

Abstract
Phosphorylation of myosin regulatory light chain (RLC) triggers contraction in smooth muscle myocytes. Dephosphorylation of phosphorylated RLC (pRLC) is mediated by myosin RLC phosphatase (MLCP), which is negatively regulated by rho-associated kinase (ROK). We have compared basal and stimulated concentrations of pRLC in myocytes from human coronary artery (hVM), which has a tonic contractile pattern to myocytes from human uterus (hUM), which has a phasic contractile pattern. Our studies reveal fundamental differences between hVM and hUM regarding the mechanisms regulating phosphorylation RLC. Whereas hVM responded to stimulation by phosphorylation of RLC at S19, hUM responded by forming diphosphorylated RLC (at T18 and S19; ppRLC), which, compared to pRLC, causes two to threefold greater activation of myosin ATPase that provides energy to power the contraction. Importantly, the conversion of pRLC to ppRLC is mediated by ROK. In hUM, MLCP has high activity for ppRLC and this is inhibited by ROK through phosphorylation of the substrate targeting subunit (MYPT1) at T853. Inhibitors of ROK significantly reduce contractility in both hVM and hUM. We demonstrated that inhibition of ppRLC in phasic myocytes (hUM) is 100-fold more sensitive to ROK inhibitors than is pRLC in tonic myocytes (hVM). We speculate that these differences in phosphorylation of RLC might reflect evolution of different contractile patterns to perform distinct physiological functions. Furthermore, our data suggest that low concentrations of ROK inhibitors might inhibit uterine contractions with minimal effects on vascular tone, thus posing a novel strategy for prevention or treatment of conditions such as preterm birth.
Keywords: uterine myocyte, contractility, preterm birth
Introduction
Smooth muscle myocyte stimulation activates a well-characterized signal transduction pathway that activates myosin regulatory light chain (RLC) kinase (MLCK), which phosphorylates RLC at S19 to yield monophosphorylated S19RLC (pRLC). Contraction of smooth muscle is determined predominantly by the concentrations of pRLC 1–3, which initiates the myosin motor to slide the actin thin filaments along the myosin thick filaments causing cell shortening. Phosphorylated RLC also activates the myosin heavy chain ATPase to provide energy for the power stroke. RLC can be phosphorylated at T18 as well as S19 to form diphosphorylated RLC (ppRLC) 4,5. In comparison to pRLC, ppRLC causes significantly greater activation of the myosin ATPase 4,5 and therefore can significantly enhance force production. The enzyme catalyzing phosphorylation at T18 is less obvious. MLCK-mediated phosphorylation at T18 is unlikely to be physiologically relevant, because it occurs at a slower rate than at S19, and requires supraphysiological concentrations of MLCK 6. Other data demonstrate that rhoA-associated kinase (ROK) can mediate phosphorylation of RLC 7–9, although the target phosphorylation site remains unknown. Here we present novel evidence that human myocytes derived from vascular smooth muscle (hVM) and uterine smooth muscle (hUM) differ significantly at the fundamental level of activation of the contractile machinery. Furthermore, the biochemical distinction can be explained by the actions of ROK.
Rho-associated kinase inhibition causes significant reduction in smooth muscle contractile activity 10–13. Using Western blots containing phos-tag, we previously noted that treatment of hUM with a ROK inhibitor (glycyl-H-1152; ‘g-H’) caused a marked reduction in ppRLC and treatment with oxytocin (OT), a potent hUM agonist, caused a significant increase in ppRLC 14. Importantly, in the face of g-H, OT induced an accumulation of pRLC, but no response in ppRLC. On the basis of these data, our primary hypothesis was that ROK mediates the OT-stimulated phosphorylation of RLC at T18 in hUM. The proportion of RLC that was diphosphorylated was considerably greater in hUM than has been demonstrated for other smooth muscles. Our second hypothesis was that the pathways favouring formation of ppRLC were predominantly important in smooth muscle such as hUM with phasic contractile patterns and would be less active in smooth muscle such as hVM with tonic contractile patterns. To address these hypotheses, we have established a quantitative and relatively high throughput in-cell Western (ICW) assay using primary cultures of smooth muscle cells from human coronary artery or human uterus to assess mono- and di-phosphorylation of RLC 15. The data presented here provide strong support for each of these hypotheses. The results suggest a novel pharmacological target to diminish contractility of uterine smooth muscle without affecting vascular smooth muscle. Development of such a pharmacological strategy could provide a major contribution towards reducing the incidence of preterm birth, which remains the most serious complication of pregnancy 16–19.
Materials and methods
Ethical approval
The protocol to obtain biopsies from the lower segment of the uterus at the time of Caesarean section was reviewed and approved by the Human Ethics Review Board of the University of Alberta and the Ethics Review Board of Capital Health, the provider of health services in the region. A research nurse at the Royal Alexandra Hospital in Edmonton obtained informed and written consent from each patient. The methodologies for production of primary myometrial cell cultures, protein extraction, SDS-PAGE (with and without phos-tag), Western immunoblotting (WB), in-cell Western and fluorescence microscopy have been thoroughly detailed previously, 14,15 and will be described only briefly here.
Pharmacological agents
Oxytocin, ET-1, glycyl-H-1152, ML7, endothall and cantharidic acid were acquired from Calbiochem (EMD Chemicals Inc., Gibbstown, NJ, USA). Calyculin A was purchased from Cell Signaling (Danvers, MA, USA). Calpeptin and the rhoA G-LISA assay were acquired from Cytoskeleton, Inc. (Denver, CO, USA). Rho-15 and SR3677 were obtained from SYNkinase (SYNthesis Med Chem, Melbourne, Australia).
Primary human myometrial cell cultures
Myometrial biopsies were obtained from the lower segment of the uterus from non-labouring patients at term (38–40 weeks) gestation during elective caesarean section. Biopsies were diced, then incubated in HBSS (Gibco, Invitrogen, Carlsbad, CA, USA) containing 1% antimycotic/antibiotic (Gibco), 2 mg/ml collagenase (Sigma-Aldrich, St. Louis, MO, USA) and 200 ng/ml DNase I (Roche, Laval, QC, Canada) in a total volume of 10 ml for 1 hr at 37°C with shaking. After 1 hr, the debris was allowed to settle, the supernatant discarded, and the remaining tissue was incubated in a fresh 10 ml of prepared HBSS (+ enzymes) at 37°C while shaking overnight (O/N). The dispersed cell mixture was filtered through a 100 μm cell strainer, centrifuged at 2000 × g for 5 min. and washed twice with PBS. Isolated hUM were grown in DMEM (Gibco) supplemented with 10% FBS (Gibco) and 1% antimycotic/antibiotic (Gibco) at 37°C in humidified 5% CO2/95% air. Cell cultures were grown to 80–100% confluence (3–5 days) in a single well of a six-well plate, post isolation, then in T25 and T75 flasks (Ultident, St. Laurent, QC, Canada). Prior to experiments, the cells were washed once with pre-warmed DMEM containing no additives, and then placed into the incubator in DMEM without additives for 2–4 hrs. All stimulants, pharmacological agents and drug diluents (DMF, DMSO, sterile ddH2O) were diluted in DMEM without additives to the desired final concentration prior to experiments.
Primary human coronary artery smooth muscle cell cultures
The hVM (purified coronary artery myocytes – ATCC PCS-100-021) were purchased from American Type Culture Collection (ATCC, Manassas VA, USA) and cultured in vascular cell basal medium (ATCC PCS-100-030) supplemented with vascular smooth muscle cell growth kit (ATCC PCS-100-042). For all experiments, hVM were seeded in this supplemented growth medium O/N, but were washed and prepared for experiments in a manner identical to that described for hUM. The hVM experiments were performed three times (passage numbers 2–5) in quadruplicate to ensure reproducibility.
Western immunoblotting
For Western immunoblotting (WB), approximately 25 μg/lane of protein was loaded onto Tris-Glycine-SDS minigels with or without phos-tag reagent (30 μM; NARD Institute, Ltd., www.phos-tag.com) combined with MnCl2 to demonstrate shifts in mobility for phosphoproteins. SDS-PAGE was carried out according to the Laemmli method. The gel was transferred to nitrocellulose for 1.5 hrs at 100 V. The final primary Ab dilutions were: total-non-phosphorylated-RLC 1:2000 (LS-C81207, LifeSpan BioSciences Seattle, WA, USA), pRLC 1:2000 (#3675; Cell Signaling), ppRLC 1:500 (#3674; Cell Signaling), total phospho-S19-RLC 1:200 (cross reacts with pRLC and ppRLC, #3671; Cell Signaling), ROKII 1:500 (#610623; BD Bio sciences, Mississauga, ON, Canada), phospho-T696-MYPT1 (#ABS45; EMD Millipore, Billerica, MA, USA), phospho-T853-MYPT1 (#4563; Cell Signaling), rhoA 1:200 (#610990; Mouse, MAb, BD Biosci.) and α-actin 1:4000 (sc-56499; Mouse, MAb, Santa-Cruz Biotechnologies, Santa Cruz, CA, USA). Secondary Abs conjugated to IRDye 800CW (Li-COR Biosciences, Lincoln NB, USA) or Alexa Fluor 680 (Molecular Probes, Invitrogen Grand Island, NY, USA) were utilized at 1:20,000 in odyssey blocking buffer (OBB) combined with PBS (1:1) and supplemented with 0.1% Tween-20 and 0.01% SDS. Membranes were scanned and analysed using an Odyssey® IR scanner with imaging software version 3.0. Ab signals were analysed as integrated intensities of regions defined around the bands of interest. Our previous study has demonstrated that this method provides excellent resolution as well as stoichiometric quantification of RLC phospho-species 14.
In-cell Westerns
Cells were plated in sterile black-walled 96 (half-area) (Greiner Bio-One, Monroe, NC, USA) plates at 600 cells/mm2 and incubated O/N at 37°C as indicated above. After treatment, cells were fixed immediately by adding formalin to a final concentration of 3.7% formaldehyde and incubating at R/T for 15 min. After fixation, the cells were permeabilized with PBS containing 0.1% Triton-X-100, and blocked using 20 μl/well OBB for 1 hr at R/T. Primary Abs were prepared as for WB and the plates were incubated O/N at 4°C using 20 μl/well. The final primary Ab dilutions were: pRLC 1:1000 (#3675; Cell Signaling), ppRLC 1:500 (#3674; Cell Signaling), total-phospho-S19-RLC 1:200 (cross reacts with pRLC and ppRLC, #3671; Cell Signaling). Secondary Abs were conjugated to IRDye 800CW (Li-COR). Ab signals were normalized to signals from DRAQ5 (Biostatus Ltd., Leicestershire, UK) combined with Sapphire700 (Li-COR) at 1:10,000 and 1:1000 respectively. Plates were scanned and analysed using an Odyssey® IR scanner using Odyssey® imaging software version 3.0, with an offset of 4.0 mm. Results are expressed as per cent relative responses [means ± standard errors of the mean (SEM)] compared to vehicle-treated controls.
Measurement of rhoA activity by G-LISA
Relative rhoA activity was estimated by a commercially available ELISA-based assay (‘G-LISA™’, BK-124; Cytoskeleton Inc.) according to the manufacturers' specifications. The hUM were treated with OT (100 nM) 20 sec., and then harvested by lysis in ice-cold buffer (provided). An aliquot was retained for protein content estimation by ADV02 reagent (Cytoskeleton, Inc.), and the rest was flash frozen in N2(l) and stored at −80°C until needed. Approximately 25–30 μg total protein was used per well, and each sample was assayed in duplicate. Data were normalized for total-rhoA content assessed by WB.
Microscopy
Eight-chamber sterile culture slides (BD Falcon) were seeded, cultured, treated, fixed and prepared for fluorescence microscopy exactly as culture plates for ICWs. The final primary Ab dilution of ppRLC (#3674; Cell Signaling) was 1:50. A secondary Ab conjugated to DyLight 488: goat-anti-rabbit-IgG (Cell Signaling) was used at 1:200. Rhodamine-phalloidin (50 nM; Cytoskeleton, Denver, CO, USA) was used to stain F-actin fibres. DAPI was used to detect nuclei. Slides were visualized under an Olympus IX81 fluorescent microscope (Carson Scientific Imaging Group; Ontario, Canada) using Slidebook 2D, 3D Timelapse Imaging Software (Intelligent Imaging Innovations Inc.; CO, USA).
Uterine myography
Biopsies were obtained from the upper edge of the low transverse incision in the uterus at the time of elective caesarean section prior to onset of spontaneous labour. Longitudinal strips approximately 2–3 mm by 10–12 mm were excised from the muscle tissue and mounted vertically in a myobath system as described in detail previously 20. With a load of one gram, concentration-response curves to OT were obtained in the absence and presence of g-H (1 μM).
Statistical Analysis
Statistical analyses were performed using Prism version 5.0 software (GraphPad Software, San Diego, CA, USA). In all text and figures, data are expressed as means ± SEM. The numbers (n) in the figure legends refer to separate experiments performed in primary cultures of hUM from individual women undergoing elective caesarean section at term (38–40 weeks gestation) prior to labour. Statistical analysis of concentration-response curves for stimulants and inhibitors was performed with one-way anova. The EC50 and IC50 values were calculated using non-linear curve fitting. Comparison between two curves (control versus treatment or two different treatments) was performed with two-way anova with the factors being treatment and concentration. If statistical significance was achieved, a post hoc Tukey's test was conducted to determine which means were significantly different from one another. Two-tailed independent t-tests were used to analyse rhoA activation and Western blot data. A P-value of ≤ 0.05 was used to establish statistical significance in all tests.
Results
Resting concentrations of pRLC are affected by ROK inhibition in both uterine and vascular myocytes
The ICW assay independently assesses monophosphorylated pRLC and ppRLC using highly specific antibodies that we have validated previously 15. A third antibody recognizes phospho-S19-RLC whether it occurs in pRLC or ppRLC, thus indicating ‘total phospho-S19-RLC,’ which we designate p19RLC to distinguish it from the pRLC and ppRLC (Fig.1). Treatment of hUM with the potent ROK inhibitor g-H 21 caused a concentration-dependent reduction in pRLC and ppRLC to 56 ± 8% and 51 ± 6% respectively of basal levels (Fig.2A). These data were consistent with reductions in total S19 phosphorylation measured using the p19RLC Ab. Similarly, in resting hVM (Fig.2B), g-H caused a reduction in pRLC and ppRLC with the effect being greater for the former. Again, this was consistent with reductions in total p19RLC. Using these data, we calculated the IC50 for g-H-induced inhibition of pRLC, ppRLC and p19RLC for hUM, which were 76 ± 63 nM, 0.2 ± 0.2 nM and 6.0 ± 15 nM respectively. In comparison, for hVM the IC50 for g-H effects on pRLC was 31 ± 23 nM. These data demonstrate that ppRLC content in hUM is >100-fold more sensitive to g-H than is the pRLC content in either hVM or hUM.
Fig 1.
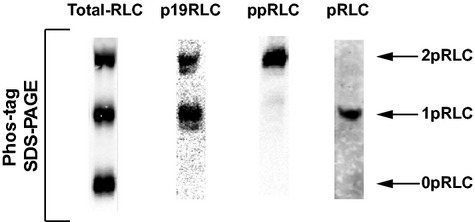
Demonstration of phospho-regulatory light chain (RLC) antibody specificity in lysates of human uterine lysates. Western blots were produced by separation of proteins in lysates from hUM using phos-tag SDS-PAGE. Single lanes were loaded with ∽25 μg of protein/lane from the same OT-stimulated cells. From left to right: Abs directed against the C-terminus of total-RLC, total phospho-S19-RLC (p19RLC, which includes pRLC + ppRLC), diphosphorylated-T18,S19- (ppRLC) and mono-S19-phosphorylated RLC. The lower, middle and upper bands in these blots correspond to non-, mono- and di-phosphorylated RLC, denoted as 0pRLC, 1pRLC and 2pRLC.
Fig 2.
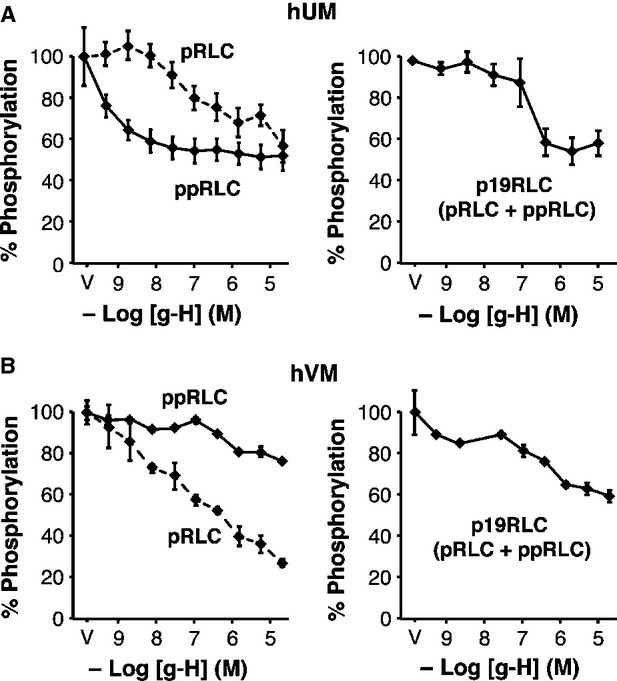
Effects of rho-associated kinase (ROK) inhibition on phosphorylation of regulatory light chain (RLC) in resting hUM and hVM. In-cell Western (ICW) assays using specific Ab measured monophospho-S19-RLC (pRLC, dashed lines), diphospho-T18/S19-RLC (ppRLC, solid lines) or total-phospho-S19-RLC [p19RLC (pRLC + ppRLC)]. The first data point in all graphs corresponds to the vehicle control (V, 100%). The specific ROK inhibitor glycyl-H-1152 (g-H) caused a concentration-dependent decrease in phosphorylated RLC in human uterine myocytes (hUM) (A) and human vascular myocytes (hVM) (B). For hUM, n = 4. As for all ICW data, results are reported as means ± SEM of% changes compared to vehicle-treated controls. For hVM, there were three independent experiments using different passages of human coronary artery myocytes from passages two to five. For hUM, the effect of g-H on ppRLC was significantly greater (P < 0.01) than on pRLC, determined by two-way anova. In contrast, for hVM, the effect of g-H was greater on pRLC compared to ppRLC.
Differential effects of ROK inhibition on stimulant-induced phosphorylation of RLC in uterine and vascular myocytes
Next we investigated the effects of physiological stimuli on concentrations of pRLC and ppRLC in hUM compared to hVM in the absence and presence of g-H. We have previously demonstrated the optimal time-point for OT-induced stimulation of p19RLC in hUM in the ICW assay is 20 sec. 15. Treatment with OT caused a concentration-dependent increase in pRLC and ppRLC (left and centre panels, Fig.3A) in agreement with the p19RLC data shown in the right panel. The responses of hUM to ET-1 were similar to OT for both pRLC and ppRLC (Fig.3B). In contrast, hVM responded to stimulation with ET-1 more slowly (maximal at 1 min. compared to 20 sec. for hUM) and with a predominant increase of pRLC (Fig.3C) but no significant response of ppRLC, again in keeping with the p19RLC data. The hVM were unresponsive to OT (data not shown).
Fig 3.
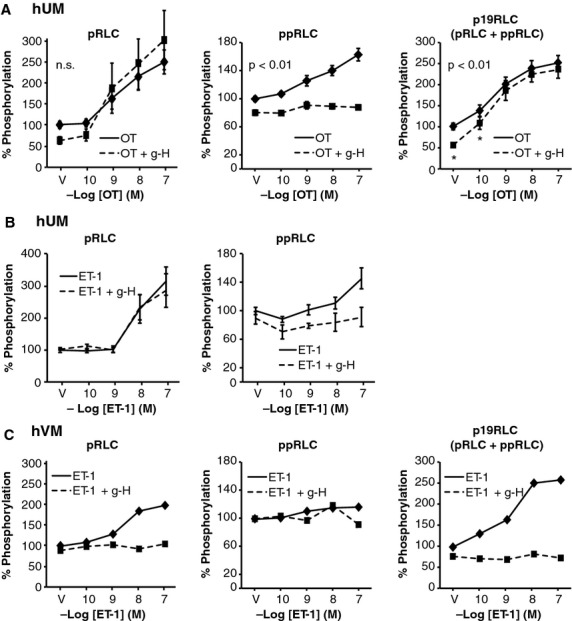
Effects of rho-associated kinase (ROK) inhibition on phosphorylation of regulatory light chain (RLC) in stimulated hUM and hVM. (A) OT caused a significant dose-dependent increase in pRLC and ppRLC in hUM. The ROK inhibitor g-H (1.0 μM) reduced basal concentrations of both pRLC and ppRLC. However, g-H had no significant effect on the pRLC response to OT, but abolished the ppRLC response. These findings were confirmed using the total p19RLC antibody (right panel). The asterisks denote that suppression of the p19RLC signal was significant (two-way anova; P ≤ 0.01), which confirms that the g-H induced suppression was due to reduction in S19 phosphorylation. (B) The responses of hUM to ET-1 in the absence or presence of g-H were similar to the responses to OT. (C) In hVM, treatment with ET-1 caused a brisk response in pRLC, but no significant response in ppRLC. Again, the p19RLC antibody confirmed that the phosphorylation in hVM was at S19.
In the presence of g-H, the increase in pRLC in hUM in response to OT or ET-1 was not affected (Fig.3A and B), but the increase in ppRLC with both agonists was abolished. The OT-induced response in p19RLC in hUM was fully retained in the presence of g-H (Fig.3A, right panel). In contrast, in hVM the ROK inhibitor abolished the ET-1-induced responses for both pRLC and total p19RLC (Fig.3C). The ROK inhibitor g-H does not distinguish between the two isoforms of ROK. However, selective isozyme inhibitors for ROK 1 or ROK 2 showed the same pattern of inhibition as g-H in basal (Fig.4A and B) and OT-stimulated (Fig.4C and D) conditions.
Fig 4.
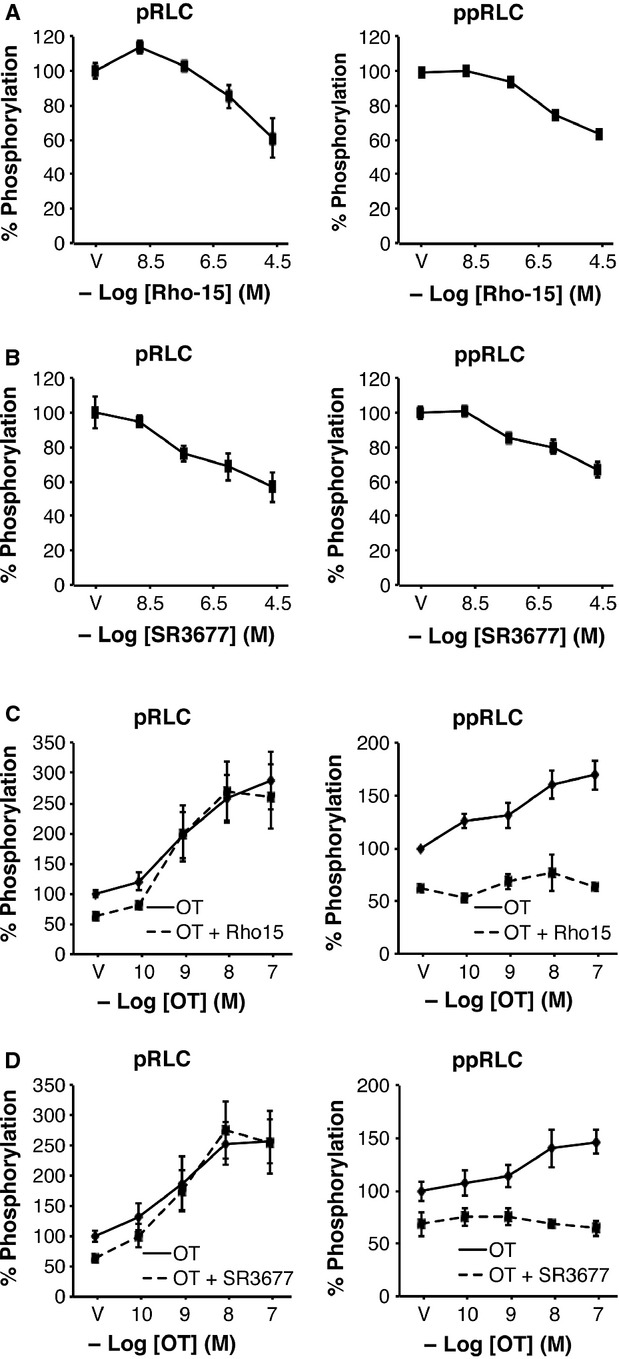
Effects of isoform-selective inhibitors of rho-associated kinase (ROK) 1 and ROK 2 on resting and stimulated phosphorylation of regulatory light chain (RLC) in hUM. Both the ROK 1-selective inhibitor (Rho-15) (A) and the ROK 2-selective inhibitor (SR3677) (B) suppressed basal pRLC and ppRLC in hUM (n = 4; P < 0.01 by one-way anova), similarly to the less selective inhibitor g-H as noted in Figure2A. Neither Rho-15 (25 μM) (C) nor SR3677 (25 μM) (D) affected the OT-induced responses in pRLC, but both abolished the responses in ppRLC (n = 4; P < 0.01 by two-way anova), in a pattern identical to g-H (Fig.3).
Comparison of the contribution of MLCK and ROK to phosphorylation of RLC in uterine myocytes
The remaining studies focused on further understanding this unique mechanism initiating contractions in hUM. First, we evaluated the relative contribution of MLCK and ROK to phosphorylation of RLC in hUM using the MLCK inhibitor ML7 22. Treatment with ML7 (25 μM) reduced both pRLC (to 71 ± 8%; P < 0.05) and ppRLC (to 65 ± 6%; P < 0.05) in resting hUM (the vehicle (V) points in Fig.5A and B, left panels). In the presence of ML7, the response to OT was maintained at a lower level for pRLC, but lost completely for ppRLC. Addition of g-H (1 μM; Fig. 5A and 5B, right panels) further reduced the basal levels of pRLC and ppRLC. In the presence of ML7 and g-H, there remained a significant response to OT for pRLC, but not ppRLC. Again, assessment using the total p19RLC Ab confirmed these findings (Fig.5C). The hUM responses to ET-1 in the presence of ML7 were identical to the responses to OT (data not shown).
Fig 5.
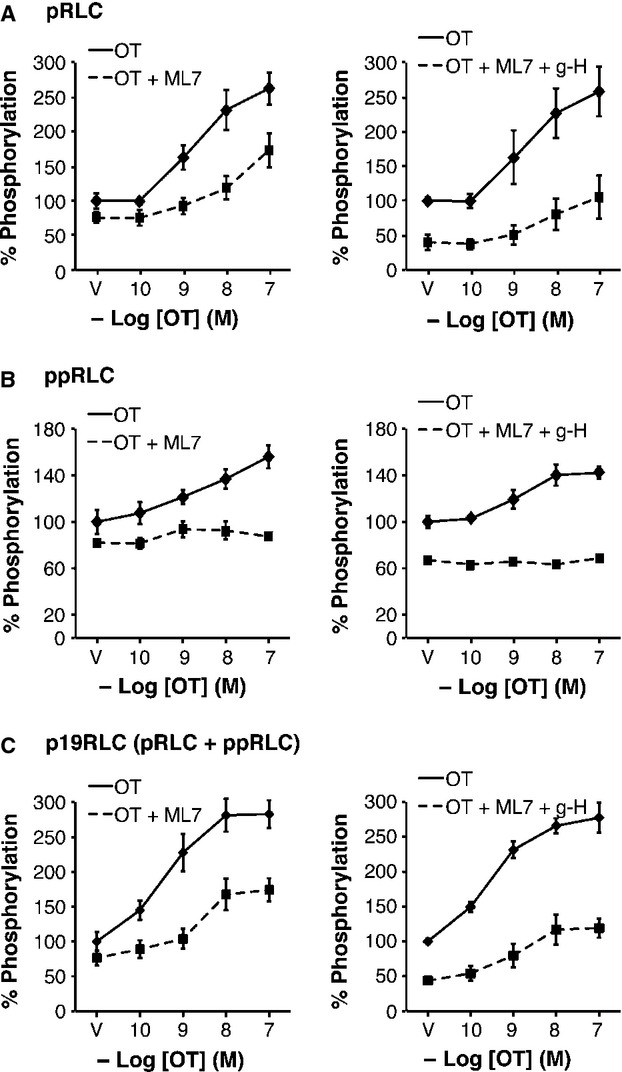
Effects of inhibition of MLCK alone and combined with inhibition of rho-associated kinase (ROK) on OT-stimulated phosphorylation of regulatory light chain (RLC) in hUM. (A) Inhibition of MLCK using ML7 (25 μM) caused significant reduction in the concentrations of pRLC in response to OT (n = 4; P < 0.01 by two-way anova). Addition of g-H (1 μM) further reduced the basal pRLC level, but the response to OT was not altered compared to ML7 alone (n = 4; P < 0.01 by two-way anova). (B) Treatment with ML7 with or without g-H abolished the OT-stimulated response in ppRLC. (C) The responses assessed using the total p19RLC antibody confirmed the findings in panels A and B, again indicating that the primary effect of inhibition of MLCK is in decreasing phosphorylation at S19.
Activation of RhoA in hUM causes increased diphosphorylation of RLC
To further explore activation of rhoA-ROK downstream of OT stimulation in hUM, we activated rhoA using the rho-protein activator calpeptin (Calp). Both OT and Calp significantly increased rhoA activity, Calp significantly more than OT (Fig.6A). Treatment with Calp caused a modest, but significant increase in pRLC (Fig.6B) and a large increase in ppRLC (Fig.6C). Treatment with ML7 had no significant effect on calp-stimulated pRLC (Fig.6B), which is in contrast to the previous studies using OT as a stimulant (Fig.5A). However, inhibition of MLCK or ROK caused a significant reduction in Calp-stimulated ppRLC (Fig.6C). ROK inhibits MLCP through phosphorylation of its targeting subunit, MYPT1, most commonly at T696 or T853 23,24. In hUM, the primary MYPT1 target for ROK appears to be T853 because both Calp and g-H significantly affected phosphorylation at this site, but not at T696 (Fig.7).
Fig 6.
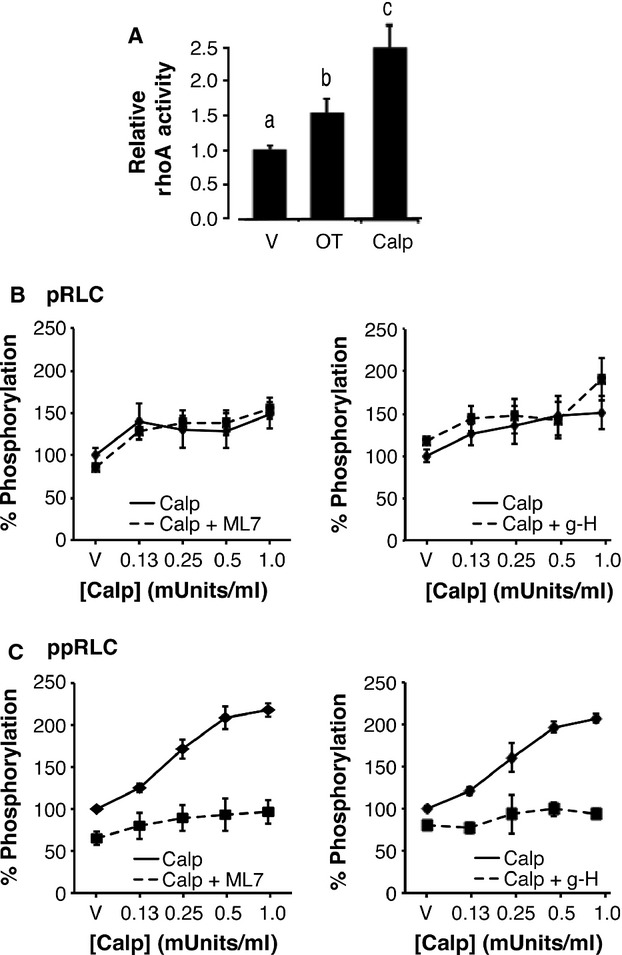
Effects of activation of rhoA on phosphorylation of regulatory light chain (RLC) in hUM. (A) Activation of rhoA, assessed by measuring GTP-bound rhoA, was significantly increased by OT (100 nM) and by the rhoA activator Calpeptin (Calp; 0.5 units/ml). These data have been normalized to rhoA peptide content assessed using Western blot. (n = 4; histograms with different superscripts are significantly different from each other using one-way anova and post hoc Tukey test). (B) Treatment with Calp caused an increase in pRLC that was not significantly affected by inhibition of MLCK using ML7 (25 μM) or inhibition of ROK using g-H (1 μM). (C) In contrast, the Calp-stimulated responses in ppRLC were significantly decreased with either ML7 or g-H (1 μM) (n = 4–8; P < 0.01 by two-way anova).
Fig 7.
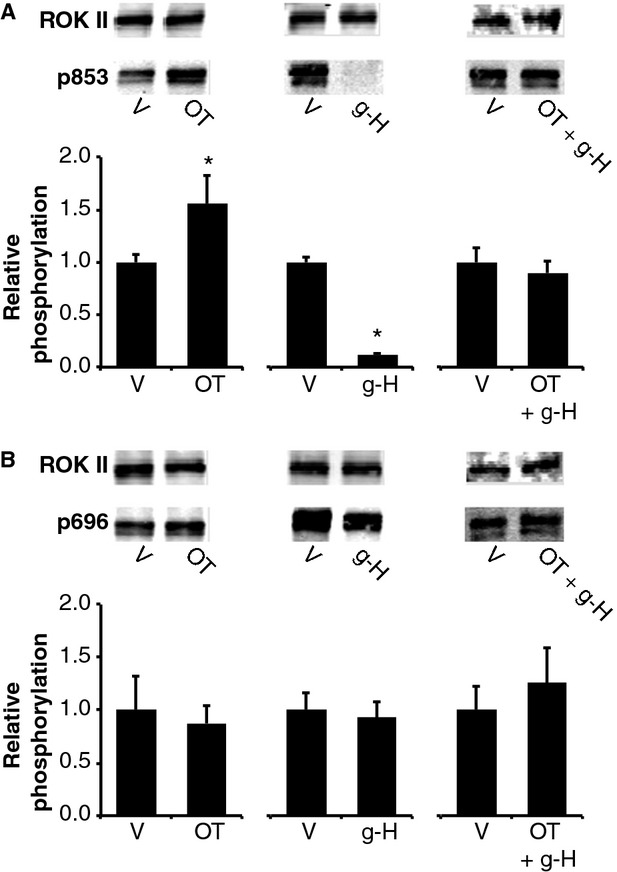
Effects of OT, Calpeptin and g-H on phosphorylation of MYPT1 in hUM. (A) Phosphorylation of MYPT1 at T853 was significantly increased by OT and Calp and was significantly decreased by the ROK inhibitor g-H (n = 4–6; P < 0.05 compared to corresponding vehicle controls (V) using unpaired t-test, as indicated by *). Representative Western blots are indicated above the histograms. The data were normalized to ROK 2, which was used as the loading control. (B) There was no effect of OT, Calpeptin or g-H on phosphorylation of MYPT1 at T696.
ROK-mediated inhibition of Protein Phosphatase 1 contributes to OT-induced diphosphorylation of RLC
To further explore the role of MLCP, we utilized the protein phosphatase inhibitor calyculin A (CalA), which inhibits phosphoprotein phosphatase 1 (PP1, including MLCP) and 2A (PP2A) 25. The resting concentration of ppRLC is particularly sensitive to CalA. At 25 nM (highest concentration used), CalA alone had relatively little effect on pRLC, but a significant enhancement in ppRLC (Fig.8A). In the presence of constant g-H at 1 μM, the basal concentrations of pRLC and ppRLC were reduced but the responses to CalA were largely unaltered. In contrast, in the presence of constant CalA (25 nM; Fig.8B), the concentration-response curves to g-H revealed a markedly different pattern. In agreement with Figure1, g-H caused a concentration-dependent reduction in pRLC and ppRLC. However, in the presence of CalA, there was a slight increase in resting pRLC (the vehicle (V) point) and g-H caused a further increase in pRLC. In contrast, 25 nM CalA caused a massive increase in resting ppRLC (as in Fig.8A). Addition of g-H caused a reduction that was similar to what occurred in the absence of CalA. Unexpectedly, the constant presence of CalA diminished the OT-induced response in pRLC (Fig.8C). In contrast, constant CalA appears to maximize the cellular capacity to generate ppRLC such that OT-stimulation cannot elevate ppRLC beyond the level of CalA alone. This is compatible with the CalA-induced effects noted in Figure8A and B. Interestingly, the CalA-induced reduction in pRLC response to OT was reversed by the addition of g-H (Fig.8D). Again, CalA maximized ppRLC concentrations and there were no significant changes with OT and g-H. To confirm that these CalA effects are attributable to protein phosphatase 1 (PP1), and therefore to MLCP activity, rather than to PP2, we tested two PP2-specific inhibitors (endothall and cantharidic acid) 26. Neither of these agents reproduced the changes in phosphorylated RLC observed with CalA (Fig.9).
Fig 8.
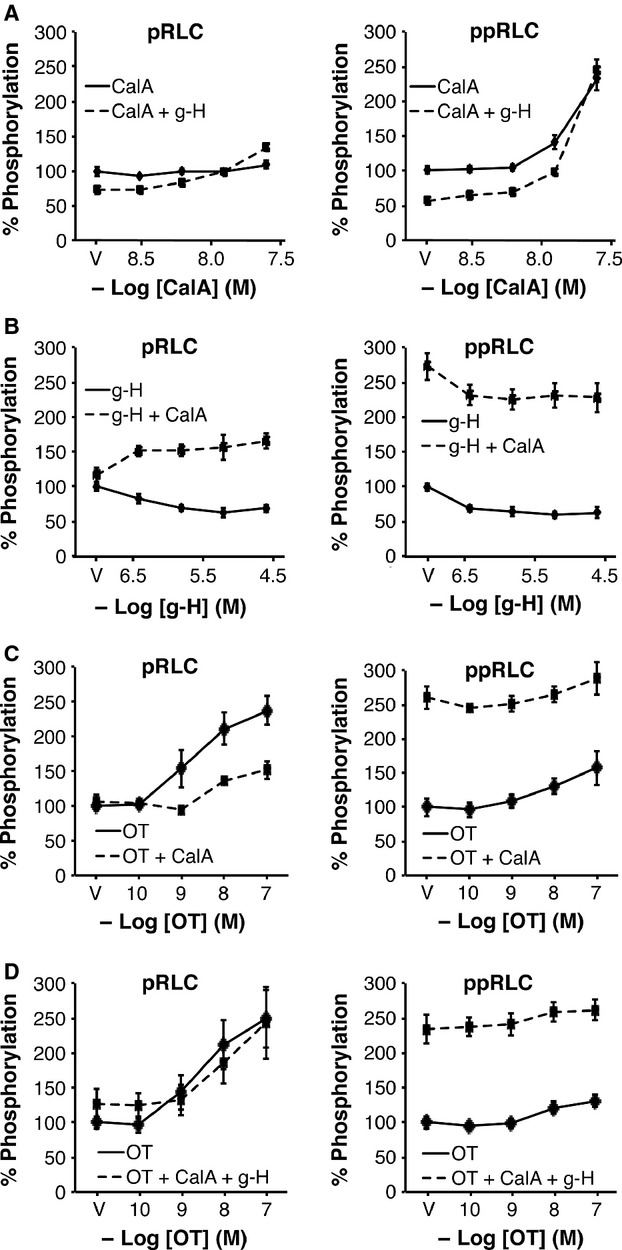
Effects of inhibition of phosphoprotein phosphatase type1 inhibition with or without ROK inhibition on basal and OT-stimulated phosphorylation of regulatory light chain (RLC) in hUM. (A) Inhibition of PP1 using Calyculin A (CalA) alone had no significant effect on basal levels of pRLC. In the presence of the ROK inhibitor g-H (1 μM), there was a slight, but significant increase in pRLC (n = 8, P < 0.01 by two-way anova). However, CalA caused a significant increase in ppRLC. Treatment with g-H suppressed basal ppRLC, but the response to increasing CalA was maintained (n = 4–8; P < 0.01 by two-way anova). (B) Treatment with g-H caused a concentration-dependent suppression of pRLC and ppRLC. However, in the presence of constant CalA (25 nM), increasing g-H caused a significant increase in pRLC. Conversely, in the absence of g-H, CalA caused a large increase in ppRLC concentrations (the vehicle (V) point in the right panel) and increasing g-H resulted in a significant suppression of this ppRLC. (C) CalA (25 nM) caused significant suppression of the OT-induced response in pRLC. CalA again cause a massive increase in ppRLC with only a small although significant increase in response to OT. (D) In the presence of inhibition of both PP1 and ROK, there was no significant change in the OT-induced increase in pRLC in response to OT. Again, treatment with CalA markedly enhanced ppRLC and OT was unable to stimulate further significant increase. (n = 4–8; all analyses used two-way anova).
Fig 9.
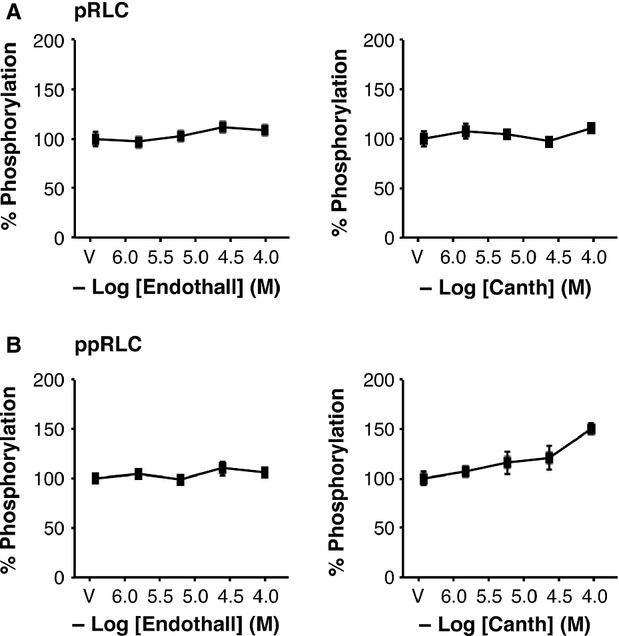
Effects of inhibition of phosphoprotein phosphatase type 2 on phosphorylation of regulatory light chain (RLC) in hUM. Inhibition of PP2 using endothall or Cantharidic acid (Canth) had no significant effect on concentrations of pRLC (A) or ppRLC (B) with the exception that the maximal concentration of Canth resulted in a small increase in ppRLC (n = 4; P < 0.05 by one-way anova).
Sub-cellular distribution of diphosphorylated RLC in uterine myocytes
The above data strongly suggest that ppRLC is an important and rapidly inducible endpoint downstream of OT in hUM. The cellular distribution of OT-stimulated ppRLC indicates that it co-localizes with filamentous actin signifying proximity to the contractile machinery (Fig. 10). Our experiments have extended and integrated previous findings obtained using ‘idealized’ cell-free systems into a novel concept regarding regulation of contractility in hUM. Obviously, additional studies are required to extend these finding from our in vitro cell-based systems to ex vivo and in vivo settings. In preliminary studies, we have used ex vivo myobath techniques with strips of human myometrium obtained from caesarean sections at term prior to labour onset to demonstrate that g-H (1 μM) caused a right shift in the concentration-response curve for OT with an increase in the EC50 from 1.3 to 27.1 nM (Fig. 11A) and a decrease in total tension generated (area under the curve: AUC) to 46 ± 15% of vehicle-treated controls (Fig. 11B).
Fig 10.
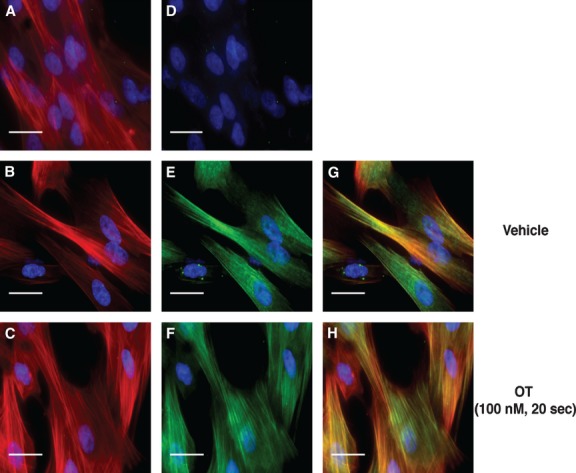
Sub-cellular localization of diphosphorylated regulatory light chain (RLC) in hUM. Immunofluorescent staining of hUM. (A–C) Filamentous actin (F-actin) stained with rhodamine-phalloidin (red). (D–F) Identical fields corresponding to panels A–C showing staining with a secondary Ab conjugated to Alexa-Fluor 488 (green). In panel D, the primary Ab has been omitted (secondary Ab only). In panels E and F, the anti-ppRLC Ab was used. (G) and (H) Merged images after combining panels B and E, or C and F respectively. Panels B, E and G correspond to vehicle (V) treatment, and panels C, F and H to cells stimulated with OT (100 nM). In all panels, nuclei are stained with DAPI (blue). Images are shown at 400× magnification. White bars represent 25 micrometres.
Fig 11.
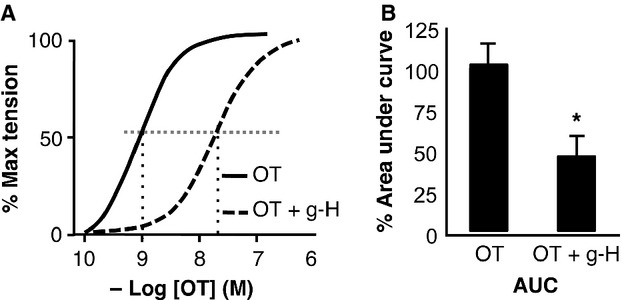
Effects of inhibition of ROK on activity of human uterine strips ex vivo. Concentration-response curves of uterine strips to OT. The calculated EC50 was 1.3 nM, but in the presence of the ROK inhibitor g-H (1 μM) the EC50 was increased to 27.1 nM. There was a corresponding significant decrease (to 46 ± 15% as indicated by *) in the area under the response curve (n = 4; unpaired t-test).
Discussion
The data from these studies strongly support the concept that there are distinct pathways that regulate the contractile mechanisms of uterine and vascular myocytes. The major difference is that, in hUM, pRLC resulting from the activity of MLCK is used as a substrate for ROK-mediated phosphorylation to ppRLC. As ppRLC generates threefold greater energy for the contractile mechanism 4,5, this unique property might represent a basic evolutionary characteristic that is generalizable to all myocytes derived from smooth muscle tissues with phasic patterns. This would facilitate the physiological phenotype of phasic contractions – high amplitude contractions occurring at episodic intervals. In contrast, myocytes with tonic contractile patterns, such as hVM require prolonged contractions with relatively small changes in amplitude over time. Such speculation is supported by reports showing that pRLC corresponds linearly with tension development in hVM 27 and that ppRLC in hVM appears to be less important than pRLC 28–30.
These pathways were elucidated through the use of combinations of physiological agonists and pharmacological inhibitors. The most informative experiments were those measuring the responses to agonist stimulation in the presence of inhibition of ROK. Glycyl-H abolished the ppRLC response to OT in hUM cells, indicating that ppRLC was dependent on ROK. However, the response of pRLC to OT was maintained, strongly supporting our interpretation that ROK is the mediator of the conversion from pRLC to ppRLC. The increase in pRLC was expected because OT is known to stimulate MLCK activity and to increase phosphorylation of RLC to pRLC. It also is known that ROK inhibits MLCP activity 23 and therefore treatment with g-H should result in greater phosphatase activity and reduced pRLC concentrations. The fact that the pRLC response to OT was normal in the presence of g-H indicates that the OT-induced, ROK-mediated increase in MLCP activity was balanced by the decreased utilization of pRLC for conversion to ppRLC. These interpretations were confirmed by use of the antibody assessing total phosphorylation of RLC at S19, indicating that g-H-induced suppression of ppRLC resulted solely from reduced phosphorylation of RLC at T18 with preservation of phosphorylation of RLC at S19.
Our previous studies of unstimulated hUM demonstrated that approximately 47% of the pool of RLC is represented by unphosphorylated RLC, 36% pRLC and 16% by ppRLC. This is compatible with other studies 31,32 that confirm the proportion of ppRLC in hUM is two to fivefold greater than the estimated 3–11% in arterial and other types of smooth muscle. Furthermore, we showed that ppRLC reaches 35–45% following stimulation with OT 14.
In contrast to the hUM, in hVM there was essentially no increase in ppRLC in response to the physiological agonist ET-1 and the increased pRLC was abolished by inhibition of ROK-limited activity of MLCP. By comparison, when ET-1 was used as the agonist for hUM, the responses were similar to those noted with OT. These results indicate that the response to the agonist is dependent on the myocyte phenotype and not on the agonist.
There are two known isoforms of ROK that appear to represent genetic duplication 33. The ROK inhibitor (g-H) that we used does not distinguish between them 21. However, ROK inhibitors designed to select for each of the ROK isoforms worked equally well indicating that the observed effects can be mediated by either isoform.
The experiments using the MLCK inhibitor ML7 confirm the importance of this pathway in synthesis of pRLC. With MLCK blockade, the OT-induced response of pRLC was decreased. The response in ppRLC even more decreased, presumably because of lack of pRLC substrate.
Together, these data suggest that in hUM, ROK participates downstream of MLCK in the sequential phosphorylation of RLC. They support previous evidence that MLCK is the primary enzyme mediating phosphorylation of RLC at S19 to yield pRLC, and here we suggest that this initial step permits a second ROK-mediated phosphorylation of T18 to yield ppRLC, which is physiologically relevant in phasic smooth muscle. Thus, we propose the concept that the primary substrate for ROK in hUM is pRLC. Furthermore, the data strongly support the concept that physiological uterine agonists signal through the classic Ca2+-dependent pathway linked through Gαq 11 subunits 34 in addition to activation of the Ca2+-independent rhoA-ROK pathway linked through Gα12/13 35.
The data also add clarity to the role of ROK in inhibition of MLCP. They demonstrate that the ROK inhibits activity of the holoenzyme by phosphorylation of MYPT1 at T853. This confirms recent studies by Hudson et al. using whole uterine tissues 36. These latter studies also noted the correlation between ROK activity, uterine contractions and concentrations of ppRLC. This suggests that the findings from our cell culture methodology likely also apply to whole intact tissues. More studies will be required to explore these pathways using ex vivo and in vivo approaches.
The effects of inhibition of PP1 on OT-stimulated phosphorylation of RLC demonstrate an unexpectedly strong ROK-dependent drive towards ppRLC in hUM that is held in balance by MLCP activity, and which is uncovered by pharmacological inhibition of MLCP. These studies suggest that the increased ppRLC observed during uterine stimulation is due primarily to ROK-mediated synthesis of ppRLC rather than inhibition of MLCP activity. On the other hand, inhibition of MLCP activity may be the predominant mechanism for regulating basal concentrations of pRLC, and by extension, ppRLC. In the presence of OT stimulation in addition to inhibition of MLCP, we interpret the data as demonstrating a diminished response in pRLC because of the increased ROK-mediated conversion of pRLC to ppRLC, thus depleting pRLC concentrations.
The experiments using Calp to activate RhoA provided greater stimulation of RhoA activity than OT and caused greater ROK-mediated stimulation of ppRLC. Stimulation of hUM using Calp in the presence of inhibition of ROK demonstrated attenuated Calp-induced ppRLC. The maintenance of the increase in pRLC in the presence of the inhibitor of MLCK can be explained by the diminished synthesis of pRLC being offset by the Calp-mediated increased inhibition of MLCP resulting in a moderate increase in pRLC. In the presence of inhibition of ROK, the decrease in ppRLC was more predominant than pRLC, this supports our interpretation above – that the principal activity of ROK is phosphorylation of pRLC at T18.
The immunofluorescence studies also point to fundamental differences between the roles of ppRLC in hUM and hVM. In our studies, OT-stimulated ppRLC co-localizes with filamentous actin, signifying proximity to the contractile machinery. This pattern is distinct from that observed in cultured rabbit aortic myocytes where stimulation with PGF2α resulted in a moderate enhancement in ppRLC that was localized at the cell periphery 37. These findings support a role for ppRLC functioning in hUM as a trigger to contraction whereas in rabbit aorta, ppRLC might act in cytoskeletal remodelling associated with cell migration 38,39.
Our preliminary ex vivo studies showing that ROK inhibition causes significant inhibition of contractile activity in human uterine strips are in agreement with previous studies in mouse, rat and human uterine smooth muscle (reviewed in 40). From the perspective of translating this to the human in vivo situation, our findings of the marked increase in sensitivity of the ppRLC response to ROK inhibition in the ICW assay compared to the pRLC response in either hVM or hUM are important and in agreement with previous studies in cell-free systems 37. They suggest that, at a specific concentration of ROK inhibitor in vivo, it is possible to significantly inhibit uterine activity without affecting vascular smooth muscle function. Obviously this will require further study in ex vivo and in vivo experiments.
In conclusion, our data support the conclusion that hUM might possess fundamentally different mechanisms regulating contractile activity than do hVM. We have attempted to illustrate our conclusions in Figure12. We propose that this might represent adaptations driven by differing physiological needs of smooth muscle that have phasic versus tonic contractile patterns. We propose that human uterine myocytes respond to stimulation predominantly by increasing concentrations of ppRLC formed by ROK-mediated phosphorylation of MLCK-derived pRLC. This mechanism does not appear to be a major pathway in vascular myocytes derived from human coronary artery. The most important human condition arising from uterine myocyte dysfunction is preterm parturition, which is associated with approximately 75% of newborn mortality and life-long disability. Current attempts to prevent or arrest preterm labour have failed to influence this poor outcome, often because vascular side effects prohibit use of effective concentration of uterine smooth muscle inhibitors. Our current findings provide new understanding of uterine myocyte function and suggest that treatment with low concentrations of ROK inhibitors might permit the management of preterm labour while avoiding significant side effects in other smooth muscle beds.
Fig 12.
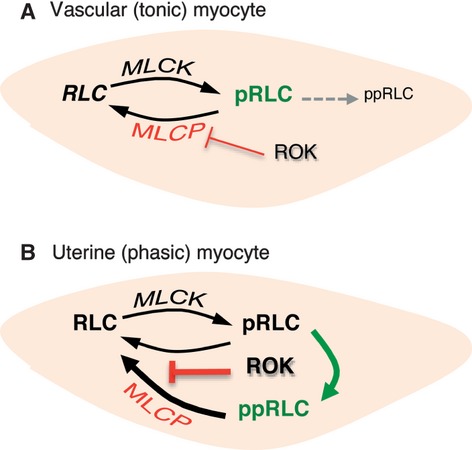
Proposed distinguishing mechanisms of phosphorylation of regulatory light chain (RLC) in tonic and phasic smooth muscle. (A) Myocytes from smooth muscle with tonic patterns of contractile activity (hVM) initiate contractions by simple phosphorylation of RLC at S19. This is predominantly regulated by Ca2+-dependent activation of MLCK. Return to a lower contractile state can be accomplished by increasing activity of MLCP, which is negatively regulated by ROK. (B) For myocytes obtained from smooth muscle with a phasic pattern of contractility (hUM), contraction is also initiated by MLCK-mediated increase in pRLC, but this is augmented by ROK-mediated phosphorylation of pRLC at T18 to form ppRLC, which enhances energy production from myosin ATPase. In addition, increasing ROK activity strongly inhibits the phosphatase activity of MLCP that is predominantly directed towards ppRLC.
Acknowledgments
This research was supported by the Canadian Institutes of Health Research (#MOP 81384, http://www.cihr-irsc.gc.ca/e/) and Alberta Innovates - Health Solutions (http://www.aihealthsolutions.ca/). The funders had no role in study design, data collection and analysis, decision to publish, or in preparation of the manuscript.
Conflict of interest statement
The authors confirm that there are no conflicts of interest.
References
- Kamm KE, Stull JT. The function of myosin and myosin light chain kinase phosphorylation in smooth muscle. Annu Rev Pharmacol Toxicol. 1985;25:593–620. doi: 10.1146/annurev.pa.25.040185.003113. [DOI] [PubMed] [Google Scholar]
- Longbottom ER, Luckas MJ, Kupittayanant S, et al. The effects of inhibiting myosin light chain kinase on contraction and calcium signalling in human and rat myometrium. Pflugers Arch. 2000;440:315–21. doi: 10.1007/s004240000305. [DOI] [PubMed] [Google Scholar]
- Takashima S. Phosphorylation of myosin regulatory light chain by myosin light chain kinase, and muscle contraction. Circ J. 2009;73:208–13. doi: 10.1253/circj.cj-08-1041. [DOI] [PubMed] [Google Scholar]
- Ikebe M, Inagaki M, Kanamaru K, et al. Phosphorylation of smooth muscle myosin light chain kinase by Ca2+-activated, phospholipid-dependent protein kinase. J Biol Chem. 1985;260:4547–50. [PubMed] [Google Scholar]
- Ikebe M, Koretz J, Hartshorne DJ. Effects of phosphorylation of light chain residues threonine 18 and serine 19 on the properties and conformation of smooth muscle myosin. J Biol Chem. 1988;263:6432–7. [PubMed] [Google Scholar]
- Ikebe M, Hartshorne DJ. Phosphorylation of smooth muscle myosin at two distinct sites by myosin light chain kinase. J Biol Chem. 1985;260:10027–31. [PubMed] [Google Scholar]
- Amano M, Ito M, Kimura K, et al. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) J Biol Chem. 1996;271:20246–9. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- Kureishi Y, Kobayashi S, Amano M, et al. Rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. J Biol Chem. 1997;272:12257–60. doi: 10.1074/jbc.272.19.12257. [DOI] [PubMed] [Google Scholar]
- Wilson DP, Sutherland C, Borman MA, et al. Integrin-linked kinase is responsible for Ca2+-independent myosin diphosphorylation and contraction of vascular smooth muscle. Biochem J. 2005;392:641–8. doi: 10.1042/BJ20051173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupittayanant S, Burdyga T, Wray S. The effects of inhibiting Rho-associated kinase with Y-27632 on force and intracellular calcium in human myometrium. Pflugers Arch. 2001;443:112–4. doi: 10.1007/s004240100668. [DOI] [PubMed] [Google Scholar]
- Woodcock NA, Taylor CW, Thornton S. Prostaglandin F2alpha increases the sensitivity of the contractile proteins to Ca2+ in human myometrium. Am J Obstet Gynecol. 2006;195:1404–6. doi: 10.1016/j.ajog.2006.03.099. [DOI] [PubMed] [Google Scholar]
- Woodcock NA, Taylor CW, Thornton S. Effect of an oxytocin receptor antagonist and rho kinase inhibitor on the [Ca++]i sensitivity of human myometrium. Am J Obstet Gynecol. 2004;190:222–8. doi: 10.1016/s0002-9378(03)00925-6. [DOI] [PubMed] [Google Scholar]
- Tahara M, Morishige K, Sawada K, et al. RhoA/Rho-kinase cascade is involved in oxytocin-induced rat uterine contraction. Endocrinology. 2002;143:920–9. doi: 10.1210/endo.143.3.8696. [DOI] [PubMed] [Google Scholar]
- Aguilar HN, Tracey CN, Tsang SC, et al. Phos-tag-based analysis of Myosin regulatory light chain phosphorylation in human uterine myocytes. PLoS ONE. 2011;6:1–10. doi: 10.1371/journal.pone.0020903. : e20903, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar HN, Zielnik B, Tracey CN, et al. Quantification of rapid Myosin regulatory light chain phosphorylation using high-throughput in-cell Western assays: comparison to Western immunoblots. PLoS ONE. 2010;5:1–11. doi: 10.1371/journal.pone.0009965. : e9965, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg RL, Culhane JF, Iams JD, et al. Epidemiology and causes of preterm birth. Lancet. 2008;371:75–84. doi: 10.1016/S0140-6736(08)60074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood NS, Marlow N, Costeloe K, et al. Neurologic and developmental disability after extremely preterm birth. EPICure Study Group. New Eng J Med. 2000;343:378–84. doi: 10.1056/NEJM200008103430601. [DOI] [PubMed] [Google Scholar]
- McCormick MC, Behrman RE. The quiet epidemic of premature birth: commentary on a recent Institute of Medicine report. Ambul Pediatr. 2007;7:8–9. doi: 10.1016/j.ambp.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Rubens CE, Cesar MGG, Victora G, Nunes TM, et al. Global report on preterm birth and stillbirth (7 of 7): mobilizing resources to accelerate innovative solutions (Global Action Agenda) BMC Preg Childbirth. 2010;10:S7–26. doi: 10.1186/1471-2393-10-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur P, Taggart MJ, Zielnik B, et al. Relationship between gene expression and function of uterotonic systems in the rat during gestation, uterine activation and both term and preterm labour. J Physiol. 2008;586:6063–76. doi: 10.1113/jphysiol.2008.164004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Suzuki M, Hidaka H. The novel and specific Rho-kinase inhibitor (S)-(+)-2-methyl-1-[(4-methyl-5-isoquinoline)sulfonyl]-homopiperazine as a probing molecule for Rho-kinase-involved pathway. Pharmacol Ther. 2002;93:225–32. doi: 10.1016/s0163-7258(02)00191-2. [DOI] [PubMed] [Google Scholar]
- Ono-Saito N, Niki I, Hidaka H. H-series protein kinase inhibitors and potential clinical applications. Pharmacol Ther. 1999;82:123–31. doi: 10.1016/s0163-7258(98)00070-9. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–8. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–58. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Martin BL, Brautigan DL, et al. Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun. 1989;159:871–7. doi: 10.1016/0006-291x(89)92189-x. [DOI] [PubMed] [Google Scholar]
- Li YM, Casida JE. Cantharidin-binding protein: identification as protein phosphatase 2A. Proc Nat Acad Sci USA. 1992;89:11867–70. doi: 10.1073/pnas.89.24.11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Itoh T. Effects of calyculin A on tension and myosin phosphorylation in skinned smooth muscle of the rabbit mesenteric artery. Br J Pharmacol. 1993;109:703–12. doi: 10.1111/j.1476-5381.1993.tb13631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnett KM, Biancani P. Calcium-dependent and calcium-independent contractions in smooth muscles. Am J Med. 2003;115(Suppl. 3A):24S–30S. doi: 10.1016/s0002-9343(03)00232-8. [DOI] [PubMed] [Google Scholar]
- Ogut O, Brozovich FV. Regulation of force in vascular smooth muscle. J Mol Cell Cardiol. 2003;35:347–55. doi: 10.1016/s0022-2828(03)00045-2. [DOI] [PubMed] [Google Scholar]
- Hirano K, Hirano M, Kanaide H. Regulation of myosin phosphorylation and myofilament Ca2+ sensitivity in vascular smooth muscle. J Smooth Muscle Res. 2004;40:219–36. doi: 10.1540/jsmr.40.219. [DOI] [PubMed] [Google Scholar]
- Miller-Hance WC, Miller JR, Wells JN, et al. Biochemical events associated with activation of smooth muscle contraction. J Biol Chem. 1988;263:13979–82. [PubMed] [Google Scholar]
- Colburn JC, Michnoff CH, Hsu LC, et al. Sites phosphorylated in myosin light chain in contracting smooth muscle. J Biol Chem. 1988;263:19166–73. [PubMed] [Google Scholar]
- Tamura N, Itoh H, Ogawa Y, et al. cDNA cloning and gene expression of human type Ialpha cGMP-dependent protein kinase. Hypertension. 1996;27:552–7. doi: 10.1161/01.hyp.27.3.552. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–25. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Chikumi H, Gutkind JS. RGS-containing RhoGEFs: the missing link between transforming G proteins and Rho? Oncogene. 2001;20:1661–8. doi: 10.1038/sj.onc.1204182. [DOI] [PubMed] [Google Scholar]
- Hudson CA, Heesom KJ, Lopez Bernal A. Phasic contractions of isolated human myometrium are associated with Rho-kinase (ROCK)-dependent phosphorylation of myosin phosphatase-targeting subunit (MYPT1) Mol Hum Reprod. 2012;18:265–79. doi: 10.1093/molehr/gar078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurada K, Seto M, Sasaki Y. Dynamics of myosin light chain phosphorylation at Ser19 and Thr18/Ser19 in smooth muscle cells in culture. Am J Physiol. 1998;274:C1563–72. doi: 10.1152/ajpcell.1998.274.6.C1563. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Horwitz AR. Myosin light chain mono- and di-phosphorylation differentially regulate adhesion and polarity in migrating cells. Biochem Biophys Res Commun. 2010;402:537–42. doi: 10.1016/j.bbrc.2010.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchimura T, Fumoto K, Yamamoto Y, et al. Spatial localization of mono-and diphosphorylated myosin II regulatory light chain at the leading edge of motile HeLa cells. Cell Struct Funct. 2002;27:479–86. doi: 10.1247/csf.27.479. [DOI] [PubMed] [Google Scholar]
- Aguilar HN, Mitchell BF. Physiological pathways and molecular mechanisms regulating uterine contractility. Hum Reprod Update. 2010;16:725–44. doi: 10.1093/humupd/dmq016. [DOI] [PubMed] [Google Scholar]