

Abstract
This study tested the reversal of subcellular remodelling in heart failure due to myocardial infarction (MI) upon treatment with losartan, an angiotensin II receptor antagonist. Twelve weeks after inducing MI, rats were treated with or without losartan (20 mg/kg; daily) for 8 weeks and assessed for cardiac function, cardiac remodelling, subcellular alterations and plasma catecholamines. Cardiac hypertrophy and lung congestion in 20 weeks MI-induced heart failure were associated with increases in plasma catecholamine levels. Haemodynamic examination revealed depressed cardiac function, whereas echocardiographic analysis showed impaired cardiac performance and marked increases in left ventricle wall thickness and chamber dilatation at 20 weeks of inducing MI. These changes in cardiac function, cardiac remodelling and plasma dopamine levels in heart failure were partially or fully reversed by losartan. Sarcoplasmic reticular (SR) Ca2+-pump activity and protein expression, protein and gene expression for phospholamban, as well as myofibrillar (MF) Ca2+-stimulated ATPase activity and α-myosin heavy chain mRNA levels were depressed, whereas β-myosin heavy chain expression was increased in failing hearts; these alterations were partially reversed by losartan. Although SR Ca2+-release activity and mRNA levels for SR Ca2+-pump were decreased in failing heart, these changes were not reversed upon losartan treatment; no changes in mRNA levels for SR Ca2+-release channels were observed in untreated or treated heart failure. These results suggest that the partial improvement of cardiac performance in heart failure due to MI by losartan treatment is associated with partial reversal of cardiac remodelling as well as partial recovery of SR and MF functions.
Keywords: cardiac dysfunction, subcellular remodelling, plasma catecholamines, renin-angiotensin blockade, cardiac gene expression
Introduction
It is now well known that cardiac dysfunction in heart failure due to myocardial infarction (MI) is associated with cardiac remodelling 1–4. Furthermore, varying degrees of defects in subcellular organelles such as sarcoplasmic reticular (SR) and myofibrils (MF) have been identified to account for impaired cardiac function in the failing heart 5–11. As both renin-angiotensin system (RAS) and sympathetic nervous system (SNS) are activated in heart failure 12–14, several agents, which produce blockade of RAS or SNS, are being used for the treatment of heart failure 15–19. In fact, different angiotensin II receptor (AT1R) antagonists and β-adrenoceptor (β-AR) blocking agents have been reported to prevent cardiac dysfunction, cardiac remodelling and subcellular alterations in heart failure due to MI 20–35. However, none of the previous studies have shown reverse cardiac remodelling, improvement in cardiac performance and attenuation of subcellular defects upon instituting the drug therapy at a well-established stage of heart failure.
By employing a rat model of heart failure due to MI, we have shown earlier that mild, moderate and advanced stages of heart failure become evident at 4, 8 and 16 weeks of inducing MI respectively 36,37. Treatment of 3 weeks MI animals for a period of 4–5 weeks with angiotensin-converting enzyme inhibitors or AT1R antagonists, including losartan, was observed to attenuate cardiac dysfunction as well as changes in SR Ca2+-pump, SR Ca2+-release and MF Ca2+-stimulated ATPase activities in the failing hearts 10,22,25,27,32. These beneficial effects of RAS blockade were demonstrated to occur at the level of cardiac gene expression because alterations in mRNA levels for SR and MF proteins were prevented by this therapy 10,22,25,26,32. The present study was undertaken to test if cardiac dysfunction, cardiac remodelling and subcellular alterations in failing hearts at moderate to advanced stages of heart failure were reversible upon starting treatment with losartan at 12 weeks after the induction of MI. Changes in mRNA levels for SR and MF proteins were examined to investigate if the beneficial effects of losartan are evident at the level of cardiac gene expression. In view of the elevated levels of plasma catecholamines due to activation of SNS in heart failure 13,14,34, alterations in plasma norepinephrine (NE), epinephrine (EPI) and dopamine were monitored upon treatment of 12 weeks MI animals with losartan.
Materials and methods
Experimental model
All experimental protocols employed in this study have been approved by the Animal Care Committee of the University of Manitoba and followed the ethical guidelines established by the Canadian Council on Animal Care. Heart failure due to MI was induced in Sprague-Dawley rats (175–200 g) by occlusion of the left anterior descending coronary artery as previously used in our laboratory 22,36–39. Briefly, rats were anaesthetized with 2.5% isoflurane gas mixed with oxygen (2 l/min.) along with intermittent positive pressure ventilation. The left anterior descending coronary artery was ligated approximately 2 mm from the origin of the aorta and the heart was then repositioned back into the chest. The sham rats were treated in the same manner, except that the coronary artery was not tied. The incidence of mortality of these MI rats was approximately 30–36% within the initial 48 hrs. The animals were assessed electrocardiographically 38 both before and after the surgery to determine the extent of coronary artery ligation, whereas echocardiography 38 was employed at 12 weeks after surgery before the commencement of drug treatment to determine that these 12 weeks MI animals were in heart failure. Both sham and 12 weeks MI animals were treated orally with or without losartan (20 mg/kg/day) for 8 weeks and used for investigations. It should be mentioned that the echocardiographic assessment of 12 weeks infarcted animals showed marked depressions in ejection fraction (EF), fractional shortening (FS) and cardiac output (CO) without any changes in heart rate (HR).
Echocardiographic assessment of cardiac performance and cardiac remodelling
Rats were anaesthetized with 2.5% isoflurane gas in 2 l/min oxygen and the echocardiographic readings using the SONOS 5500 ultrasonograph (Agilent Technologies Inc., Andover, MA, USA) were recorded with animals lying on their left side. The following parameters were measured by this method as described previously 38: interventricular septum diastole/systole thickness (IVSd, IVSs), left ventricular internal diameter diastole/systole (LVIDd, LVIDs), left ventricular posterior wall diastole/systole thickness (LVPWd, LVPWd), EF, FS, CO and HR.
Haemodynamic measurement and tissue preparation
For assessment of left ventricular function, animals were anaesthetized using an intraperitoneal injection containing a cocktail of ketamine:xylazine (90:10 mg/kg). By employing a micromanometer-tipped catheter (Millar SPR-249; Millar Instruments Inc, Houston, TX, USA) inserted into the left ventricle (LV), the following parameters were measured: systolic pressure (LVSP), diastolic pressure (LVEDP), mean arterial pressure (MAP), as well as rates of pressure development (+dP/dt) and pressure decay (−dP/dt) 38,39. Haemodynamic data were recorded using the AcqKnowledge program for Windows 3.03 (MP100, BIOPAC Systems, Inc., Goleta, CA, USA).
The hearts were quickly excised from the chest and the LV along with the septum, the right ventricle (RV) and the scar tissues were carefully dissected, washed, weighed and frozen in liquid nitrogen and stored at −80°C. Both lung and liver were washed, weighed and dried to give a wet/dry ratio which represented an index of pulmonary and liver congestion. In addition, the ratio of heart wt (both ventricles and infarct scar tissue) to total body wt provided an index of cardiac hypertrophy. The ratio of scar wt/total LV wt which indicated a fairly linear association with the size of the infarct was also determined 25,38.
RNA isolation and Northern blot analysis
Total RNA was isolated from the viable LV tissue including septum using TRIzol Reagent (Life Technologies Inc., Burlington, ON, Canada) according to the method described earlier 25,29. Total RNA was denatured, subjected to electrophoresis to size fractionate the mRNA transcripts and transferred to a positively charge-modified nylon filter (NYTRAN PLUS, Schleider and Schuell, Keene, NH, USA). The nylon membrane was then promptly UV cross-linked covalently and the blots were pre-hybridized to random primed cDNA or oligonucleotide probes. SERCA2a was probed with a 0.762 kb cDNA fragment from the rabbit heart Ca2+-pump ATPase (courtesy of Dr. A.K. Grover, McMaster University, Hamilton, Canada). PLB was probed with a 0.153 kb cDNA fragment from the rabbit heart (courtesy of Dr. D.H. MacLennan, University of Toronto, Toronto, Canada). CQS was probed with a 2.5 kb cDNA fragment from the rabbit heart (courtesy of Dr. A. Zilverman, University of Cincinnati, Cincinnati, OH, USA). For myocardial MF α-MHC, a 39-meroligonucleotide derived from the 3′-untranslated region of the rat α-MHC gene and for the myocardial MF β-MHC probe, a 30-meroligonucleotide was derived from the 3′-untranslated region of the gene (specifically rat genome). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a 1.2 kb cDNA fragment of the human GAPDH (American Type Culture Collection, Rockville, MD, USA), as well as a 24 base oligonucleotide probe of rat 18S ribosomal RNA, were used as an internal standard to account for the differences in nucleic acid loading and transfer of the RNA. This procedure of Northern blot analysis is the same as described previously 25–27,39.
Preparation of SR and measurements of Ca2+-uptake and Ca2+-release activities
Sarcoplasmic reticular vesicles from the cardiac muscle were isolated as previously described 22,39. The SR Ca2+-uptake activity was measured using a procedure described elsewhere 22,39. The determination of the Ca2+-release activity of the isolated SR vesicles was carried out by a modified procedure 22,39. The relative protein contents of the SR Ca2+-ATPase (SERCA2a), phospholamban (PLB) and calsequestrin (CQS) were measured using Western blot analysis 22,39. The blots were stained with Ponceau S solution to ensure uniform protein loading in all groups 22,39.
Isolation of MF and determination of ATPase activity
The isolation of the MF fraction was carried out as previously described 27,39. The Mg2+-ATPase activity and total ATPase activity were measured 27,39. The Ca2+-stimulated ATPase activity was determined as the difference between the values obtained for the total and Mg2+-ATPase activities.
Plasma catecholamine determination
The plasma catecholamine levels of NE, EPI and dopamine were measured using high-performance liquid chromatography and electrochemical detection as described previously 34.
Statistical analysis
All results were expressed as mean ± S.E. Differences between the sham, MI- and drug-treated animal groups were evaluated by the analysis of variance (anova) test, followed by the Newman-Keuls test. Statistical analysis was performed with Origin 6.0 (Microcal Software, Northampton. MA, USA), and a probability level of P < 0.05 was considered the threshold for statistical significance.
Results
General characteristics due to MI
Occlusion of the left anterior coronary artery for 20 weeks produced a large scar in the left ventricular chamber, whereas the surrounding cardiac muscle underwent hypertrophy as reflected by increased values for the heart wt compared to control (Table1). The heart wt, which consisted of the LV, RV and scar, in addition to the heart wt/body wt ratio were significantly increased due to the development of cardiac hypertrophy 20 weeks after MI. The cardiac hypertrophy parameters were significantly attenuated by losartan treatment (Table1). The mean scar wt varied from 700 to 800 mg with no major difference amongst the various groups of the MI rats, which equates to a mean scar size value of approximately 44%, as reflected by the scar wt/LV wt ratio. The cardiac architecture also revealed key changes in chamber dimensions, where the RV wt showed marked thickening (increase of 69%), which was greatly diminished with losartan to suggest a reduction in the degree of overall cardiac remodelling. Table1 indicates no significant changes in body wt, scar wt and scar wt/LV wt ratio between infarcted and drug-treated groups when compared with sham rats. Although heart wt of sham animals was significantly increased upon treatment with losartan, the heart wt/body wt ratio was not altered (Table1). The data in Table1 also show an alteration in the lung wet/dry wt ratio, with an increase of 25% of the sham value to suggest a considerable amount of pulmonary congestion and oedema in MI animals. Treatment with losartan demonstrated substantial recovery of the lung congestion during cardiac failure. However, no changes were observed for the liver wet/dry wt ratio due to MI in drug-treated or untreated groups (Table1).
Table 1.
General characteristics of sham and infarcted rats with and without losartan treatment for 8 weeks starting at 12 weeks after coronary artery occlusion
| Parameters | Sham | MI | Sham + LOS | MI + LOS |
|---|---|---|---|---|
| Body wt (g) | 680 ± 29 | 684 ± 30 | 709 ± 45 | 692 ± 16 |
| Scar wt (mg) | ND | 750 ± 130 | ND | 516 ± 87 |
| Scar wt/LV wt (%) | ND | 44 ± 6 | ND | 38 ± 6 |
| Heart wt (mg) | 1616 ± 64 | 2600 ± 86* | 2166 ± 37* | 2129 ± 64# |
| Heart wt/body wt (mg/g) | 2.85 ± 0.11 | 4.18 ± 0.19* | 3.11 ± 0.50 | 3.26 ± 0.07# |
| RV wt (mg) | 375 ± 20 | 635 ± 60* | 283 ± 31 | 371 ± 18# |
| Lung wet/dry wt ratio | 4.08 ± 0.18 | 5.13 ± 0.24* | 4.15 ± 0.10 | 3.56 ± 0.28# |
| Liver wet/dry wt ratio | 2.68 ± 0.07 | 2.90 ± 0.03 | 3.07 ± 0.16 | 2.82 ± 0.05 |
Values are mean ± S.E. of seven animals in each group. MI: myocardial infarction; LOS: Losartan (20 mg/kg/day); ND: not detected; wt: weight; LV: left ventricle; RV: right ventricle; *P < 0.05 compared with the 20 weeks sham group; #P < 0.05 compared with the 20 weeks MI group.
Cardiac performance and plasma catecholamines
The 20 weeks infarcted animals showed a depression in contractile function that was evident by a marked increase in LVEDP (4.4-fold) with an accompanying 48% decrease in +dP/dt, 58% decrease in −dP/dt and 43% decrease in LVSP (Table2A). The contractile function in the infarcted animals was improved with losartan treatment, as the following parameters illustrated noticeable changes: LVEDP elevation was lowered from 4.4-fold to 1.6-fold, +dP/dt increased from 52% to 76%, −dP/dt increased from 42% to 76% and LVSP increased from 57% to 81%. No significant alterations in HR or MAP were observed amongst the sham and MI groups with or without drug treatment (Table2A). Plasma NE and EPI were markedly higher in the infarcted rats as compared with the sham rats (1.9-fold increase and 1.7-fold increase respectively; Table2B). Treatment of MI animals with losartan showed a further increase in the circulating levels of NE (1.3-fold increase), without any change in EPI levels. Plasma dopamine levels were also markedly elevated in MI rats (3.1-fold increase); however, treatment with losartan showed a dramatic reduction (from 203 to 79 pg/ml) in dopamine levels in the infarcted rats. Treatment of control animals with losartan showed no significant effect on plasma levels of catecholamines.
Table 2.
Haemodynamic parameters and plasma catecholamines in sham and myocardial infarcted rats with and without losartan treatment for 8 weeks beginning at 12 weeks after coronary artery occlusion
| Parameters | Sham | MI | Sham + LOS | MI + LOS |
|---|---|---|---|---|
| (A) Haemodynamic parameters | ||||
| Heart rate (bpm) | 220 ± 8 | 230 ± 6 | 237 ± 3 | 232 ± 11 |
| LVSP (mm Hg) | 134 ± 2.5 | 76 ± 2.1* | 125 ± 6 | 109 ± 4# |
| LVEDP (mm Hg) | 4.7 ± 0.11 | 20.8 ± 0.70* | 5.4 ± 0.63 | 7.6 ± 0.54# |
| +dP/dt (mm Hg/sec.) | 7350 ± 400 | 3827 ± 130* | 6529 ± 290 | 5598 ± 346# |
| −dP/dt (mm Hg/sec.) | 5620 ± 155 | 2350 ± 230* | 5253 ± 81 | 4263 ± 250# |
| MAP (mm Hg) | 151 ± 15 | 135 ± 10 | 149 ± 13 | 146 ± 12 |
| (B) Plasma catecholamines | ||||
| Norepinephrine (pg/ml) | 182 ± 4.8 | 355 ± 13.2* | 197 ± 6.0 | 455 ± 15# |
| Epinephrine (pg/ml) | 78 ± 7.0 | 132 ± 5.6* | 77 ± 6.2 | 131 ± 4.5 |
| Dopamine (pg/ml) | 66 ± 7.3 | 203 ± 19.7* | 77 ± 4.5 | 79 ± 5.0# |
Values are mean ± S.E. of five animals in each group. LVSP: left ventricular systolic pressure; LVEDP: left ventricular end diastolic pressure; MI: myocardial infarction; MAPL: mean arterial pressure; +dP/dt: rate of pressure development; −dP/dt: rate of pressure decay; bpm: beats per min; LOS: Losartan (20 mg/kg/day); *P < 0.05 compared with the 20 weeks sham group; #P < 0.05 compared with the 20 weeks MI group.
Ventricular remodelling and performance
Echocardiographic examination of internal cardiac diastolic and systolic dimensions revealed marked changes in the structure of the heart due to MI (Table3A). The MI hearts showed a 26% decrease in the thickness of IVSs, a 23% increase in LVIDd, a 78% increase in LVIDs, a 55% increase in the thickness of LVPWs and a 28% increase in the thickness of LVPWd. Attenuation of changes in these parameters was observed with the treatment of MI animals with losartan, as IVSs increased from 74% to 90%, LVIDs showed a reduction from 178% to 145%, LVPWs showed a reduction from 155% to 107% and LVPWd showed a decrease from 128% to 90%. Interestingly, there was no significant reduction in the LVIDd value after treatment with losartan. Furthermore, there were no alterations in the thickness of IVSd in any group (Table3A). The infarcted animals also showed reductions in cardiac performance parameters to reflect a 40% decrease in EF, a 50% decrease in FS and a 54% decline in CO (Table3B). The 8-week treatment period with losartan revealed improvement of cardiac function, as these values were enhanced with an increase in EF from 60% to 73%, an increase in FS from 50% to 63%, and an increase in CO from 54% to 65%. The HR did not show any considerable variation in values. Furthermore, treatment of sham control animals with losartan did not produce any significant changes in parameters for internal cardiac dimensions or cardiac function (Table3A and B).
Table 3.
Internal cardiac diastolic and systolic dimensions and cardiac performance parameters by echocardiography of sham and infarcted animals with and without losartan treatment for 8 weeks beginning at 12 weeks after coronary artery occlusion
| Parameters | Sham | MI | Sham + LOS | MI + LOS |
|---|---|---|---|---|
| (A) Cardiac remodelling parameters | ||||
| IVSd (cm) | 0.220 ± 0.01 | 0.227 ± 0.02 | 0.254 ± 0.03 | 0.239 ± 0.03 |
| IVSs (cm) | 0.387 ± 0.02 | 0.287 ± 0.01* | 0.387 ± 0.03 | 0.351 ± 0.02# |
| LVIDd (cm) | 0.878 ± 0.03 | 1.080 ± 0.02* | 0.845 ± 0.03 | 1.060 ± 0.07 |
| LVIDs (cm) | 0.468 ± 0.04 | 0.834 ± 0.05* | 0.533 ± 0.04 | 0.681 ± 0.02# |
| LVPWs (cm) | 0.241 ± 0.02 | 0.374 ± 0.03* | 0.270 ± 0.03 | 0.259 ± 0.01# |
| LVPWd (cm) | 0.346 ± 0.02 | 0.444 ± 0.03* | 0.305 ± 0.03 | 0.313 ± 0.03# |
| (B) Cardiac performance parameters | ||||
| Ejection fraction (%) | 80 ± 5.1 | 48 ± 3.2* | 70 ± 3.8 | 58 ± 2.2# |
| Fractional shortening (%) | 46 ± 3.5 | 23 ± 1.3* | 37 ± 2.7 | 29 ± 1.4# |
| Cardiac output (l/min.) | 0.461 ± 0.04 | 0.255 ± 0.02* | 0.457 ± 0.04 | 0.301 ± 0.04# |
| Heart rate (bpm) | 345 ± 10 | 360 ± 11 | 318 ± 7 | 338 ± 14 |
Values are mean ± S.E. of seven animals in each group. MI: myocardial infarction; LOS: Losartan (20 mg/kg/day); IVS: internal ventricular septum; LVID: left ventricular internal diameter; LVPW: left ventricular posterior wall; d: diastolic measurement; s: systolic measurement. *P < 0.05 compared with the 20 weeks sham group. #P < 0.05 compared with the 20 weeks MI group.
Alterations in subcellular activities
The SR function was markedly depressed in heart failure as the data in Table4A show a decrease in Ca2+ uptake activity by 68% in MI hearts. However, there was an attenuation of this change showing an improvement from 32% to 58% upon losartan treatment. SR Ca2+-release activity was depressed in the untreated MI hearts by 53%, but this change was not affected by treatment with losartan (Table4A). MI hearts showed a 39% decrease in MF Ca2+-stimulated ATPase activity without any changes in Mg2+-ATPase activity when compared with the 20 week sham hearts (Table4B). Treatment of MI animals with losartan, showed a significant reversal from 61% to 79% in MF Ca2+-stimulated ATPase activity without any changes in Mg2+-ATPase activity. There were no alterations observed in SR Ca2+-transport and MF ATPase activities in control rats upon treatment with losartan (Table4).
Table 4.
Sarcoplasmic reticular and myofibrillar activities in sham and infarcted animals with and without losartan treatment for 8 weeks beginning at 12 weeks after coronary occlusion
| Parameter | Sham | MI | Sham + LOS | MI + LOS |
|---|---|---|---|---|
| (A) Sarcoplasmic reticulum activities | ||||
| Ca2+-uptake (nmol Ca2+/mg/min.) | 53.1 ± 2.01 | 17.3 ± 2.70* | 51.9 ± 3.03 | 30.6 ± 3.30# |
| Ca2+-release (nmol Ca2+/mg/15 sec.) | 8.9 ± 0.20 | 4.2 ± 0.15* | 8.4 ± 0.09 | 4.3 ± 0.11 |
| (B) Myofibrillar ATPase activities | ||||
| Mg2+ATPase (μmol Pi/mg/hr) | 3.1 ± 0.07 | 3.0 ± 0.10 | 3.2 ± 0.12 | 2.9 ± 0.11 |
| Ca2+-stimulated ATPase (μmol Pi/mg/hr) | 13.2 ± 0.80 | 8.1 ± 0.61* | 12.9 ± 0.73 | 10.5 ± 0.50# |
Values are mean ± S.E. of seven animals in each group. MI: myocardial infarction; LOS: Losartan (20 mg/kg/day); *P < 0.05 compared with the 20 weeks sham group; #P < 0.05 compared with the 20 weeks MI group.
Modification of SR protein expression
To test if changes in SR Ca2+-uptake activity in the failing heart are associated with alterations in the expression of Ca2+-cycling and regulatory proteins, SR PLB, SERCA2a and CQS content were measured using Western blot analysis (Fig.1). The failing heart showed a reduction in the expression of PLB by 41% and SERCA2a by 68%, without any significant decrease in the protein expression of CQS. Upon the treatment of MI animals with losartan, these hearts revealed a significant amount of recovery in protein expression as values increased from 59% to 71% for PLB and from 32% to 74% for SERCA2a, without any apparent change in the value for CQS. Treatment of control animals with losartan had no effect on SR protein content.
Fig 1.
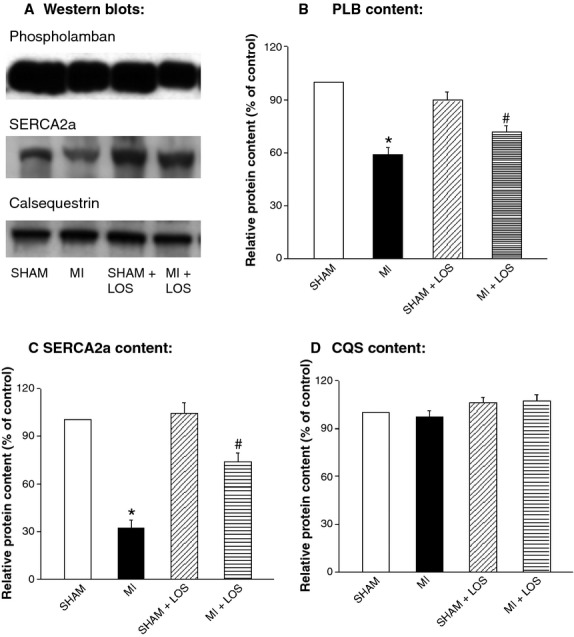
Typical Western blots (A) and relative protein content of sarcoplasmic reticular phospholamban (PLB; B), Ca2+-pump (SERCA2a; C) and calsequestrin (CQS; D) from sham and infarcted (MI) rat hearts with or without losartan (LOS) treatment. Each value is a mean ± S.E. of five animals. *P < 0.05 compared with sham. #P < 0.05 compared with MI group.
Alterations in SR and MHC mRNA expression
To understand the mechanisms of changes in subcellular activities in the failing and drug-treated rat hearts, the steady-state mRNA levels for SR and MF proteins were examined using Northern blot analysis (Figs2 and 3). The failing hearts following MI showed a reduction in mRNA levels for SR proteins with a 21% decline in SERCA2a and a 22% decline in PLB (Fig.2). These changes were not significantly reversed by losartan treatment, although there was a slight improvement in the levels of SERCA2a mRNA from 79% to 88%, and of PLB mRNA from 78% to 93% when compared with the control values of the sham rats. Furthermore, there was no alteration in the mRNA level for RyR in failing hearts with or without losartan treatment (Fig.3B). On the other hand, a significant 39% reduction in the α-MHC mRNA was observed in the MI hearts, whereas an increase in the β-MHC mRNA level of 200% was noted (Fig.3C and D). Treatment with losartan significantly attenuated the changes in the mRNA levels for MHC as β-MHC mRNA decreased from 300% to 212%, whereas that for α-MHC mRNA increased from 61% to 77% of the control hearts.
Fig 2.
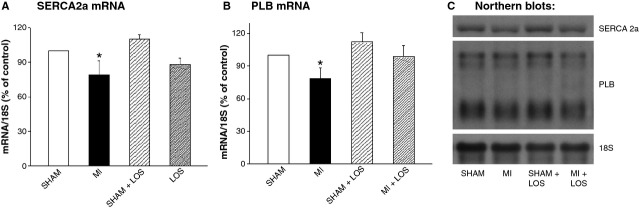
Relative mRNA levels for sarcoplasmic reticular Ca2+-pump ATPase (SERCA2a; A) and phospholamban (PLB; B), as well as their corresponding Northern blots (C) from sham and infarcted (MI) rats, with or without losartan (LOS) treatment. Each value is a mean ± S.E. of seven animals. *P < 0.05 compared with sham.
Fig 3.
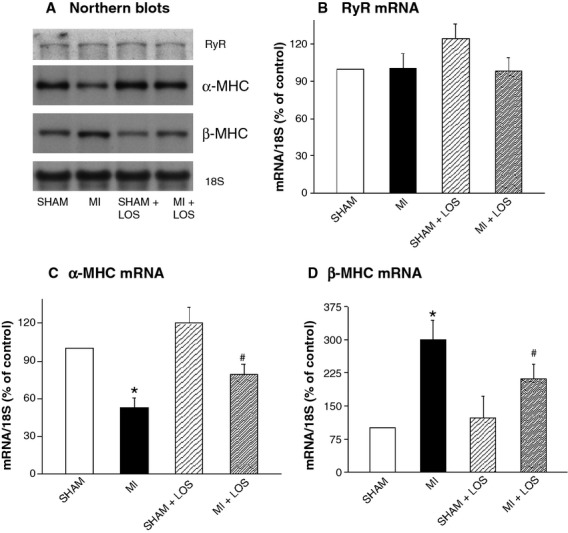
Typical Northern blots (A) and relative mRNA levels for sarcoplasmic reticular ryanodine receptor (RyR; B), alpha myosin heavy chain (α-MHC; C) and beta myosin heavy chain (β-MHC; D) isozymes from sham and infarcted (MI) rat hearts with or without losartan (LOS) treatment. Each value is a mean ± S.E. of seven animals. *P < 0.05 compared with sham. #P < 0.05 compared with MI group.
Discussion
In this study, cardiac dysfunction in rats upon inducing MI for 20 weeks was evident from depressed LVSP, +dP/dt and −dP/dt as well as increased LVEDP. Furthermore, impairment of cardiac performance in these animals was seen because EF, FS and CO were markedly decreased. The infarcted animals showed cardiac hypertrophy as heart wt and heart wt/body wt ratio were increased. Furthermore, these animals were at congestive heart stage because the lung wet wt/dry wt ratio was increased. All these observations are in agreement with previous reports indicating the development of congestive heart failure at different times of inducing MI 22,25,27,39,40. Such alterations in cardiac function are most likely due to cardiac remodelling as various parameters including increased LVIDd, LVIDs, LVPWs and LVPWd, as well as decreased IVSs were evident in 20 weeks infarcted animals. These observations are consistent with other reports showing cardiac remodelling in heart failure due to MI of different durations 31,33,39,41,42. In view of the role of the prolonged activation of SNS in the development of heart failure 13,14, the elevated levels of plasma, NE, EPI and dopamine can be seen to produce cardiac dysfunction and cause cardiac hypertrophy, as well as cardiac dilatation in the infarcted animals. Treatment of 12 weeks infarcted animals (exhibiting cardiac dysfunction) with losartan for a period of 8 week was found to reverse cardiac hypertrophy, cardiac remodelling and cardiac dysfunction partially. On the other hand, increases in RV wt, LVPWd and lung congestion were reversed fully by treatment of infarcted animals with losartan. It is pointed out that partial to complete prevention of these changes in failing heart have also been reported when the infarcted animals at pre-failure stage were treated with blockers of the RAS 22,33,43–46.
In spite of marked alterations in cardiac diastolic and systolic dimensions as well as LV pressures due to MI, no changes in HR or MAP were observed in this experimental model of heart failure. Such differences in the response of various haemodynamic and remodelling parameters to MI are surprising because the levels of plasma catecholamines were elevated as a consequence of prolonged activation of the sympathetic nervous system. This discrepancy may be due to haemodynamic adjustments under chronic conditions and/or release of some factors, which may have opposing effects (on specific sites) to those of the elevated levels of catecholamines, in the circulation. The partial reversal of cardiac dysfunction and cardiac remodelling in animals with heart failure by losartan treatment was not due to changes in work-load or after-load on the heart because no alterations in infarct size or mean arterial blood pressure were seen in the infarcted rats upon treatment with losartan. Likewise, changes in the plasma catecholamines cannot be considered to account for the reversal of cardiac dysfunction and cardiac remodelling in heart failure because plasma NE level was increased and that for EPI was unaltered upon the treatment of MI animals with losartan. It should be pointed out that because the high plasma angiotensin II level in the infarcted animals was further increased upon treatment with losartan 22, it is possible that the increase in plasma NE level in the losartan-treated infarcted animals may be due to the effect of angiotensin II on the sympathetic nerve terminals. However, it is also noteworthy that the observed increase in plasma dopamine in MI animals with heart failure was fully reversed upon losartan treatment. In view of the lack of information concerning the role of dopamine in heart failure, the exact significance of changes in plasma dopamine remains to be a matter of speculation. Furthermore, the mechanisms for the differential effects of losartan in heart failure due to MI need to be investigated.
Cardiac dysfunction in the failing heart is not only explained on the basis of cardiac remodelling but subcellular remodelling has also been suggested to play a critical role in the development of abnormalities in cardiac contraction and cardiac relaxation 6,10,32. Particularly, alterations in SR Ca2+-release and SR Ca2+-pump, as well as MF Ca2+-stimulated ATPase activities have been shown to occur in heart failure due to MI 20,22,26,27,47. Likewise, we have observed that both SR Ca2+-release and Ca2+-uptake activities, as well as MF Ca2+-stimulated ATPase activities were depressed in 20 weeks infarcted hearts. Depression in SR Ca2+-uptake activity in failing hearts was not only accompanied by corresponding changes in gene and protein expressions of SERCA2a but was also associated with gene and protein expressions for PLB, which is known to regulate the SR Ca2+-pump activity. Furthermore, the depression in MF Ca2+-stimulated ATPase activity appears to be due to a shift in myosin isozymes because mRNA level for α-MHC was decreased and that for β-MHC was increased in the failing hearts. Such a remodelling of SR and MF with respect to SR Ca2+-pump and MF Ca2+-stimulated ATPase proteins has been reported in failing hearts due to MI for 8 weeks 20,22,26,27. On the other hand, the observed decrease in SR Ca2+-release activity in 20 weeks infarcted animals was not associated with a corresponding depression in mRNA levels for Ca2+-release channel protein; this observation is in contrast to what was seen in the 8 weeks infarcted hearts 20,22. As no change in mRNA level for Ca2+-release channel protein was observed in 40 weeks infarcted hearts 48, it is likely that alterations in cardiac gene expression are biphasic where mRNA levels for Ca2+-handling proteins are depressed at early stages and are normalized or increased at late stages of heart failure. This view is consistent with our earlier observations showing biphasic changes in gene expression for SL Na+-Ca2+ exchanger at different times of inducing MI 21,48.
The results in this study have revealed that alterations in SR Ca2+-uptake, Ca2+-pump protein, PLB protein and mRNA levels, as well as MF Ca2+-stimulated ATPase, α-MHC mRNA and β-mRNA levels due to heart failure were partially reversible upon treatment of 12 weeks infarcted hearts with losartan. Blockade of RAS with agents such as losartan has been shown to partially or fully prevent changes in SR and MF remodelling upon starting the treatment at the pre-failure stage in 3 weeks infarcted animals 22,25–27. Because the beneficial effects of losartan on SR Ca2+-pump protein, unlike SR PLB protein, were not evident at the level of its gene expression in 20 weeks infarcted hearts; it appears that losartan may affect post-translational sites for SR remodelling. Nonetheless, partial recovery of SR Ca2+-pump and MF Ca2+-stimulated ATPase activities by losartan treatment can be seen to account for the partial improvement of cardiac function in 12 weeks infarcted animals. It should be noted that losartan treatment showed no effects on the depressed Ca2+-release activity or unaltered gene expression for Ca2+-channel protein in 12 weeks infarcted animals. As the elevated plasma NE and EPI levels in infarcted animals were not decreased by losartan treatment, it is possible that the high levels of plasma catecholamines may induce the observed irreversible defect in Ca2+-channel proteins. Such a defect in SR Ca2+-release channels may suggest irreversibility of subcellular components in cardiomyocytes and/or ineffectiveness of different cardiac drugs to improve heart function at advanced stages of heart failure.
Because AT1R antagonists have been shown to improve cardiac function and attenuate cardiac remodelling in patients with heart failure 12,17,18, it can be argued that the conceptual novelty of this study to test reverse remodelling by losartan is quite limited. However, this does not seem to be the case because this study provides the first experimental evidence that the improvement of cardiac performance and reverse cardiac remodelling by any pharmacological intervention is associated with reversal of subcellular defects in the failing hearts. From the foregoing discussion, it is evident that partial reversal of cardiac dysfunction by losartan in MI animals at advanced stages of heart failure is associated with partial reversal of cardiac remodelling and partial attenuation of changes in SR Ca2+-transport and MF Ca2+-stimulated ATPase activities. Because losartan as well as different ACE inhibitors have also been demonstrated to prevent MI-induced alterations in SR and MF functions 20,22–27, it is likely that the mechanism for the prevention and reversal of subcellular defects in heart failure due to MI may be similar to each other. Furthermore, because blockade of RAS has also been shown to prevent SL remodelling, restructuring of extracellular matrix and defects in signal transduction as well as metabolic and regulatory systems during early stages of heart failure 45,46,49–51, the possibility of their contribution in reversing cardiac dysfunction and remodelling at advanced stages of heart failure cannot be ruled out. Nonetheless, it should be noted that as losartan treatment of control animals did not affect any parameter of cardiac function, ventricular remodelling and subcellular activities, the beneficial effects of losartan in preventing or reverse remodelling in failing heart may not be due to its direct action on the myocardium. In view of the role of both oxidative stress and intracellular Ca2+-overload in the pathogenesis of cardiac remodelling, cardiac dysfunction and subcellular defects in heart failure 5,6,10,32, it appears that agents such as losartan may both prevent and reverse cardiac dysfunction by reducing the development of oxidative stress and/or the occurrence of intracellular Ca2+-overload in the failing cardiomyocytes. Although different studies have provided evidence in this regard with respect to the prevention by losartan 19,22,46,50,51, extensive studies are required to understand the mechanisms of reverse cardiac remodelling, attenuation of subcellular defects and improvement of cardiac function by AT1R antagonists in the failing heart.
Acknowledgments
The research work in this study was supported by a grant from the Canadian Institutes of Health Research in partnership with Manitoba Health Research Council. The infrastructural support for this project was provided by the St. Boniface Hospital Research Foundation.
Conflict of Interest
There is no conflict of interest.
References
- Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling – concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol. 2000;35:569–82. doi: 10.1016/s0735-1097(99)00630-0. [DOI] [PubMed] [Google Scholar]
- Fedak PWM, Verma S, Weisel RD, et al. Cardiac remodeling and failure from molecules to man (Part I) Cardiovasc Physiol. 2005;12:1–11. doi: 10.1016/j.carpath.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–62. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction: experimental observations and clinical implications. Circulation. 1990;81:1161–72. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G. Alterations of calcium-regulatory proteins in heart failure. Cardiovasc Res. 1998;37:279–89. doi: 10.1016/s0008-6363(97)00277-0. [DOI] [PubMed] [Google Scholar]
- Dhalla NS, Rangi S, Babick AP, et al. Cardiac remodeling and subcellular defects in heart failure due to myocardial infarction and aging. Heart Fail Rev. 2011 doi: 10.1007/s10741-011-9278-7. ; doi: 10.1007/s10741-011-9278-7. [DOI] [PubMed] [Google Scholar]
- Morano I, Hadicke K, Haase H, et al. Changes in essential myosin light chain isoform expression provide a molecular basis for isometric force regulation in the failing human heart. J Mol Cell Cardiol. 1997;29:1177–87. doi: 10.1006/jmcc.1996.0353. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G. Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovasc Res. 1998;39:60–76. doi: 10.1016/s0008-6363(98)00110-2. [DOI] [PubMed] [Google Scholar]
- Prestle J, Quinn FR, Smith GL. Ca2+-handling proteins and heart failure: novel molecular targets? Curr Med Chem. 2003;10:967–81. doi: 10.2174/0929867033457656. [DOI] [PubMed] [Google Scholar]
- Dhalla NS, Saini-Chohan HK, Rodriguez-Leyva D, et al. Subcellular remodelling may induce cardiac dysfunction in congestive heart failure. Cardiovasc Res. 2009;81:429–38. doi: 10.1093/cvr/cvn281. [DOI] [PubMed] [Google Scholar]
- Machackova J, Barta J, Dhalla NS. Myofibrillar remodeling in cardiac hypertrophy, heart failure and cardiomyopathies. Can J Cardiol. 2006;22:953–68. doi: 10.1016/s0828-282x(06)70315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir MR, Dzau VJ. The renin-angiotensin-aldosterone system: a specific target for hypertension management. Am J Hypertens. 1995;12:205S–13S. doi: 10.1016/s0895-7061(99)00103-x. [DOI] [PubMed] [Google Scholar]
- Francis GS, Cohn JN, Johnson G, et al. Plasma norepinephrine, plasma-renin activity, and congestive heart failure. Relations to survival and the effects of therapy in V-HeFT II. The V-HeFT VA Cooperative Studies Group. Circulation. 1993;87(Suppl. VI):40–8. [PubMed] [Google Scholar]
- Esler M, Lambert G, Brunner-La Rocca HP, et al. Sympathetic nerve activity and neurotransmitter release in humans: translation from pathophysiology into clinical practice. Acta Physiol Scand. 2003;177:275–84. doi: 10.1046/j.1365-201X.2003.01089.x. [DOI] [PubMed] [Google Scholar]
- Sallach JA, Goldstein S. Use of beta-blockers in congestive heart failure. Ann Med. 2003;35:259–66. doi: 10.1080/14734220310011716. [DOI] [PubMed] [Google Scholar]
- Krum H. Beta-blockers in heart failure. The ‘new wave’ of clinical trials. Drugs. 1999;58:203–10. doi: 10.2165/00003495-199958020-00001. [DOI] [PubMed] [Google Scholar]
- Dickstein K, Kjekshus J. OPTIMAAL Steering Committee of the OPTIMAAL Study Group. Effects of losartan and captopril on mortality and morbidity in high-risk patients after acute myocardial infarction: the OPTIMAAL randomised trial. Optimal Trial in Myocardial Infarction with Angiotensin II Antagonist Losartan. Lancet. 2002;360:752–60. doi: 10.1016/s0140-6736(02)09895-1. [DOI] [PubMed] [Google Scholar]
- Yusuf S, Pfeffer MA, Swedberg K, et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet. 2003;362:777–81. doi: 10.1016/S0140-6736(03)14285-7. [DOI] [PubMed] [Google Scholar]
- Guo X, Saini HK, Wang J, et al. Prevention of remodeling in congestive heart failure due to myocardial infarction by blockade of renin angiotensin system. Expert Rev Cardiovasc Therap. 2005;3:717–32. doi: 10.1586/14779072.3.4.717. [DOI] [PubMed] [Google Scholar]
- Shao Q, Ren B, Zarain-Herzberg A, et al. Captopril treatment improves the sarcoplasmic reticular Ca2+ transport in heart failure due to myocardial infarction. J Mol Cell Cardiol. 1999;31:1663–72. doi: 10.1006/jmcc.1999.1000. [DOI] [PubMed] [Google Scholar]
- Shao Q, Ren B, Elimban V, et al. Modification of sarcolemmal Na+-K+ ATPase and Na+/Ca2+ exchanger expression in heart failure by blockade of renin-angiotensin system. Am J Physiol Heart Circ Physiol. 2005;288:H2637–46. doi: 10.1152/ajpheart.01304.2004. [DOI] [PubMed] [Google Scholar]
- Shao Q, Ren B, Saini HK, et al. Sarcoplasmic reticulum Ca2+-transport and gene expression in congestive heart failure are modified by imidapril treatment. Am J Physiol Heart Circ Physiol. 2005;288:H1674–82. doi: 10.1152/ajpheart.00945.2004. [DOI] [PubMed] [Google Scholar]
- Semb SO, Lunde PK, Holt E, et al. Reduced myocardial Na+, K+-pump capacity in congestive heart failure following myocardial infarction in rats. J Mol Cell Cardiol. 1998;30:1311–28. doi: 10.1006/jmcc.1998.0696. [DOI] [PubMed] [Google Scholar]
- Hanatani A, Yoshiyama M, Takeuchi K, et al. Angiotensin II type 1-receptor antagonist candesartan cilexitil prevents left ventricular dysfunction in myocardial infarcted rats. Jpn J Pharmacol. 1998;78:45–54. doi: 10.1254/jjp.78.45. [DOI] [PubMed] [Google Scholar]
- Guo X, Chapman D, Dhalla NS. Partial prevention of changes in SR gene expression in congestive heart failure due to myocardial infarction by enalapril or losartan. Mol Cell Biochem. 2003;254:163–72. doi: 10.1023/a:1027321130997. [DOI] [PubMed] [Google Scholar]
- Wang J, Guo X, Dhalla NS. Modification of myosin protein and gene expression in failing hearts due to myocardial infarction by enalapril or losartan. Biochim Biophys Acta. 2004;1690:177–84. doi: 10.1016/j.bbadis.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu X, Ren B, et al. Modification of myosin gene expression by imidapril in failing heart due to myocardial infarction. J Mol Cell Cardiol. 2002;34:847–57. doi: 10.1006/jmcc.2002.2023. [DOI] [PubMed] [Google Scholar]
- Maczewski M, Mackiewicz U. Effect of metoprolol and ivabradine on left ventricular remodelling and Ca2+ handling in the post-infarction rat heart. Cardiovasc Res. 2008;79:42–51. doi: 10.1093/cvr/cvn057. [DOI] [PubMed] [Google Scholar]
- Sun YL, Hu SJ, Wang LH, et al. Effect of beta-blockers on cardiac function and calcium handling protein in postinfarction heart failure rats. Chest. 2005;128:1812–21. doi: 10.1378/chest.128.3.1812. [DOI] [PubMed] [Google Scholar]
- Omerovic E, Bollano E, Soussi B, et al. Selective beta1-blockade attenuates post-infarct remodelling without improvement in myocardial energy metabolism and function in rats with heart failure. Eur J Heart Fail. 2003;5:725–32. doi: 10.1016/s1388-9842(03)00153-3. [DOI] [PubMed] [Google Scholar]
- Omerovic E, Bollano E, Mobini R, et al. Selective beta(1)-blockade improves cardiac bioenergetics and function and decreases neuroendocrine activation in rats during early postinfarct remodeling. Biochem Biophys Res Commun. 2001;281:491–8. doi: 10.1006/bbrc.2001.4336. [DOI] [PubMed] [Google Scholar]
- Dhalla NS, Dent MR, Tappia PS, et al. Subcellular remodeling as a viable target for the treatment of congestive heart failure. J Cardiovasc Pharmacol Therapeut. 2006;11:31–45. doi: 10.1177/107424840601100103. [DOI] [PubMed] [Google Scholar]
- Gurevich AK, Falk SA, Nemenoff RA, et al. Effects of angiotensin receptor blockade on haemodynamics and gene expression after myocardial infarction. Drugs R D. 2002;3:239–49. doi: 10.2165/00126839-200203040-00004. [DOI] [PubMed] [Google Scholar]
- Machackova J, Sanganalmath SK, Barta J, et al. Amelioration of cardiac remodelling in congestive heart failure by beta-adrenoceptor blockade is associated with depression in sympathetic activity. Cardiovasc Toxicol. 2009;10:9–16. doi: 10.1007/s12012-009-9058-y. [DOI] [PubMed] [Google Scholar]
- Machackova J, Sanganalmath SK, Elimban V, et al. β-adrenergic blockade attenuates cardiac dysfunction and myofibrillar remodeling in congestive heart failure. J Cell Mol Med. 2011;15:545–54. doi: 10.1111/j.1582-4934.2010.01015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon IM, Lee SL, Dhalla NS. Nitrendipine binding in congestive heart failure due to myocardial infarction. Circ Res. 1990;66:782–8. doi: 10.1161/01.res.66.3.782. [DOI] [PubMed] [Google Scholar]
- Dixon IM, Hata T, Dhalla NS. Sarcolemmal calcium transport in congestive heart failure due to myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 1992;31:H1387–94. doi: 10.1152/ajpheart.1992.262.5.H1387. [DOI] [PubMed] [Google Scholar]
- Sanganalmath SK, Barta J, Takeda N, et al. Antiplatelet therapy mitigates cardiac remodeling and dysfunction in congestive heart failure due to myocardial infarction. Can J Physiol Pharmacol. 2008;86:180–9. doi: 10.1139/Y08-005. [DOI] [PubMed] [Google Scholar]
- Sanganalmath SK, Babick AP, Barta J, et al. Antiplatelet therapy attenuates subcellular remodelling in congestive heart failure. J Cell Mol Med. 2008;12:1728–38. doi: 10.1111/j.1582-4934.2007.00197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Liu X, Sentex E, et al. Increased expression of protein kinase C isoforms in heart failure due to myocardial infarction. Am J Physiol Heart Circ Physiol. 2003;284:H2277–87. doi: 10.1152/ajpheart.00142.2002. [DOI] [PubMed] [Google Scholar]
- Sjaastad I, Sejersted OM, Ilebekk A, et al. Echocardiographic criteria for detection of postinfarction congestive heart failure in rats. J Appl Physiol. 2000;89:1445–54. doi: 10.1152/jappl.2000.89.4.1445. [DOI] [PubMed] [Google Scholar]
- Birkeland JA, Sjaastad I, Brattelid T, et al. Effects of treatment with a 5-HT4 receptor antagonist in heart failure. Br J Pharmacol. 2007;150:143–52. doi: 10.1038/sj.bjp.0706966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang BS, Ahmad M, Tan J, et al. Chronic central versus systemic blockade of AT1 receptors and cardiac dysfunction in rats post myocardial infarction. Am J Physiol Heart Circ Physiol. 2009;297:H968–75. doi: 10.1152/ajpheart.00317.2009. [DOI] [PubMed] [Google Scholar]
- Zhu YZ, Zhu YC, Li J, et al. Effects of losartan on haemodynamic parameters and angiotensin receptor mRNA levels in rat heart after myocardial infarction. J Renin Angiotensin Aldosterone Syst. 2000;1:257–62. doi: 10.3317/jraas.2000.039. [DOI] [PubMed] [Google Scholar]
- Sethi R, Shao Q, Ren B, et al. Changes in beta-adrenoceptors in heart failure due to myocardial infarction are attenuated by blockade of renin-angiotensin system. Mol Cell Biochem. 2004;263:11–20. [PubMed] [Google Scholar]
- Khaper N, Singal PK. Modulation of oxidative stress by a selective inhibition of angiotensin II type 1 receptors in MI rats. J Am Coll Cardiol. 2001;37:1461–6. doi: 10.1016/s0735-1097(01)01126-3. [DOI] [PubMed] [Google Scholar]
- Afzal N, Dhalla NS. Differential changes in left and right ventricular SR calcium transport in congestive heart failure. Am J Physiol Heart Circ Physiol. 1992;262:H868–74. doi: 10.1152/ajpheart.1992.262.3.H868. [DOI] [PubMed] [Google Scholar]
- Ren B, Shao Q, Ganguly PK, et al. Influence of long-term treatment of imidapril on mortality, cardiac function, and gene expression in congestive heart failure due to myocardial infarction. Can J Physiol Pharmacol. 2004;82:1118–27. doi: 10.1139/y04-115. [DOI] [PubMed] [Google Scholar]
- Ju H, Zhao S, Jassal DS, et al. Effect of AT1 receptor blockade on cardiac collagen remodeling after myocardial infarction. Cardiovasc Res. 1997;35:223–32. doi: 10.1016/s0008-6363(97)00130-2. [DOI] [PubMed] [Google Scholar]
- Guo X, Wang J, Elimban V, et al. Both enalapril and losartan attenuate sarcolemmal Na+-K+-ATPase remodeling in failing rat heart due to myocardial infarction. Can J Physiol Pharmacol. 2008;86:139–47. doi: 10.1139/Y08-006. [DOI] [PubMed] [Google Scholar]
- Saini HK, Shao Q, Musat S, et al. Imidapril treatment improves the attenuated inotropic and intracellular calcium responses to ATP in heart failure due to myocardial infarction. Br J Pharmacol. 2005;144:202–11. doi: 10.1038/sj.bjp.0705867. [DOI] [PMC free article] [PubMed] [Google Scholar]