research in the field. In fact, in clinical practise, a complex recovery outcome is evaluated that is generated by all of the protection and recovery processes of EDA in an inseparable way.
Randomized control trials must be internally valid (i.e. the design and conduct must minimize the possibility of bias), but to be clinically useful, the result must also be relevant to a definable group of patients in a particular clinical setting. This relevance is generally termed external validity, applicability or generalizability 5,6. There is compelling evidence that RCTs often lack external validity. However, the assessment of external validity is complex and requires clinical rather than statistical expertise 7.
The proper design of RCTs is difficult, particularly in neurorehabilitation, because of several significant limitations: RCTs results are not applicable to small heterogeneous samples, which is similar to most neurorehabilitation studies; difficulties in subgroup analyses; not all relevant information is used in meta-analyses (e.g. language); individual (e.g. genetic) dispositions for treatment (i.e. responsivity) are often not addressed; difficult to approach ‘complex situations’, which are frequent in neurorehabilitation; and the high costs of properly running RCTs.
Not many RCTs or systematic reviews have analyzed the deleterious effects of different drugs used as concomitant treatments in patients during the recovery phase and in neurorehabilitation in various pathologies.
Understanding the neurobiology of neuroprotection, neurorecovery and related concepts
Cell death pathways
Two main types of cell death, passive and active, have been described. Necrosis is a process caused by almost any pathological insult, including physical, chemical and biological agents. The sequence of events leading to necrotic cell death is the same every time: osmolysis, which is caused by cellular oedema, leads to the passive death of the damaged cell. Necrosis not only affects the dying cell itself, but inflammation is also triggered by the release of the cell's contents as a secondary effect, which is accompanied by cytokine discharge.
The other mechanism of cell death is apoptosis, which, in contrast with necrosis, requires adenosine triphosphoric acid (ATP). Derived from an ancient Greek word, the term ‘apoptosis’ in modern terminology designates a form of cell death with specific morphological characteristics that is used by the organism to control the number and quality of cells to maintain functioning organs. The nervous system is one of the best examples where developmental cell death shapes its structure.
In addition to providing support to neurons throughout their lifetime, neurotrophic factors regulate apoptotic cell death and, therefore, form an important protective mechanism. Events similar to this form of cell death, including cellular signalling, have been observed in both neurodegenerative diseases and acute injury processes, such as trauma or stroke, in which neurons degenerate and die. There are clear differences between the two processes, the most evident being the time span involved. Apoptosis-like cell death takes longer, whereas necrosis is usually faster. However, dying cells occasionally display characteristics of both necrosis and apoptosis. In neurons, membrane rupture and DNA fragmentation, which indicate necrosis and apoptosis (respectively), can sometimes affect the same cell. Thus, it is clear that the current vocabulary of cell death is inadequate. The term ‘active cell death’ (ACD) has been suggested to designate cell death involving the activation of intracellular mechanisms regardless of cellular morphology. In contrast, ‘passive cell death’ (PCD) should replace the historical term necrosis 8. The relationship between some pathophysiological mechanisms and the types of cell death can be briefly summarized as follows: excitotoxicity can lead to both necrosis and apoptosis-like death. Inflammation can also result in necrosis and apoptosis-like death, whereas protein misfolding usually induces only apoptosis-like death.
The concept of a neurovascular unit was recently described. This unit consists of endothelial cells, pericytes, neurons, glial cells and matrix proteins that function together using biochemical signalling 9.
This unit exists everywhere in the brain, including both the grey and white matter. Neurovascular unit dysfunction can explain the occurrence and evolution of several brain conditions, such as stroke, vascular dementia, migraine, trauma, all neurodegenerative disorders and even normal ageing. Because the neurovascular unit is unique to the human body, the way cells die in these units is also unique.
The lack of support from any component of the vascular unit cells or matrix causes a particular apoptosis-like phenomenon known as anoikis 10.
Currently, the entire therapeutical approach in brain lesion and recovery has been developed around the neurovascular unit.
Damage mechanism and endogenous defence activity
Different pathophysiological mechanisms are triggered by various aetiological agents or biological events and produce a range of both acute and chronic neurological disorders.
There are a limited number of pathophysiological processes (see Fig.1) that have many similarities to various central nervous system (CNS) diseases. In stroke, excitotoxicity, oxidative stress, inflammation and apoptotic-like processes are predominant. In neurodegenerative disorders, protein misfolding plays an important role, in addition to the aforementioned processes. Excitotoxicity, inflammation and apoptosis-like processes represent the backbone common to most neuropathologies, and the modulation of these processes is the key to efficient neuroprotection in all neurological disorders.
Fig 1.
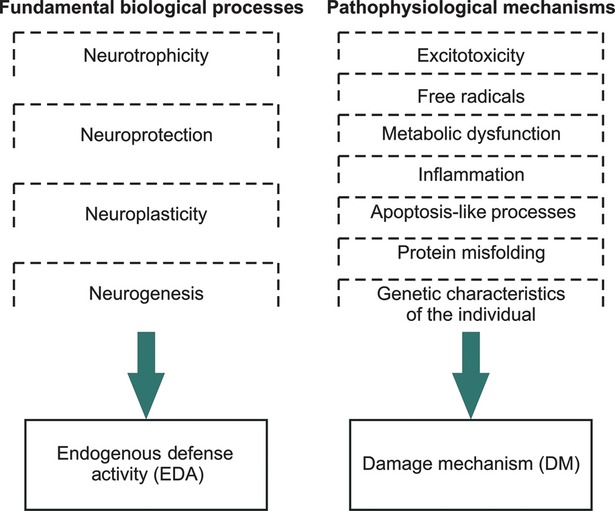
Endogenous defence activity and damage mechanism.
The classic perception of the effects of brain lesions is that DM is a linear sum of independent events (e.g. excitotoxicity, inflammation, apoptosis-like and oxidative stress) that generate the pathways of pathological cascades. This concept led to the development of the pharmacologic subtractive suppressive strategy for neuroprotection therapies. The rationale for this strategy is the presumption that if a certain pathophysiological process is pharmacologically suppressed, this will simply subtract that specific amount of damage, from the total amount created by all pathophysiological mechanisms. Then, it can be suppose that remaining damage amount will be below the death threshold. Current basic and clinical data do not confirm this assumption, and for this reason, the subtractive suppressive strategy for neuroprotection therapies has failed. This failure occurs because the interference between DM and EDA has not been properly considered but not because the key cell death pathway has not yet been discovered 11. Even from empirical molecular pathways models, we know that it is impossible to interfere with the DM without influencing EDA.
Therefore, the key issue is that the neurobiological processes of EDA share common biological background with the pathophysiological mechanisms of DM. Pathophysiological mechanisms (DM) have a dual character due to common biological background with EDA. For example, excitotoxicity (a pathophysiological mechanism) and neuroplasticity (a neuroreparatory process) share N-methyl-D-aspartate receptors (NMDAR) activity as their common important driver. Furthermore, inflammation is an important contributor to neuroregeneration because it stimulates neuroplasticity via trophic factors 12.
The historical concept that neuroprotection means the suppression of pathophysiological processes, the idea that a single mechanism molecule may be effective in clinical practise, is now obsolete and represents the root cause of failure of clinical neuroprotection. This indicates a failure of the reductionist approach to the problem. Therefore, when one acute pathophysiological process is pharmacologically suppressed (e.g. excitotoxicity or inflammation) by a chemical drug with a single mechanism of action, the long-term endogenous drivers of recovery (e.g. neuroplasticity and neurotrophicity) are disturbed (Fig.2).
Fig 2.
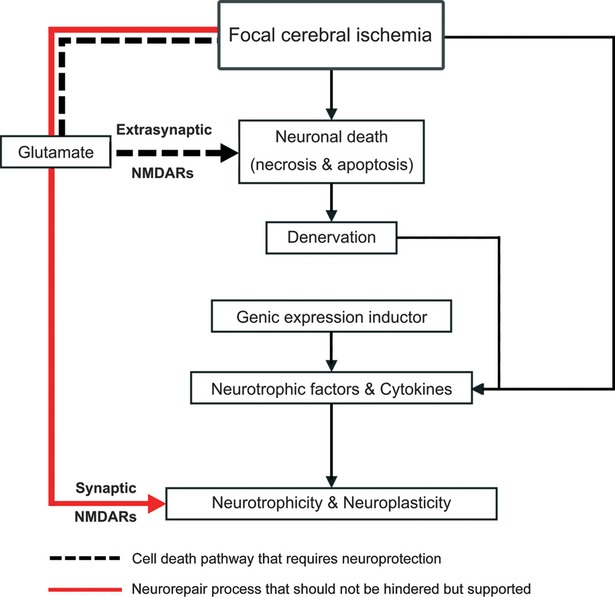
The dual roles performed by glutamate makes it a potent and indispensable factor for neurotrophicity and neuroplasticity processes.
Neurotrophicity, neuroprotection, neuroplasticity and neurogenesis are the most important neurobiological processes that act together under genetic control towards EDA, which attempts to counteract pathophysiological processes (i.e. DM) and stimulate endogenous recovery (Fig.1).
Definitions
Neurotrophicity denotes a natural biological process by which the continuous effort of the cell maintains correct DNA expression and a normal phenotype.
Neuroprotection represents the sum of all of the mechanisms directed against harmful factors and is a short-term endogenous neurobiological process.
Neuroplasticity is the permanent adaptation to new functional horizons and responsibilities. This concept describes the brain's ability to change extant structures in response to environmental stimuli, such as learning, new experiences or injury 13,14.
Neurogenesis is the process by which new nervous tissue cells, such as neurons, astrocytes and oligodendrocytes, are formed from stem cells. In a strict sense, neurogenesis is defined as the formation of new neurons.
The EDA of the nervous system is a continuous process that simultaneously performs and integrates the neurobiological processes of neurotrophicity, neuroprotection, neuroplasticity and neurogenesis.
When studying endogenous neuroprotection, we must distinguish between two different aspects: the so-called absolute and relative mechanisms. The absolute aspect refers to all of the mechanisms that determine the activation of DNA expression, followed by protein synthesis induction. The relative aspect refers to all of the mechanisms that ultimately determine neuroprotective activities with preponderant expression in the membrane, cytosol and organelles.
Pharmacological neuroprotection involves the same patterns. Pharmacologically, the absolute mechanisms are predominantly controlled by neurotrophic factors and neurotrophic-like molecules, but the relative mechanisms mainly utilize ion channel blockers, agonists and antagonists of specific receptors, antioxidants, chelators of various metals and many others agents 1.
Therefore, all of these biological mechanisms can be endogenously or pharmacologically activated. To successfully counteract pathophysiological mechanisms, and stimulate recovery, the effect of EDA must be pharmacologically enhanced.
Disturbances in the regulation of each of the four major players of EDA are themselves causes of some pathological conditions. For instance, a neurotrophicity deficit will always increase susceptibility to lesions. So far, no pathologies have been discovered that arise due to an excess of neurotrophicity or neuroprotection. For neuroplasticity, both up-regulation and down-regulation generate pathologies. Down-regulation generates a deficit of recovery, whereas up-regulation could generate hundreds of neuropathological patterns of pathological plasticity, usually involved in the pathogenesis of neuropathic pain, multiple sclerosis, movement disorders, tinnitus, impulse control disorders, obsessive-compulsive disorders and more. With regard to neurogenesis, both up-regulation and down-regulation generate pathological conditions, including down-regulation in Alzheimer's disease. Up-regulation of oligodendrogenesis and astrogenesis beyond normal regeneration is responsible for neuroproliferative disorders.
The dual character of DM
Excitotoxicity
Excitotoxicity is the pathological process by which nerve cells are damaged by excess glutamate and similar substances. NMDA receptor activation is one of the key features of excitotoxicity. The continuous activity of NMDARs is crucial for cell survival, and this is achieved through the regulation of neurotrophicity and neuroplasticity via calcium-controlled proteolytic systems (e.g. the calpain system) 15.
Physiological patterns of synaptic NMDAR activity actually promote neuronal survival by controlling the minimum calcium influx into the neuron. These small quantities of calcium activate ‘high affinity calcium molecules’ (e.g. μ calpain) and play the physiological role of conducting proteolytic activity, which is an important factor in neurotrophicity and neuroplasticity. This process is highly regulated by neurotrophic factors 16. Key pro-survival pathways involving NMDA receptors are essential for neurotrophicity and neuroplasticity 17.
The phosphoinositide 3-kinase (PI3K)–Akt cascade is strongly activated by NMDARs in many but not all neuronal types. Synaptic NMDARs signalling also activates the Ras–extracellular signal-regulated kinase (1/2 ERK) 1/2 cascade with pro-survival consequences, including cAMP-response element-binding protein (CREB) activation, BAD inactivation and antagonism of glycogen synthase kinase 3 (GSK3) β–induced apoptosis 18.
Furthermore, synaptic NMDAR-dependent calcium transients trigger a number of transcriptional changes that mediate long-lasting neuroplasticity via CRE-dependent gene expression 19.
When NMDARs are over-activated by glutamate in pathological conditions, such as stroke or trauma, large quantities of calcium enter the cells and activate ‘low affinity calcium molecules’, such as m-calpain. These low affinity calcium molecules have non-selective and uncontrolled proteolytic activities that lead to cell death. There are several fundamental mechanisms implicated in NMDAR-dependent cell death 20–22.
Cleavage of the plasma membrane Na+/Ca2+ exchanger by the Ca2+-dependent protease, calpain, leads to necrosis. Mitochondrial dysfunction caused by excessive Ca2+ uptake through the uniporter also leads to apoptosis-like processes. Finally, the overactivation of the Ca2+-dependent neuronal nitric oxide synthase (nNOS) by NMDAR activity has toxic downstream effects: p38 mitogen-activated protein kinase (MAPK) signalling, mitochondrial dysfunction and transient receptor potential melastatin channel (TRPM) activation, leading to apoptosis-like processes.
Next, we examine NMDAR involvement in pro-survival (e.g. neurotrophicity and neuroplasticity) and pro-death signalling (excitotoxicity) (Fig.3).
Fig 3.
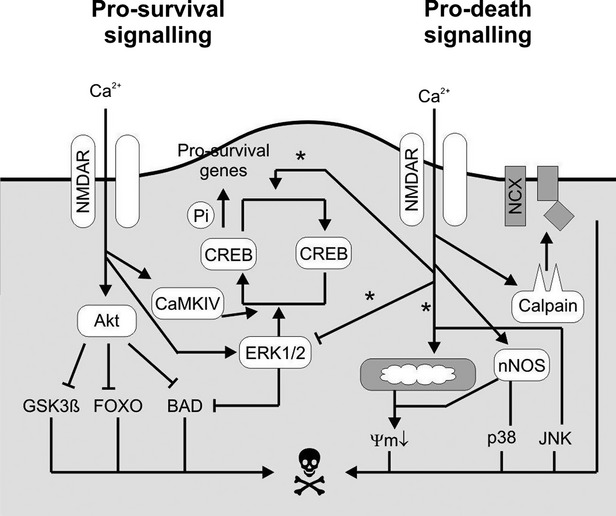
NMDAR pro-survival and pro-death signalling.
The first factor that influences NMDAR receptor activation is the magnitude of stimulation (e.g. intensity or duration); low levels of activation are protective. Ca2+ effectors of survival have considerably lower requirements for Ca2+ than death effectors. Therefore, the [Ca2+] threshold for the activation of pro-survival signalling by PI3K, ERK1/2 and CaMKIV–CREB must be lower than the [Ca2+] that triggers toxic levels of calpain activation, mitochondrial uptake or nitric oxide production. Therefore, the pro-survival effectors affinity for Ca2+ is high. Certain potential death effectors, such as m-calpain and the mitochondrial uniporter, have higher thresholds for Ca2+ and intrinsically low Ca2+ affinity 23.
The second important factor is NMDA receptor location. Extrasynaptic NMDAR activity promotes the inactivation of CREB by dephosphorylation and early excitotoxic events (e.g. mitochondrial depolarization), concomitant with the inactivation of the ERK1/2 pathway, which causes necrosis and induces apoptosis-like processes. However, synaptic NMDAR activity promotes the activation of CREB and the ERK1/2 pathway. Synaptic NMDAR activity does not disturb mitochondrial function, and it offers overall neuroprotective activity and promotes neurotrophicity and neuroplasticity.
Inflammation
The pathological role of inflammation has been recognized in almost all neurological conditions. This well-orchestrated process, situated on the borderline between physiology and pathology, tends to become highly destructive when prolonged or deregulated. However, inflammatory cells and mediators may also have beneficial functions and contribute to tissue repair processes.
There is evidence demonstrating that inflammation plays a positive role in neuroprotection and neuroplasticity 24,25.
Neurotrophic factors are the major players in this process. The neurotrophic factors produced by activated immune cells participate in neuronal protection and neuroplasticity 26.
Neurotrophic factors either bind directly to their receptors or act by modulating the local immune response. Even a very potent pro-inflammatory molecule such as TNF-α has neuroprotective and neurotrophic effects via trophic factors when activating R2 27,28.
The very low permeability of the blood–brain barrier (BBB) extends to immune cells and molecules. Usually, resident cells in the CNS (particularly astrocytes and microglia) regulate immune reactivity within the CNS. Other alien immune entities enter the CNS only through highly regulated processes mediated by adhesion molecules, chemokines, cytokines and matrix metalloproteinases 29–31.
Apoptosis and apoptosis-like processes
Apoptosis is a positive process that maintains the number and quality of cells. If a cell has a DNA lesion, it activates the p53 gene. Then, the cell will halt in the G1 phase of the cell cycle by bcl-2 activation, repair its DNA and recommence division. Alternatively, altered DNA repair may cause the activation of ‘bax’, which leads to apoptotic death. If apoptosis is not effective, then malignant clone formation will occur. An apoptosis-like process is a pathological apoptosis. From morphological and a biochemical point of view there is no difference between normal apoptosis and pathological apoptosis. It is only the trigger which is different (physiological or pathological).
Conclusions
From the above highlights of the links between DM and EDA, we can draw the following conclusions:
NMDAR activity plays a positive role during physiological activation by generating neurotrophicity and neuroplasticity or a deleterious role by overactivation-induced excitotoxicity generating pathological cascades in different conditions (e.g. stroke, trauma and neurodegenerative disorders).
Inflammation generally has a negative impact, but it can positively influence neuroprotection and neuroplasticity via neurotrophic factors.
Apoptosis is a positive process, but apoptotic-like processes are always negative. Apoptotic-like processes must be endogenously and therapeutically controlled.
In this light, there is a compelling body of basic and clinical evidence indicating that even the pharmacological subtractive suppressive strategy was an important achievement that generated beneficial results for neuroprotection in animal models, such as reducing the volume of ischaemic lesions in experimental stroke, this strategy failed from the clinical neurorecovery perspective.
Pharmacological modulation in brain protection and recovery
It is becoming evident that pharmacological intervention should address modulation not suppression. The more pathophysiological processes are modulated, the better the chances are for therapeutic success in brain protection and recovery.
Therefore, drugs with pleiotropic neuroprotective mechanisms of action are the best candidates for acute neuroprotection.
The concept of a neuroprotective pleiotropic effect is related to DM and represents the capacity of a pharmacological agent to interfere in more than one pathophysiological process (Fig.1).
The best approach for clinical neuroprotection is modulating (not suppressive) pleiotropic drugs that down-regulate extrasynaptic NMDAR excitotoxicity and, at the same time, decrease the negative effects of inflammation, increase the positive effects of inflammation and prevent apoptotic-like processes. In this way, the valuable processes that support recovery (e.g. neuroplasticity and neurotrophicity) are not hindered.
Important biological molecules such as neurotrophic factors may have a concomitant supportive effect, beside the pleiotropic neuroprotective effect, on the EDA side by stimulating also neurotrophicity, neuroplasticity and neurogenesis. This capacity of a molecular agent is described as a multimodal effect (Figs1 and 4).
Fig 4.
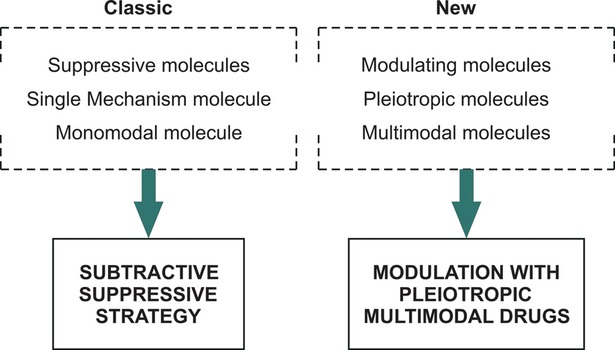
Classic versus new pharmacological strategies for neuroprotection and neurorecovery.
The capacity of a drug to pharmacologically influence, in the post-lesional brain only one neurobiological process of EDA (e.g. neuroprotection or neuroplasticity), is described as monomodal effect. The capacity to simultaneously regulate, in the post-lesional brain, two or more endogenous neurobiological processes of EDA is described as the multimodal effect, so similar with the real sequence of endogenous post-lesional regulation. The concept of multimodality is related to EDA whereas the concept of pleiotropic effect is related to DM.
There are suppressive chemical agents that cause pleiotropic effects, but these agents are not very helpful in clinical practise because they lack the multimodal effect. Biological agents (e.g. neurotrophic factors and related molecules) with modulating and pleiotropic neuroprotective effects are better pharmacological agents for brain protection and recovery because they usually have also multimodal effect. That is why they are capable of pharmacologically bridging acute neuroprotective processes with the long-term recovery processes. The pharmacological consequences of the evolution of ‘neuroprotection and neurorecovery’ concepts are depicted in Figure5.
Fig 5.
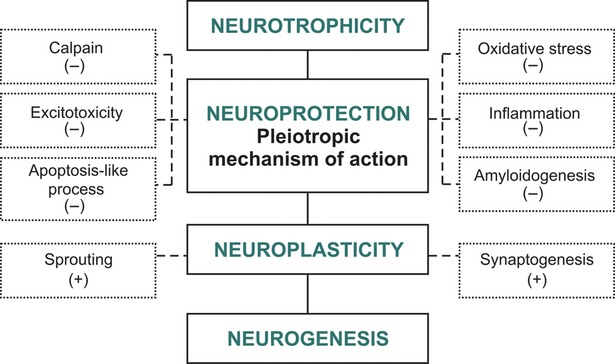
Multimodal drugs with pleiotropic neuroprotective effect—mechanism of action.
The concept and mechanism of action of multimodal drugs with pleiotropic neuroprotective effect (e.g. neurotrophic factors) are presented in Figure5.
We can better understand the mechanism of action of these drugs by following the example of how neuroprotection and neuroplasticity are endogenously integrated during an acute injury, such as stroke or trauma, where glutamate plays a dual role (Fig.2).
From this intuitive example, we learned that in the first hour after an acute injury, the large amount of glutamate (which acts predominantly on extrasynaptic NMDARs) is deleterious due to its final cell death effect. Beyond 48–72 hrs, glutamate becomes the key player in controlling neurorecovery via synaptic NMDARs that stimulate neuroplasticity and neurotrophicity.
Only a multimodal pharmacological agent modulates both protection and plasticity within the continuous EDA process.
All of these concepts and data are supported by the results of RCTs in neuroprotection and neurorecovery. In one of the most accurate meta-analyses, ‘Clinical Trials for Cytoprotection in Stroke’, Labiche and Grotta 3 underscored the fact that all of the trials that have studied the effect of monomodal suppressive single mechanism drugs have negative or neutral results. Only two drugs, Cerebrolysin and Citicoline, which are non-suppressive, multimodal drugs, with pleiotropic neuroprotective effect, demonstrated positive trends (Table1). Cerebrolysin is the only drug available for clinical use containing active fragments of some important neurotrophic factors. Larger RCTs are necessary to ultimately confirm these positive results. We have similar situations in acute traumatic brain injury clinical trials for neuroprotection. In a systematic review written by Maas et al. 4, the only drug that showed significant results was progesterone, which is also a non-suppressive, multimodal agent with pleiotropic neuroprotective effect 32.
Table 1.
Past and current cytoprotective clinical trials
| Drugs | Phase | Latest extent of time window | Adeq. power ∞ | Adeq dose | Dose-limiting AEs | Homogen patient population | Linked to TPA | Biologic imaging marker | Results |
|---|---|---|---|---|---|---|---|---|---|
| Calcium antagonists | |||||||||
| Nimodipine | 3 | 6–48 hrs | + | Hypotension | Neutral | ||||
| Nicardipine | 2 | 12 hrs | Hypotension | Neutral | |||||
| Glutamate antagonists | |||||||||
| Selfotel | 3 | 6–12 hrs | + | No | Neuropsych | Negative | |||
| Dextrorphan | 2 | 48 hrs | Yes | Neuropsych | Neutral | ||||
| Cerestat | 3 | 6–24 hrs | + | Yes | Hypertension | Negative | |||
| AR-R15696 | 2 | 12 hrs | Yes | Neuropsych | Neutral | ||||
| Magnesium | 3* | 2–12 hrs | + | Yes | No | + | + | ? | |
| AMPA antagonists | |||||||||
| YM872 | 2b | 3–6 hrs | + | ? | ? | + | + | + | Neutral |
| ZK200775 | 2 | 24 hrs | ? | Sedation | + | Negative | |||
| Indirect glutamate modulators | |||||||||
| Eliprodil | 3 | ? | ? | ? | ? | ? | ? | ? | Negative |
| Gavestinel | 3 | 6 hrs | +† | Yes | No | + | Neutral | ||
| Sipatrigine | 2 | 12 hrs | ? | Neuropsych | + | Negative | |||
| Fosphenytoin | 2/3 | 4 hrs | + | ? | No | Neutral | |||
| BMSS-204352 | 3 | 6 hrs | + | ? | No | + | + | Neutral | |
| Lifarizin | 2 | ? | ? | Hypotension | Neutral | ||||
| Lubeluzole | 3 | 4–8 hrs | +† | No | Cardiac | + | + | Neutral | |
| Other neurotrans modulators | |||||||||
| Trazadone | 2 | ? | ? | ? | ? | ? | ? | ? | Neutral |
| Repinotan | 3* | 6 hrs | +† | Yes | ? | + | ? | ||
| ONO-2506 | 2/3* | 6 hrs | + | ? | ? | + | ? | ||
| Opioid antagonists | |||||||||
| Naloxone | 2 | 8–60 hrs | ? | No | Neutral | ||||
| Nalmefene | 3 | 6 hrs | +† | ? | No | + | Neutral | ||
| GABA agonist | |||||||||
| Clonethiazole | 3 | 12 hrs | +† | Yes | Sedation | + | Neutral | ||
| Diazepam | 3* | 12 hrs | + | ? | ? | ? | |||
| Free radical scavengers | |||||||||
| Tirilazad | 3 | 6 hrs | + | ? | No | + | Negative | ||
| Ebselen | 3* | 48 hrs | + | ? | ? | + | ? | ||
| NXY-059 | 2b/3* | 6 hrs | +† | ? | ? | + | Negative | ||
| Anti-inflammatory agents | |||||||||
| Enlimomab | 3 | 6 hrs | + | Yes | Fever | + | Negative | ||
| LeukArrest | 3 | 12 hrs | ? | ? | ? | Neutral | |||
| FK-506 | 2* | 12 hrs | ? | ? | + | ? | |||
| Steroids | 2 | 48 hrs | ? | Infection | Negative | ||||
| Membrane stabilizers/trophic factor | |||||||||
| GM1 | 3 | 72 hrs | + | ? | No | Neutral | |||
| Cerebrolysin | 2 | 12–24 hrs | ? | No | Positive Trend | ||||
| Citicoline | 3 | 24 hrs | +† | ? | No | + | + | Positive Post hoc | |
| EPO | 2a* | ||||||||
| bFGF | 2/3 | 6 hrs | + | ? | Hypotension | + | Negative | ||
| Hypothermia | 2* | 5–24 hrs | Yes | Pneumonia, arrhythmias, hypotension | + | + | ? | ||
| Caffeinol oxygen delivery | 2* | 4–6 hrs | Yes | No | + | ? | |||
| DCLHb | 2 | 18 hrs | ? | HTN | Negative | ||||
| Nimodipine HBO | 2/3* | 24 hrs | ? | ? | Neutral | ||||
Only relevant to phase 2b or 3 efficacy trials.
Currently enrolling.
Not adequately powered for TPA subgroup.
+, positive; HTN, hypertension; AE, adverse effects; Neuropsych, Neuropsychiatric side effects.
Neurorecovery in acute and chronic neurological disorders
Endogenous defence activity of the nervous system is a continuous process that simultaneously performs and integrates the neurobiological processes of neurotrophicity, neuroprotection, neuroplasticity and neurogenesis. Neuroregeneration is the morphological outcome of the interactions between these basic neurobiological processes that develops in a particular biological and individual context. Neurorecovery is the positive outcome that produces clinically relevant results with immediate functional and late structural effects. Immediate and late effects generate two types of changes: restitution and substitution. Restitution is an intrinsic process involving biochemically and genetically-induced events, such as a reduction of oedema, the absorption of haeme and the restoration of axonal transport and ionic currents. Substitution depends on external stimuli, such as practise, which drives activity-dependent plasticity through learning 33.
Neurorecovery—biological background
All neurobiological processes can be endogenously or exogenously activated. The altered regulation of any of the four components of EDA may generate pathological conditions. Furthermore, altered neuroplasticity, in the form of both up- and down-regulation, generates pathologies 34.
Normally, these processes are regulated via key endogenous players, such as neurotrophic factors, neurotrophic-like molecules and other biological agents. To successfully compete with pathological processes and support neurorecovery, EDA should be exogenously enhanced by pharmacological intervention, physical activity, electromagnetic stimulation, psychological support, environmental stimulation or any demonstrated combinations of these factors capable of improving a patient's condition. From the pharmacological perspective, it is clear that the focusing on molecules that are capable of mimicking the structure and function of endogenous molecules with multimodal and pleiotropic neuroprotective is the best approach in neuroprotection and neuroplasticity. Both processes occur in a particular sequence in the continuous process of EDA. Indeed, the brain uses the same endogenous molecules for both neuroprotection and neuroplasticity, although in different combinations. These molecules are activated during altered gene expression induced by lesioning; brain ischaemia regulates the expression of more genes than any other condition 35,36.
However, many activated genes are not translated into proteins after injury. Endogenous neuroprotection is maximally effective approximately 72 hrs following an insult. Further positive clinical outcome is driven by the processes of neuroplasticity and neurogenesis. A brief overview of post-lesional regulation is presented in Table2.
Table 2.
Post-lesional regulation (J Neuropathol Exp Neurol, Vol 61, October, 2002)
| Early regulation ≤3 days post-lesion | Late regulation >3 days post-lesion | ||||
|---|---|---|---|---|---|
| Molecule | Ipsi-lesional | Contra-lesional | Ipsi-lesional | Contra-lesional | Involvement in experience—induced plasticity |
| Immediate early gene/transcription factor | |||||
| c-Fos | ↑ | – | ↑ | ↑ | Environmental enrichment |
| c-Jun | ↑ | – | ↑ | – | Learning |
| JunB | ↑ | – | – | – | Environmental enrichment |
| NGFI-A | ↑ | – | ↑ | – | Environmental enrichment |
| NGFI-B | ↑ | – | – | ↑ | Learning |
| NGFI-C | ↑ | – | – | – | Used-induced |
| Krox-20 | ↑ | – | – | – | – |
| Arc | ↑ | – | – | ↑ | Environmental enrichment |
| CREB (increased phosphorylation | ↑ | – | ↑ in c. callosum | ↑ | Used-induced |
| NF-kB | Controversial results | Learning | |||
| Kinase network molecules | |||||
| MAP kinase | ↑ | – | – | ↑ | Learning |
| CaM kinase | ↓ | – | ↑ | – | Physical exercise |
| Neurotransmitter receptors | |||||
| GluR1 | ↓ | – | ↓ | – | Environmental enrichment |
| GluR2 | ↓ | – | ↓ | – | – |
| GluR3 | ↓ | – | ↓ | – | – |
| NMDAR (receptor binding) | – | ↑ | ↑ | ↑ | Environmental enrichment |
| mGluR3 | ↓ | – | – | – | Learning |
| mGluR2 | ↓ | – | ↓ | ↓ | Learning |
| GABAR (receptor binding) | ↓ | – | ↓ | ↓ | Learning |
| Growth factors/receptors | |||||
| NGF | ↑ | – | ↑ | ↑ | Environmental enrichment |
| BDNF | ↑ | ↑ | – | – | Environmental enrichment |
| NT3 | ↓ | – | ↓ | – | Environmental enrichment |
| BFGF | ↑ | – | ↑ | ↑ | Learning/physical exercise |
| GDNF | ↑ | – | – | – | Environmental enrichment |
| PDGF | ↑ | – | ↑ | – | – |
| IGF | – | – | ↑ | ↑ | – |
| TGF-β1 | ↑ | – | ↑ | – | – |
| Trk B | ↑ | ↑ | – | – | Learning |
| Neuropilin -1, -2 | ↑ | – | ↑ | – | – |
| TNF-α | ↑ | ↑ | – | – | – |
| APP | – | – | ↑ | – | Learning |
| Growth—associated/cytoskeletal molecules | |||||
| GAP—43 | ↑ | – | ↑ | ↑ | Learning |
| SCG—10 | – | ↑ | – | ↑ | – |
| α—tubulin | – | – | ↑ | – | Learning |
| MAP—2 | ↑ | – | ↑ | – | Learning |
| apoE | ↓ | – | ↑ | – | Learning |
| apoD | ↑ | – | ↑ | ↑ | – |
| Synapse-related molecules | |||||
| Synaptophysin | – | – | ↑ | ↑ | Environmental enrichment |
| synapsin-I | ↑ | – | ↑ | – | Learning |
| SNAP-25 | – | – | ↑ | – | – |
| Adhesion molecules | |||||
| PSA-NCAM | ↑ | – | ↑ | – | Learning |
| L1 | ↓ | – | ↑ | – | Learning |
| F3 | ↓ | – | ↓ up to 1 week then ↑ | ↑ | – |
| Tenascin-C | ↑ | – | ↑ | – | – |
–, no report; ↑, up-regulation; ↓, down-regulation.
The identification of when particular changes occur (early or late) and the interpretation of their influence are often difficult. Early changes reflect neuroprotective efforts induced by cell damage and have little relevance to recovery potential. Late changes generally suggest recovery processes, but simultaneous overlapping events add complexity to the identification of individual changes. With regard to gene expression, patterns of gene expression changes and not simply individual genes must be considered. It is important to acknowledge the remarkable flexibility of endogenous programmes 36.
For simplicity, 72 hrs is considered as a distinguishing time point: the first 72 hrs after insult represents the early time window, and anything after 72 hrs represents the late time window.
Pharmacological agents with single mechanism of action are unlikely to demonstrate a clinical relevant neuroprotective effect. They can even hinder the spontaneous recovery process. A change of concept is required in pharmacological brain protection and recovery. In fact, in clinical practise, a complex recovery outcome is evaluated, that is generated by all of the protection and recovery processes of EDA in an inseparable way. Prospective considerations include an integrated pharmacological approach in neuroprotection and neurorecovery, focusing on drugs with multimodal activity and pleiotropic neuroprotective effect.
Conflicts of interest
The authors confirm that there are no conflicts of interest.
References
- Muresanu DF. Neuroprotection and neuroplasticity—a holistic approach and future perspectives. J Neurol Sci. 2007;257:38–43. doi: 10.1016/j.jns.2007.01.041. [DOI] [PubMed] [Google Scholar]
- Ahn AC, Tewari M, Poon C-S, et al. The limits of reductionism in medicine: could systems biology offer an alternative? PLoS Med. 2006;3:709–13. doi: 10.1371/journal.pmed.0030208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labiche LA, Grotta JC. Clinical trials for cytoprotection in stroke. NeuroRx. 2004;1:46–70. doi: 10.1602/neurorx.1.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas AI, Roozenbeek B, Manley GT. Clinical trials in traumatic brain injury: past experience and current developments. Neurotherapeutics. 2010;7:115–26. doi: 10.1016/j.nurt.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocock SJ. Clinical Trials—A Practical Approach. Chichester, New York, Brisbane, Toronto, Singapore: John Wiley & Sons; 1983. p. 265 S. [Google Scholar]
- Friedman M, Furberg CD, DeMets L. Fundamentals of Clinical Trials. 3rd ed. New York: Springer-Verlag; 1998. p. 361. [Google Scholar]
- Rothwell P. Treating Individuals—From Randomised Trials to Personalised Medicine. London: Elsevier Health Sciences; 2007. [Google Scholar]
- Sloviter R. Apoptosis: a guide for perplexed. Trends Pharmacol Sci. 2002;23:19–24. doi: 10.1016/s0165-6147(00)01867-8. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–85. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13:555–62. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- DeGracia DJ. Towards a dynamical network view of brain ischemia and reperfusion. Part I: background and preliminaries. J Exp Stroke Transl Med. 2010;3:59–71. doi: 10.6030/1939-067x-3.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseda T, Nishio T, Kawaguchi S, et al. Spontaneous regeneration of the corticospinal tract after transection inyoung rats: a key role of reactive astrocytes in making favourable andunfavourable conditions for regeneration. Neuroscience. 2004;126:365–74. doi: 10.1016/j.neuroscience.2004.03.056. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow ND. The neural basis of addiction: pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–13. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- Muresanu DF. Neurotrophic Factors. Bucuresti: Libripress; 2003. [Google Scholar]
- Wu HY, Yuen EY, Lu YF, et al. Regulation of N-Methyl-D-aspartate receptors by calpain in cortical neurons. J Biol Chem. 2005;280:21588–93. doi: 10.1074/jbc.M501603200. [DOI] [PubMed] [Google Scholar]
- Hutter-Paier B, Grygar E, Fruhwirth M, et al. Further evidence that Cerebrolysin protects cortical neurons from neurodegeneration in vitro. J Neural Transm Suppl. 1998;53:363–72. doi: 10.1007/978-3-7091-6467-9_32. [DOI] [PubMed] [Google Scholar]
- Hetman M, Kharebava G. Survival signaling pathways activated by NMDA receptors. Curr Top Med Chem. 2006;6:787–99. doi: 10.2174/156802606777057553. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H. A calcium microdomain near NMDA receptors: on switch for ERK-dependent synapse-tonucleus communication. Nat Neurosci. 2001;4:565–6. doi: 10.1038/88380. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–14. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004;61:657–68. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bano D, Young KW, Guerin CJ, et al. Calpain cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–85. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, et al. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–73. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Hardingham GE. Compartmentalized NMDA receptor signalling to survival and death. J Physiol. 2007;584-2:381–7. doi: 10.1113/jphysiol.2007.138875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseda T, Nishio T, Kawaguchi S, et al. Spontaneous regeneration of the corticospinal tract after transection in young rats: a key role of reactive astrocytes in making favourable and unfavourable conditions for regeneration. Neuroscience. 2004;126:365–74. doi: 10.1016/j.neuroscience.2004.03.056. [DOI] [PubMed] [Google Scholar]
- Kotter MR, Setzu A, Sim FJ, et al. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia. 2001;35:204–12. doi: 10.1002/glia.1085. [DOI] [PubMed] [Google Scholar]
- Masson JL, Jones JJ, Taniike M, et al. Mature oligodendrocytes apoptosis precedes IGF-I production: an oligodendrocyte progenitor accumulation and differentiation during demyelination/remyelination. J Neurosci Res. 2000;61:251–62. doi: 10.1002/1097-4547(20000801)61:3<251::AID-JNR3>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Arnett HA, Wang Y, Matsushima GK, et al. Functional genomic analysis of remyelination reveals importance of infl ammation in oligodendrocytes regeneration. J Neurosci. 2003;23:9824–32. doi: 10.1523/JNEUROSCI.23-30-09824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti L, Klein M, Schlett K, et al. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyld-aspartate receptor activation. J Biol Chem. 2004;279:32869–81. doi: 10.1074/jbc.M311766200. [DOI] [PubMed] [Google Scholar]
- Becher B, Antel JP. Comparison of phenotypic and functional properties of immediately ex vivo and cultured human adult microglia. Glia. 1996;18:1–10. doi: 10.1002/(SICI)1098-1136(199609)18:1<1::AID-GLIA1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Windhangen A, Newcombe J, Dan-gond F, et al. Expression of costimulatory molecules B7–1 (CD80), B7–2 (CD86), and interleukin 12 cytokine in multiple sclerosis lesions. J Exp Med. 1995;182:1985–96. doi: 10.1084/jem.182.6.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachter JS, de Vries HE, Fabry Z. The blood–brain barrier and its role in immune privilege in the central nervous system. J Neuropathol Exp Neurol. 2003;62:593–604. doi: 10.1093/jnen/62.6.593. [DOI] [PubMed] [Google Scholar]
- Xiao G, Wei J, Yan W, et al. Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: a randomized controlled trial. Crit Care. 2008;12:R61. doi: 10.1186/cc6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes MP, Dobkin BH, Bogousslavsky J. Recovery after Stroke. New York: Cambridge University Press; 2003. [Google Scholar]
- Read SJ, Parsons AA, Harrison DC, et al. Stroke genomics: approaches to identify, validate, and understand ischemic stroke gene expression. J Cereb Blood Flow Metab. 2001;21:755–78. doi: 10.1097/00004647-200107000-00001. [DOI] [PubMed] [Google Scholar]
- Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol. 2003;62:127–36. doi: 10.1093/jnen/62.2.127. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD. Injury mechanisms in the ischemic penumbra. Approaches to neuroprotection in acute ischemic stroke. Cerebrovasc Dis. 1997;7:7–12. [Google Scholar]