

Abstract
Progressive cardiomyopathy is a major cause of death in Duchenne muscular dystrophy (DMD) patients. Coupling between Ca2+ handling and contractile properties in dystrophic hearts is poorly understood. It is also not clear whether developing cardiac failure is dominated by alterations in Ca2+ pathways or more related to the contractile apparatus. We simultaneously recorded force and Ca2+ transients in field-stimulated papillary muscles from young (10–14 weeks) wild-type (wt) and dystrophic mdx mice. Force amplitudes were fivefold reduced in mdx muscles despite only 30 % reduction in fura-2 ratio amplitudes. This indicated mechanisms other than systolic Ca2+ to additionally account for force decrements in mdx muscles. pCa-force relations revealed decreased mdx myofibrillar Ca2+ sensitivity. ‘In vitro’ motility assays, studied in mdx hearts here for the first time, showed significantly slower sliding velocities. mdx MLC/MHC isoforms were not grossly altered. Dystrophic hearts showed echocardiography signs of early ventricular wall hypertrophy with a significantly enlarged end-diastolic diameter ‘in vivo’. However, fractional shortening was still comparable to wt mice. Changes in the contractile apparatus satisfactorily explained force drop in mdx hearts. We give first evidence of early hypertrophy in mdx mice and possible mechanisms for already functional impairment of cardiac muscle in DMD.
Keywords: papillary muscle, muscular dystrophy, calcium, force transients, motility assay
Introduction
Duchenne muscular dystrophy is a progressive, wasting muscle disease that originates from numerous point mutations in the X-chromosomal DMD gene. Its product, dystrophin, is predominantly expressed in skeletal, cardiac and smooth muscle. Lack of dystrophin renders muscle fibres more susceptible to membrane damage during mechanical stress, especially for stretched contractions. Although skeletal muscle wasting clinically predominates, heart failure from progressive dilative cardiomyopathy (DCM) accounts for at least 30 % of deaths. However, many individuals show subclinical cardiac involvement presumably because of reduced cardiac workload in the mobility compromised patients 1. Also, a direct cardiac correlate with eccentric contractions found in skeletal muscle is usually not present during pump cycles. A distinct effect of dystrophin deficiency can be expected in the heart, as its distribution is different from the one in skeletal muscle, i.e. a large proportion at the t-system level. It has long been thought that clinical manifestations of dystrophic cardiomyopathy were primarily a matter in aged DMD patients 1. Accordingly, studies in young dystrophic mdx mice (2–3 mo) showed low levels of cardiac fibrosis, but a several-fold increase at older age (9–12 mo, 2; 17 mo, 3). Although no signs of heart failure were observed in young mdx mice 2, the disruption of the cardiac and skeletal muscle patterning process priming the dystrophic phenotype already begins early in embryonic development 4. In the post-natal phase, mdx mice undergo a sudden onset of massive skeletal muscle degeneration (necrotic phase) at ∽3–5 weeks 5 followed by a stable phase of degeneration/regeneration cycles and a terminal phase of exhausted regenerative capacity from ∽15 mo 6. Such systematic studies are not available for cardiac muscle. Echocardiography studies have detected beginning signs of ventricular hypertrophy in mdx hearts from 29 weeks which were not seen in 8 weeks old mice 3. At 42 weeks, dilated cardiomyopathy with decreased fractional shortening was already apparent 3.
A couple of recent studies point towards altered cardiac Ca2+ handling in adult mdx mice (5–9 months [‘mo’], 7; 9–12 mo, 2), and in mdx mice in the early post-necrotic phase (2–3 mo, 2). Surprisingly, only few studies addressed contractile properties of cardiac muscle from mdx mice, where specific force in young (8–14 weeks) animals was compromised 8,9. Likewise, there are even less studies that have addressed the frequency response of Ca2+ dynamics or force responses in stimulated dystrophic cardiac muscle 8. This is important to know, as diastole progressively shortens with heart rate and may compromise intracellular Ca2+ removal.
To our knowledge, there is no study available that explains compromised cardiac contractile performance in direct correlation with myoplasmic Ca2+ fluctuations or provides further details on the molecular level of the contractile apparatus. Here, we simultaneously recorded force and Ca2+ transients in paced papillary muscle from 10 to 14 weeks old mdx and wt mice to test the hypothesis that impaired Ca2+ amplitudes were the main determinant of compromised force in mdx preparations. Reduced force output of mdx preparations was only partially explained by reduced Ca2+ transients, but rather by reduced myofibrillar Ca2+ sensitivity. Moreover, we detected slower sliding velocities in cardiac ‘in vitro’ motility assays. Although morphological signs of hypertrophy can be already anticipated at this early stage, ‘in vivo’ echocardiography still showed a compensated situation with balanced fractional shortening.
Materials and methods
Papillary muscle preparations
Papillary heart muscle from young 10–14 weeks old mdx and wt mice were used. All experiments complied with guidelines laid down by the Local Animal Care Facilities (Universities Heidelberg & Essen; TG-27/09; IFORES 107-55604). Details on preparation procedures are given in the supporting information (SI Methods).
Force and Ca2+ fluorescence recordings in papillary muscle preparations during external field stimulation
After fura-2 loading, muscle strips were horizontally mounted in a temperature-controlled (37°C) recording chamber with a built-in force transducer (OPT1L; Scientific Instruments, Heidelberg, Germany). Muscle strips were field stimulated by trains of pulses at frequencies from 1 to 4 Hz. Prior to the first stimulation, papillary muscles were allowed to equilibrate for ∽20 min. Initial stimulations were performed at 1 Hz with a pulse duration of 6–10 ms at 3 V. At the time of stimulation, resting metabolism was expected to be already at a very low steady-state level 10. The muscle was gradually stretched until force transient amplitudes reached a maximum. All subsequent recordings were made at this length.
Repetitive trains of stimuli starting at 1 Hz and increasing frequency at 0.5 Hz intervals up to 4 Hz were applied and force and fura-2 twitch transients recorded for 10 sec. at each stimulation frequency. In between each train, a 5 min. rest period was allowed to ensure sufficient oxygenation to the muscle core 11. Maximum twitch force amplitudes, time-to-peak (TTP) and the time constant τdec of exponential force relaxation were evaluated (10–90% rise time or exponential fit to the decay of the traces, respectively).
Fura-2 Ca2+ transients were simultaneously recorded via an epifluorescence system (PH1A; Scientific Instruments) by dual wavelength excitation. The collected net fluorescence covered a region of interest ∽0.3 mm² in the middle of the muscle. Fura-2 ratio amplitudes, systolic and diastolic levels, activation kinetics (time-to-peak) and inactivation kinetics (τdec) were analysed for each frequency. As we were primarily interested in the frequency dependence of Ca2+ dynamics in the same papillary muscle, intracellular [Ca2+]i is presented by the fura-2 ratios rather than converting to absolute Ca2+ concentrations which normally requires careful ‘in situ’ calibration 12. As buffer properties are not supposed to change within the same preparation for different stimulation regimes, it is practical to represent Ca2+ levels by fura-2 ratios, for which we did not attempt to perform further calibration. Even if there was some difference in fura-2 Kd between genotypes 12, where appropriate, we compare frequency dependence of fura-2 signals normalized to the values obtained at 1 Hz to eliminate such a difference.
Myofibrillar Ca2+ sensitivity in skinned papillary muscle preparations
pCa-force experiments were carried out in skinned papillary muscles bathed in solutions mimicking the intracellular environment of varying pCa and recordings of steady-state force. Further details are given in the supporting information (SI Methods).
In vitro motility assay of ventricular muscle myosin extracts
Ventricles were dissected from the heart of mdx or wt mice and ∽150 mg pieces transferred to a Guba-Straub phosphate extraction buffer at pH 6.5, immediately frozen in liquid N2 and stored until experimentation. The pyrophopsphate-based myosin extraction followed a multi-step high-speed centrifugation procedure with solution composition described earlier 13. Briefly, small pieces of heart were stirred for 20 min. in ice-cold protein extraction buffer (mM: NaCl 300, NaH2PO4 100, Na2HPO4 50, MgCl2 1, Na2P2O7 10, EDTA 10, DTT 10, pH 6.5) containing 0.1% NaN3 and 10 μg/ml and then centrifuged at 30,000 g for 10 min. The supernatant was diluted 1:12 in low salt buffer LSB (EDTA 1, Tris 10, pH 7.0) and stirred for another 30 min. followed by a second spin down at 30,000 g for 10 min. Pellets were dissolved in ATP-LSB (EGTA 1, MgCl2 2, ATP 1, DTT 1, Tris 10, ph 7.0), incubated on ice for 30 min. and spun down for another 10 min. The extracted myosin was pipetted onto a flow cell coated with nitrocellulose. The motility assay procedure followed the one given in Svensson et al. 13. After extraction and removal of rigor bridges, 100 ng/ml rhodamin-phalloidin labelled actin was flushed into the chamber and washed out after a 2 min. incubation period. Cross-bridge cycling was initiated by adding 3 mM ATP. The filament sliding was visualized on an inverted epifluorescence microscope. Recordings and automated analysis were performed as before 14. Several independent recordings were made from each flow cell (total of XYT movies sequences: wt 24; mdx 10) from two mdx and wt animals. With several hundred filaments being tracked during each movie sequence, our analysis gives a robust representation of sliding filaments from more than 5000 single filaments. From each movie sequence, the velocity distribution was constructed and fitted by a Gaussian function from which the median velocity values were further analysed for statistical differences. Velocity bins <1 μm/sec. were not included in the fitting procedure as within this range also a number of myosin heads in the rigor configuration may be contained.
SDS PAGE and myosin light and heavy chain distributions
Gel electrophoresis on homogenates of either dissected heart ventricles or atria from wt or mdx mice was performed as described for skeletal muscle 15 with the exception that total protein content added to stacking gels was ∽50 μg for light chain separation (12% polyacrylamide) and ∽10 μg for heavy chains (8%). Samples from wt skeletal edl and soleus muscle were also run on the same gels.
In vivo transthoracic echocardiography in conscious wt and mdx mice
Transthoracic echocardiography was performed in young conscious wt and mdx mice (10–16 weeks) as described in Goehringer et al. 16. Briefly, parasternal short-axis M-mode images were obtained using a 15 MHz linear transducer (Sonos 5500, Philips Medical Syst., Boeblingen, Germany). At least three consecutive beats were used to measure end-diastolic internal diameter (LVEDD), end-systolic internal diameter (LVESD), diastolic posterior wall thickness and diastolic septum thickness. Fractional shortening (FS) is given as (LVEDD-LVESD)/LVEDD.
Statistical analysis
Details on the statistical analysis are given in the supporting information (SI Methods).
Results
Simultaneously recorded force and Ca2+-frequency relations in field-stimulated papillary muscles of young wt and mdx mice
Figure1A shows recordings of force and fura-2 Ca2+ transients in a papillary muscle from a 12-week-old wt mouse stimulated at 1 and 4 Hz. At the higher frequency, force amplitudes drop by 40% while Ca2+ transients are still comparable to low frequency stimulation. The three consecutive force transients shown at 1 and 4 Hz were taken from a series of 10 and 40 successive transients during the 10 sec. stimulations train, respectively, which are both shown in the insets. As can be seen, during the train force amplitudes remained fairly constant arguing against the development of hypoxic cores even at highest stimulation frequencies. Already from 1.5 Hz, force was significantly reduced and further dropped in a sigmoidal manner (Fig.1B). Ca2+ transients showed a different behaviour: although absolute fura-2 ratio amplitudes were about 40% reduced in mdx papillary muscles, their frequency dependence was rather flat, whereas in wt they showed a positive bell shape (Fig.1C). For smaller frequencies, decay time constants of fura-2 transients were somewhat larger in mdx versus wt preparations but approached the wt values for larger frequencies (Fig.1C). In contrast, there was virtually no difference on transient rise time between strains in the frequency range (not shown).
Fig 1.

Simultaneous force and calcium transients in papillary muscle strips from young wild-type (wt) and mdx mice. (A) Example of simultaneously recorded force and fura-2 Ca2+ transients in a wt papillary muscle stimulated at 1 and 4 Hz. The insets show all the force transients during the 10 sec. recording train on an enlarged scale to demonstrate the robustness of the recording during the train. The larger plots show the superposed force and fura-2 ratio transients at the same scale. Note the almost twofold decrease in force amplitudes at 4 Hz while fura-2 ratio amplitudes change very little. Scaling is the same in both cases to emphasize the drop in force amplitudes with larger frequencies. Note that even during the highest stimulation frequency of 4 Hz, the force amplitudes remained on a robust level during the 10 sec. stimulation train as shown in the inset from where the selected magnified transients were taken (white circle). (B) Negative sigmoidal absolute and relative force-frequency relations in wt and mdx papillary muscle preparations show a marked right shift in mdx mice. (C) Fura-2 ratio amplitudes showing a positively bell-shaped relation in wt mice but a flat relationship in mdx mice with amplitudes also only 50% of those in wt mice. For smaller stimulation frequencies, Ca2+ transients decay significantly slower but approach those for wt preparations in mdx mice. *: P < 0.05 at a given frequency within each strain compared with 1 Hz. #: P < 0.05 at a given frequency mdx versus wt.
Myofibrillar Ca2+ sensitivity is reduced in mdx papillary muscles
To search for possible explanations for vastly reduced force despite only a 40% decrease in Ca2+ amplitudes in the mdx samples, we tested a set of different possible contractile parameters. Myofibrillar Ca2+ sensitivity of papillary muscle strips was assessed in different pCa-containing solutions. The steady-state pCa-force relations from 8 wt and 20 mdx preparations clearly showed a marked right shift towards smaller pCa values for papillary muscles from dystrophic mice in the early pre-fibrotic phase (Fig.2). pCa50 values were significantly smaller in mdx muscles: 5.548 ± 0.04 for wt and 5.310 ± 0.03 for mdx (P < 0.001), whereas Hill coefficients were 2.572 ± 0.02 for wt and 2.795 ± 0.27 for mdx (P = 0.63). Maximum forces at pCa = 4.28 were 2.17 ± 0.51 mN in wt and 2.42 ± 0.18 mN in mdx papillary muscles (P =0.22, one-way anova on ranks).
Fig 2.
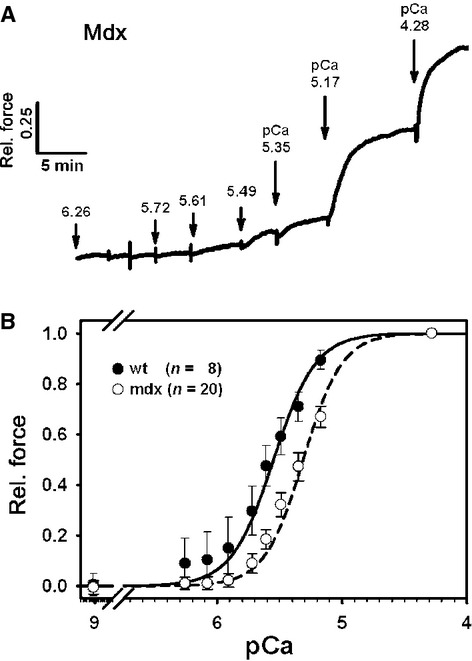
pCa-force recordings in papillary muscle strips from normal and dystrophic pre-fibrotic mice. (A) Example of original recordings of force responses in solutions of varying pCa. Note that force is given relative to maximum force at pCa ∽ 4.2 for better comparison between different preparations. (B) mdx papillary muscles show a significant right shift of pCa-force relations indicative of reduced myofibrillar Ca2+ sensitivity.
In vitro motility assays reveal slower actomyosin filament sliding in mdx hearts
Recordings of the molecular interactions of fluorescently labelled actin with ventricular myosin extracts in an ‘in vitro motility assay’ showed a markedly impaired molecular interaction of the motor proteins. Figure3A shows image sequences from representative recordings tracking two individual filaments in successive images from a wt and an mdx ventricular myosin extract. Velocity histograms from individual recordings showed a marked left shift in mdx hearts that was statistically highly significant, as judged from the much decreased mean sliding velocities in mdx hearts (Fig.3B).
Fig 3.
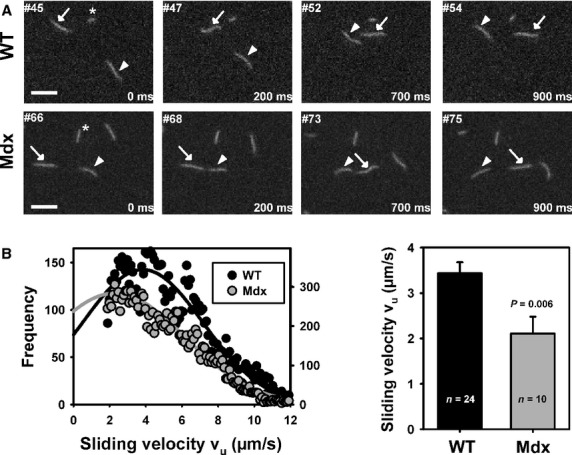
In vitro motility assay in ventricular myosin extracts from young wild-type (wt) and mdx mice. (A) Example images from two motility assay XYT stacks of wt and mdx myosin extracts. Arrows and arrowheads point towards individual actin filaments tracked in successive images. Image frames and times relative to first frame are indicated. The asterisk points towards a standing filament that does not move in successive images indicative of rigour bridges that introduce artificially high counts velocities <1.5 μm/sec. (B) Velocity distributions from two individual XYT recording sequences (n = 24 for wt; n = 10 for mdx) showing the Gaussian fits for the velocities from several hundreds of tracked filaments. There is a prominent left shift of the mdx curve. The results from several thousand filaments confirmed a significantly shifted velocity towards smaller values in mdx hearts.
Myosin heavy and light chain distributions are unchanged in mdx ventricles
To test whether the slower propelling velocities in the mdx ventricles were also reflected by changes in the myosin isoforms expression, SDS PAGE analysis on several mdx and wt hearts was performed. In ventricles and atria extracts, the ventricular and atrial light chain isoforms were clearly distinguishable (ALC, VLC) showing the two well-known isoforms 13,17 (Fig.4A). In MHC gels, a clear single band of ∽185 kD was seen reflecting the overall prominent α-MHC in mouse atria and ventricles 17. Densitometric analysis of gels from three wt and four mdx hearts of similar age showed unchanged MHC expression (given as atria-to-ventricle signal density ratios) and VLC profiles in the ventricles. Interestingly, atrial ALC isoforms were significantly increased in mdx cardiac ventricles (Fig.4B).
Fig 4.

Myosin heavy (MHC) and light chain (MLC) distributions are similar in mdx and wild-type (wt) hearts. (A) 8% (MHC) and 12% (MLC) SDS PAGE gels from atrial and ventricular preparations of two wt and mdx mice, each. For the MLC gels, the atrial ALC and ventricular VLC isoforms are shown. Protein loading was ∽10 μg (MHC) or 50 μg (MLC) for each lane. (B) Densitometric analysis shows unaltered atrial-to-ventricular MHC ratios in mdx papillary muscle. For MLCs, signals were doubled in atria but the slower ventricular isoforms in the ventricles were similar in wt and mdx hearts. S.D.: standard marker (17 kD band, MLC; 200 kD, MHC). Analysis from three to four wt and mdx hearts. *: P < 0.05.
Mdx hearts already show morphological signs of cardiomyopathy in the pre-fibrotic phase with preserved fractional shortening ‘in vivo’
In vivo echocardiography sequences of beating hearts in M-mode were obtained from three conscious wt and four mdx mice. Quantification of ventricular wall diameters already showed significantly increased thickness of the posterior wall (wt: 0.57 ± 0.02 mm, mdx: 0.87 ± 0.05 mm; P < 0.01) and septum (wt: 0.75 ± 0.05 mm, mdx: 1.09 ± 0.10 mm; P < 0.05) and an increased left ventricular end-diastolic diameter (wt: 2.39 ± 0.03 mm, mdx: 2.56 ± 0.05 mm; P < 0.05) in mdx (n =4) over wt (n = 3) mice. However, at this stage of the disease, fractional shortening of the hearts was not yet compromised (wt: 0.19 ± 0.1, mdx: 0.17 ± 0.02 mm; P = 0.59).
Discussion
In dystrophic cardiomyopathy, it is still not known which cellular events precede the morphological hypertrophic/dilative changes and to which extent force output is compromised at early stages before the fibrotic onset. Intracellular Ca2+ overload has been linked to amplified positive feedback loops between mitochondrial dysfunction, ROS production and cell death that could explain the progressive ongoing fibrotic remodelling during ageing 18. Contractile recordings during interposed beats from a basal 1 Hz stimulation (force-interval curves) showed that force amplitudes were markedly depressed in atrial preparations from young mdx mice (10–14 weeks) 9. In papillary-like muscles from 8 weeks old mdx mice, force-frequency relations were markedly reduced compared with wt muscles stimulated between 4 and 12 Hz 8. However, none of those studies provided a mechanistic explanation. Altered Ca2+ homeostasis may be a strong candidate, however, other parameters on the molecular level of the contractile filaments also need to be considered. There are a couple of studies at hand that addressed Ca2+ transients, resting myoplasmic Ca2+ levels or Ca2+ sparks under various conditions 2,7,19. However, none of these studies simultaneously caught up on Ca2+ regulation in papillary muscle where mdx force recordings were obtained or, alternatively, simultaneously assessed force in cases where Ca2+ was monitored in single cardiomyocytes. Our study is the first to simultaneously report force and Ca2+ recordings in the same papillary muscle preparation from young mdx mice in the early pre-fibrotic phase.
Force-Ca2+ transients in papillary muscles from young mdx mice
Under our basal conditions of 1 Hz stimulation, isometric twitch force amplitudes were fivefold reduced in mdx papillary muscle which is more than the approx. two times reduction found in ‘papillary-like’ mdx preparations by Janssen et al. 8 under similar experimental conditions (37°C, 1.5 mM external Ca2+, BDM omitted). Their specific force amplitudes at 4 Hz stimulation in wt muscle was around 30 mN/mm² which seems to be substantially larger than in our wt papillary muscles (∽1 mN at 1 Hz; ∽0.6 mN at 4 Hz with cross-sectional area, CSA, values between 0.3 and 0.7 mm², Methods). Therefore, our specific values would be roughly estimated to account for 1–3 mN/mm², which is a factor 10 smaller than in Janssen et al. (2005) but only five times smaller than in a more recent paper from the same group 20. However, under optimum conditions, paced intact heart muscle operates in the range of maximum force and, indeed, our maximum isometric force in skinned papillary muscle preparations at pCa = 4.28 accounted for ∽2.2 mN which would translate to ∽3–6 mN/mm² which is closer to the values given by Endoh for rabbit papillary muscle 21. Development of hypoxic cores in papillary muscles could aggravate a putatively smaller force compromise 8. However, from modelling studies and recordings of resting metabolism, it is known that a maximum diffusion radius of 0.4 mm (equivalent to 0.8 mm diameter) at 37°C is sufficient to maintain adequate oxygenation in our preparation (diameter <0.6 mm) at a contraction frequency up to ∽5 Hz 10,11. This is also supported by prolonged resting times until experimentation after which diffusive oxygen supply will remain adequate under our experimental conditions 10. Mörner & Wohlfahrt also gave a critical diameter of 0.65 mm above which thin papillary muscle function declined 22. The most direct argument against hypoxic cores developing during our ∽10 sec. stimulation train period, even for highest frequencies of 4 Hz used, is reflected by rather stable force amplitudes during the whole train period (Fig.1A). It is interesting that, in our hands, force-frequency curves were negative even for wt papillary muscles from young mice. Force-frequency relations (FFR) are an important intrinsic property to judge chronotropic efficiency of cardiac contractility 23. Normal human cardiac papillary muscle exhibits a positive FFR between 100 and 160 bpm 24. wt mouse papillary muscle usually shows a positive FFR at physiological external Ca2+ conditions but also strongly depends on temperature. At 37°C and 1.5 mM external Ca2+, FFR is more or less flat between 1 and 5 Hz 25 and can also turn negative for larger Ca2+ levels 26. Force production usually declines at higher stimulation frequencies, i.e. a negative ‘treppe’ in wt mouse papillary-like muscle from 8 Hz stimulation or in rat papillary muscle already at <6 Hz 24. However, as stated by Redel et al. 23, FFRs in mouse papillary muscles can range from negative to strongly positive relations. At ∽2 mM external Ca2+, force-frequency relations were negative 26,27 and supposedly are flat under conditions that most closely resemble the physiological situation in mouse papillary muscle 25. To further rule out possible artefacts from potential hypoxic cores that could result in production of reactive oxygen species 7, we concentrated on stimulation frequencies between 1 and 4 Hz only.
The most obvious reason for compromised force in mdx hearts would be apparent from altered Ca2+ handling. In failing hearts, it is well known that a negative FFR is often reflected by alteration in Ca2+ handling and/or ec-coupling 21. Ca2+ handling abnormalities in mdx mice are related to aberrant mechanosensitive channel activity (e.g. TRPC1, 2), increased Na+/Ca2+ exchanger function 2, ROS production and inflammatory activity 7,18 that maintain Ca2+ overload and fibrotic remodelling in dystrophic heart. Several Ca2+ handling proteins, e.g. expression patterns of SERCA2, are known to be altered in mdx mice also in an age-dependent manner 2,28. In the age group used in our study, Williams and Allen 2 found a slight reduction in SERCA2 expression that turned into a marked increase in old mice (12 mo). Alongside with hypernitrosylation of RyR2 and increased SR leakage 19, all these Ca2+ handling mechanisms compete to favour or limit the increase in myoplasmic Ca2+ that was found in resting or stressed cardiomyocytes 2. The strong t-tubular localization of dystrophin in cardiomyocytes might also point towards its involvement in tubular mechanosensitive channels that could contribute to aberrant mdx cardiac Ca2+ homeostasis 7. As the time for Ca2+ extrusion shortens with higher stimulation frequencies, it was important to us to quantify the frequency dependence of the Ca2+-transient properties to validate the hypothesis that Ca2+ homeostasis was solely (or largely) responsible for compromised force. This hypothesis does not seem to hold given that the reduction in force amplitudes was larger than corresponding reductions in Ca2+ amplitudes. Clearly, Ca2+ was less markedly affected (about 40% reduction in mdx papillary muscles) compared with force and transient decay times were larger in mdx preparations compared with wt which would additionally favour smaller amplitudes (owing to a higher diastolic level). A previous study in isolated mdx ventricular cardiomyocytes found markedly increased Ca2+-transient amplitudes at basal 1 Hz stimulation compared with wt 2. A direct comparison, however, does not hold because we evaluated the fura-2 ratio amplitudes rather than calibrating to absolute [Ca2+]i values. This is valid when comparing the Ca2+-transient responses at different stimulation frequencies within the same wt or mdx muscles because buffer properties are not expected to change upon stimulation regimes. Nevertheless, differences in buffer properties of Ca2+-binding proteins in the two strains are likely to be rather subtle 12,29. Although, to our knowledge, we provide the first frequency responses of Ca2+ transients in mdx papillary muscles, there must be additional mechanisms to explain the marked force decrements in the early pre-fibrotic phase.
Alterations in motorprotein properties seem to explain impaired force in early dystrophic hearts
pCa-force recordings in our preparations showed a significant shift towards larger [Ca2+] values in mdx papillary muscles with a pCa of ∽5.3. In wt preparations, this value was ∽5.5 which is still somewhat smaller than the values given by Dong Gao et al. 30 in a study on wt trabeculae muscles also using combined force-calcium recording technique with fura-2 as in our study. If we assumed similar absolute Ca2+ amplitudes of 1.5–1.6 μM for 2–4 Hz stimulations in the wt 30 in our study, these would translate to a relative force of 0.179. A ∽40% reduction in mdx Ca2+ amplitudes in our setting (Fig.1C) would reflect a value of ∽0.95 μM for Ca2+ amplitude or 0.017 for relative force and thus predict an averaged 10-fold lower force in mdx papillary muscle in the range of physiological Ca2+ amplitudes. Even if the Ca2+ transients were actually larger as in Williams & Allen 2, a similar calculation would still predict a three times smaller force output taking our pCa-force curves. It should be noted that maximum force at saturating Ca2+ conditions was not different in wt and mdx papillary muscles under our conditions. The explanation of the reduced myofibrillar Ca2+ sensitivity may be embedded in previous findings of subverted cAMP/PKA pathways and abolished cAMP microdomain separation in mdx skeletal muscle 31. As a matter of fact, increased protein kinase A (PKA)-mediated phosphorylation of RyR2 has been shown to be associated with the development of dystrophic cardiomyopathy which could be prevented by genetic inhibition of the PKA-RyR2 phosphorylation 32. PKA phosphorylation of troponin I (TnI) at serine residues is known to decrease the myofibrillar Ca2+ sensitivity and increased phosphorylation levels will survive the skinning procedure and be detectable in our pCa-force experiments 33,34. Such an increased PKA-mediated TnI phosphorylation would also explain a disproportionate decrease of force in mdx papillary muscle despite a much lesser drop in Ca2+-transient amplitude.
In addition to the already compromised myofibrillar Ca2+ sensitivity in early pre-fibrotic mdx papillary muscle preparations, we present first evidence for altered actomyosin interaction in mdx hearts using a cardiac in vitro motility assay. Actin sliding velocities were significantly slowed down in ventricular mdx myosin extracts which may relate to impaired contractile parameters of reduced maximum myosin ATPase activity of unloaded speed of shortening. Our values for wt extracts are somewhat larger (∽3.2 μm/sec.) than those given by Svensson et al. 13 in pig hearts (∽2.1 μm/sec.) which might be explained most likely by differences in myosin isoforms. Song et al. 35 reported similar values to ours in regulated filaments at pCa 5. Separated for cardiac myosin isoforms, ∽5 μm/sec. for adult mouse α-MHC and ∽2.5 μm/sec. for adult mouse β-MHC have been given 36. Previous studies reported that velocity of actomyosin sliding was impaired in the diaphragm muscle from mdx mice unlike in semitendinosus muscle 37. This may point towards a common defect in the contractile apparatus in mdx mice selectively affecting muscle that experience large mechanical stress loads, like the diaphragm but also, as shown in our study, the heart.
Unlike the slowed down filament velocity, there was no shift of ventricular myosin isoforms towards β-MHC or higher VLC expression in mdx hearts. In fact, we could not detect ß-MHC in neither wt nor mdx ventricles, which is consistent with a very low, if present at all, ß-MHC expression in mouse hearts, in contrast to rat hearts 17.
Implications for dystrophic hearts: in vivo recordings show signs of hypertrophy in the pre-fibrotic phase
So far, the mechanisms found that would reduce isometric force fivefold in papillary muscle preparations would predict a rather non-sustainable situation in vivo. However, although mdx mice have a reduced lifespan, signs of beginning cardiomyopathy were established as soon as week 16 38. No differences between wt and mdx hearts were recently found from echocardiography recordings in 8-week-old mice, but there was a significantly increased LV mass at 29–42 weeks 3. Our echocardiography data in young conscious mdx mice showed unaltered fractional shortening which is considered a global clinical parameter for cardiac contractile function 16. This seems in contrast to the vast reduction in force production in the isolated mdx preparations. However, in a very recent clinical follow-up study of 11 DMD patients who developed severe DCM over 9 years, all patients showed clear LVEDD enlargements within the observation period with almost no impairment of left ventricular function and only modest reduction in the fractional shortening index, as assessed by echocardiography 39. This indicates that despite clear signs of DCM ‘in vivo’, presumably paralleled by force decrements on the organ muscular level, cardiac output may appear balanced for long times. One explanation, which still awaits further experimental evidence, could be reflected by biophysical considerations. Using Laplace's law and our values for the significantly increased ventricular wall diameters and enlarged ventricular cavity, a force (tension) decrement of ∽1.5 still yields the same pressure level for mdx hearts. Therefore, early hypertrophy in the pre-fibrotic phase as soon as ∽10–16 weeks in our study can be an attempt to compensate for an already decreased wall tension. However, such calculations are not static and fractional shortening can still appear normal despite substantially suppressed contractility on the cellular level. For example, in a study on cardiac performance following aortic banding hypertrophy in mice, a decrease in fractional shortening also showed some time lag to the increase in left ventricular wall thickness suggesting that despite already developed morphological signs of cardiac hypertrophy, global function can remain well compensated 40.
In summary, we present the very first evidence for early hypertrophy in mdx hearts in the pre-fibrotic phase at <16 weeks and possible cellular mechanisms for impaired force output in dystrophic hearts.
Acknowledgments
Supported by grants from DFG to OF (FR1499/7-1), Stiftung Landesbank Baden-Württemberg (2009020004) and University Heidelberg to SW (3612, 3613) and University of Essen to SK (IFORES 107-55604). OF held a senior research international fellowship (Australian Research Council; LX0882427). OF acknowledges ongoing support by the German Research Foundation (DFG) through the ‘Erlangen Graduate School in Advanced Optical Technologies (SAOT)’ within the German Excellence Initiative. The authors are indebted to HG Jakob (Essen) and RHA Fink (Heidelberg) for generously providing laboratory facilities.
Conflicts of interest
We declare that there are no potential conflicts of interest. All funding agencies had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Data S1 Supporting Materials and Methods related to preparation, in vitro motility assays and biochemistry.
References
- Finsterer J, Stöllberger C. The heart in human dystrophinopathies. Cardiology. 2003;99:1–19. doi: 10.1159/000068446. [DOI] [PubMed] [Google Scholar]
- Williams IA, Allen DG. Intracellular calcium handling in ventricular myocytes from mdx mice. Am J Physiol Heart Circ Physiol. 2007;292:H846–55. doi: 10.1152/ajpheart.00688.2006. [DOI] [PubMed] [Google Scholar]
- Quinlan JG, Hahn HS, Wong BL, et al. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromusc Disord. 2004;14:491–6. doi: 10.1016/j.nmd.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Merrick D, Stadler LKJ, Larner D, et al. Muscular dystrophy begins early in embryonic development deriving from stem cell loss and disrupted skeletal muscle formation. Dis Model Mech. 2009;2:374–88. doi: 10.1242/dmm.001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe Y, Esaki K, Nomura T. Skeletal muscle pathology in X-chromosome-linked muscular dystrophy (mdx) mouse. Acta Neuropathol. 1986;69:91–5. doi: 10.1007/BF00687043. [DOI] [PubMed] [Google Scholar]
- Pastoret C, Sebille A. Further aspects of muscular dystrophy in mdx mice. Neuromusc Disord. 1993;3:471–5. doi: 10.1016/0960-8966(93)90099-6. [DOI] [PubMed] [Google Scholar]
- Fanchaouy M, Polakova E, Jung C, et al. Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes. Cell Calcium. 2009;46:114–21. doi: 10.1016/j.ceca.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen PML, Hiranandani N, Mays TA, et al. Utrophin deficiency worsens cardiac contractile dysfunction present in dystrophin-deficient mdx mice. Am J Physiol Heart Circ Physiol. 2005;289:H2373–8. doi: 10.1152/ajpheart.00448.2005. [DOI] [PubMed] [Google Scholar]
- Sapp JL, Bobet J, Howlett SE. Contractile properties of myocardium are altered in dystrophin-deficient mdx mice. J Neurol Sci. 1996;142:17–24. doi: 10.1016/0022-510x(96)00167-0. [DOI] [PubMed] [Google Scholar]
- Widen C, Barclay CJ. Resting metabolism of mouse papillary muscle. Pflugers Arch. 2005;450:209–16. doi: 10.1007/s00424-005-1408-4. [DOI] [PubMed] [Google Scholar]
- Barclay CJ. Modelling diffusive O2 supply to isolated preparations of mammalian skeletal and cardiac muscle. J Muscle Res Cell Motil. 2005;26:225–35. doi: 10.1007/s10974-005-9013-x. [DOI] [PubMed] [Google Scholar]
- Gailly P, Boland B, Himpens B, et al. Critical evaluation of cytosolic calcium determination in resting muscle fibres from normal and dystrophic (mdx) mice. Cell Calcium. 1993;14:473–83. doi: 10.1016/0143-4160(93)90006-r. [DOI] [PubMed] [Google Scholar]
- Svensson C, Morano I, Arner A. In vitro motility assay of atrial and ventricular myosin from pig. J Cell Biochem. 1997;67:241–7. doi: 10.1002/(sici)1097-4644(19971101)67:2<241::aid-jcb9>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Friedrich O, Both M, Weber C, et al. Microarchitecture is severely compromised but motor protein function is preserved in dystrophic mdx skeletal muscle. Biophys J. 2010;98:606–16. doi: 10.1016/j.bpj.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich O, Weber C, v WegnerF, et al. Unloaded speed of shortening in voltage-clamped skeletal muscle fibers from wt, mdx and transgenic minidystrophin mice using a novel high-speed acquisition system. Biophys J. 2008;94:4751–65. doi: 10.1529/biophysj.107.126557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehringer C, Rutschow D, Bauer R, et al. Prevention of cardiomyopathy in δ-sarcoglycan knockout mice after systemic transfer of targeted adeno-associated viral vectors. Cardiovasc Res. 2009;82:404–10. doi: 10.1093/cvr/cvp061. [DOI] [PubMed] [Google Scholar]
- Andruchov O, Andruchova O, Galler S. Fine-tuning of cross-bridge kinetics in cardiac muscle of rat and mouse by myosin light chain isoforms. Pflugers Arch. 2006;452:667–73. doi: 10.1007/s00424-006-0080-7. [DOI] [PubMed] [Google Scholar]
- Williams IA, Allen DG. The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am J Physiol Heart Circ Physiol. 2007;293:H1969–77. doi: 10.1152/ajpheart.00489.2007. [DOI] [PubMed] [Google Scholar]
- Fouconnier J, Thireau J, Reiken S, et al. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2010;107:1559–64. doi: 10.1073/pnas.0908540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Delfin DA, Rafael-Fortney JA, et al. Lengthening-contractions in isolated myocardium impact force development and worsen cardiac contractile function in the mdx mouse model of muscular dystrophy. J Appl Physiol. 2011;110:512–9. doi: 10.1152/japplphysiol.00253.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh M. Force-frequency relationship in intact mammalian ventricular myocardium: physiological and pathophysiological relevance. Eur J Pharmacol. 2004;500:73–86. doi: 10.1016/j.ejphar.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Moerner SE, Wohlfahrt B. Myocardial force interval relationships: influence of external sodium and calcium, muscle length, muscle diameter and stimulation frequency. Acta Physiol Scand. 1992;145:323–32. doi: 10.1111/j.1748-1716.1992.tb09372.x. [DOI] [PubMed] [Google Scholar]
- Ross J, Jr, Miura T, Kambayashi M, et al. Adrenergic control of the force-frequency relation. Circulation. 1995;92:2327–32. doi: 10.1161/01.cir.92.8.2327. [DOI] [PubMed] [Google Scholar]
- Parilak LD, Taylor DG, Song Y, et al. Contribution of frequency-augmented inward Ca2+ current to myocardial contractility. Can J Physiol Pharmacol. 2009;87:69–75. doi: 10.1139/Y08-087. [DOI] [PubMed] [Google Scholar]
- Redel A, Baumgartner W, Golenhofen K, et al. Mechanical activity and force-frequency relationship of isolated mouse papillary muscle: effects of extracellular calcium concentration, temperature and contraction type. Pflugers Arch. 2002;445:297–304. doi: 10.1007/s00424-002-0931-9. [DOI] [PubMed] [Google Scholar]
- Bluhm WF, Kranias EG, Dillmann WH, et al. Phospholamban: a major determinant of the cardiac force-frequency relationship. Am J Physiol. 2000;278:H245–55. doi: 10.1152/ajpheart.2000.278.1.H249. [DOI] [PubMed] [Google Scholar]
- Meyer M, Bluhm WF, He H, et al. Phospholamban-to-SERCA2 ratio controls the force-frequency relationship. Am J Physiol Heart Circ Physiol. 1999;276:H779–85. doi: 10.1152/ajpheart.1999.276.3.H779. [DOI] [PubMed] [Google Scholar]
- Lohan J, Ohlendieck K. Drastic reduction in the luminal Ca2+ binding proteins calsequestrin and sarcalumenin in dystrophin-deficient cardiac muscle. Biochim Biophys Acta. 2004;1689:252–8. doi: 10.1016/j.bbadis.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Hopf FW, Turner PR, Denetclaw WF, et al. A critical evaluation of resting intracellular free calcium regulation in dystrophic mdx muscle. Am J Physiol Cell Physiol. 1996;40:C1325–39. doi: 10.1152/ajpcell.1996.271.4.C1325. [DOI] [PubMed] [Google Scholar]
- Dong Gao W, Perez NG, Marban E. Calcium cycling and contractile activation in intact mouse cardiac muscle. J Physiol. 1998;507:175–84. doi: 10.1111/j.1469-7793.1998.175bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röder IV, Lissandron V, Martin J, et al. PKA microdomain organisation and cAMP handling in healthy and dystrophic muscle in vivo. Cell Signal. 2009;21:819–26. doi: 10.1016/j.cellsig.2009.01.029. [DOI] [PubMed] [Google Scholar]
- Sarma S, Li N, van Oort RJ, et al. Genetic inhibition of PKA phosphorylation of RyR2 prevents dystrophic cardiomyopathy. Proc Natl Acad Sci USA. 2010;107:13165–70. doi: 10.1073/pnas.1004509107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verduyn SC, Zaremba R, van der Velden J, et al. Effects of contractile protein phosphorylation on force development in permeabilized rat cardiac myocytes. Basic Res Cardiol. 2007;102:476–87. doi: 10.1007/s00395-007-0663-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson BA, Locher MR, Bekyarova T, et al. Differential roles of regulatory light chain and myosin binding protein-C phosphorylations in the modulation of cardiac force development. J Physiol. 2010;588:981–93. doi: 10.1113/jphysiol.2009.183897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Dyer E, Stuckey D, et al. Investigation of a transgenic mouse model of familial dilated cardiomyopathy. J Mol Cell Cardiol. 2010;49:380–9. doi: 10.1016/j.yjmcc.2010.05.009. [DOI] [PubMed] [Google Scholar]
- Malmqvist UP, Aronshtam A, Lowey S. Cardiac myosin isoforms from different species have unique enzymatic and mechanical properties. Biochemistry. 2004;43:15058–65. doi: 10.1021/bi0495329. [DOI] [PubMed] [Google Scholar]
- Coirault C, Lambert F, Poruny JC, et al. Velocity of actomyosin sliding in vitro is reduced in dystrophic mouse diaphragm. Am J Respir Crit Care Med. 2002;165:250–3. doi: 10.1164/ajrccm.165.2.2105088. [DOI] [PubMed] [Google Scholar]
- Jearawiriyapaisarn N, Moulton HM, Kole R, et al. Long-term improvement in mdx cardiomyopathy after therapy with peptide-conjugated morpholino oligomers. Cardiovasc Res. 2010;85:444–53. doi: 10.1093/cvr/cvp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schade van Westrum SM, Hoogerwaard EM, Dekker L, et al. Cardiac abnormalities in a follow-up study on carriers of Duchenne and Becker muscular dystrophy. Neurology. 2011;77:62–6. doi: 10.1212/WNL.0b013e318221ad14. [DOI] [PubMed] [Google Scholar]
- Liao Y, Ihsikura F, Beppu S, et al. Echocardiographic assessment of LV hypertrophy and function in aortic-banded mice: necropsy validation. Am J Physiol Heart Circ Physiol. 2001;282:H1703–8. doi: 10.1152/ajpheart.00238.2001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Supporting Materials and Methods related to preparation, in vitro motility assays and biochemistry.