

Abstract
There are controversies concerning the capacity of Rosuvastatin to attenuate heart failure in end-stage hypertension. The aim of the study was to show whether the Rosuvastatin might be effective or not for the heart failure treatment. Twenty-one spontaneously hypertensive rats (SHRs) aged 52 weeks with heart failure were randomly divided into three groups: two receiving Rosuvastatin at 20 and 40 mg/kg/day, respectively, and the third, placebo for comparison with seven Wistar-Kyoto rats (WKYs) as controls. After an 8-week treatment, the systolic blood pressure (SBP) and echocardiographic features were evaluated; mRNA level of B-type natriuretic peptide (BNP) and plasma NT-proBNP concentration were measured; the heart tissues were observed under electron microscope (EM); myocardial sarcoplasmic reticulum Ca2+ pump (SERCA-2) activity and mitochondria cytochrome C oxidase (CCO) activity were measured; the expressions of SERCA-2a, phospholamban (PLB), ryanodine receptor2 (RyR2), sodium–calcium exchanger 1 (NCX1), Ca2+/calmodulin-dependent protein kinase II (CaMKII) and protein phosphatase inhibitor-1 (PPI-1) were detected by Western blot and RT-qPCR; and the total and phosphorylation of protein kinase Cα/β (PKCα/β) were measured. Aged SHRs with heart failure was characterized by significantly decreased left ventricular ejection fraction and left ventricular fraction shortening, enhanced left ventricular end-diastolic diameter and LV Volume, accompanied by increased plasma NT-proBNP and elevated BNP gene expression. Damaged myofibrils, vacuolated mitochondria and swollen sarcoplasmic reticulum were observed by EM. Myocardium mitochondria CCO and SERCA-2 activity decreased. The expressions of PLB and NCX1 increased significantly with up-regulation of PPI-1 and down-regulation of CaMKII, whereas that of RyR2 decreased. Rosuvastatin was found to ameliorate the heart failure in aged SHRs and to improve changes in SERCA-2a, PLB, RyR2, NCX1, CaMKII and PPI-1; PKCα/β2 signal pathway to be suppressed; the protective effect of Rosuvastatin to be dose dependent. In conclusion, the heart failure of aged SHRs that was developed during the end stage of hypertension could be ameliorated by Rosuvastatin.
Keywords: spontaneously hypertensive rat, heart failure, Rosuvastatin, protein kinase Cα/β2, sarcoplasmic reticulum calcium pump, phospholamban, ryanodine receptor2, sodium–calcium exchanger
Introduction
Hypertension has become the most familiar ‘epidemic disease’ in the world. Because of its harmful complications in the cardiovascular system, it has been identified as the mayor risk factor for high cardiac mortality. Although plenty of new anti-hypertensive medications have been developed 1,2, ventricular hypertrophy and the subsequent congestive heart failure are still the common occurrences in most aged hypertensives 3. Therefore, an effective, safe and widely used agent is urgently needed to attenuate the progress of heart failure.
The HMG-CoA-reductase inhibitors (statins) exert a protective effect on the cardiovascular system 4,5, in addition to their lipid-lowering action 6–8, as indicated by the heart function restoration in the hypertensive patients with or without coronary diseases 9–11. Furthermore, the JUPITER trial suggested that Rosuvastatin could effectively decrease C-reactive protein (CRP) level, thereby reducing cardiovascular event rates 12,13. Braunwald 14 doubted that this effect might be based on reducing inflammation per se, without an effect on LDL-C. However, the exact role and underlying mechanism of these protective effects are not completely clear.
The purpose of this study was to observe the impairment of cardiac systolic function in aged spontaneously hypertensive rats (SHRs). We reported here that Rosuvastatin could ameliorate the impairment that occurs at the end stage of hypertension in the aged SHRs; and provide evidence indicating signalling pathways are involved.
Material and methods
Animals
Twenty-one male SHRs, aged 52 weeks and weighing approximately 400 g, and seven age-matched normotensive male Wistar-Kyoto rats (WKY) as controls (Animal Administration Center of Fudan University, Shanghai, China) were bred ad libitum. SHRs were randomly divided into three groups, the first two receiving Rosuvastatin at a dose of 20 mg/kg/day (SHR+LD, n = 7) and 40 mg/kg/day (SHR+HD, n = 7) respectively; the third, placebo (SHR, n = 7) for comparison with seven WKYs as controls. Rosuvastatin and vehicle were daily administered through an intra-gastric tube for 8 weeks. All animal experimental procedures were approved by the Animal Care and Use Committee of Fudan University and performed in accordance with the guide for the care and use of laboratory animals (NIH publication no. 85-23, National Academy Press, Washington, DC, USA, revised 1996).
Measurement of systolic blood pressure
All the rats were trained to adapt themselves to the restraining cages and tail-cuff apparatus for the standard non-invasive tail-cuff method before SBP measurement 15, which was performed at the beginning without exception, and then every 4 weeks under their conscious conditions. Each measurement was repeated thrice.
Determination of plasma NT-proBNP concentration
The animals were killed so that their blood samples were collected from the abdominal aorta, the blood serum separated and NT-proBNP concentration determined by ELISA according to the manufacturer's instructions (R & D Systems, Minneapolis, MN, USA).
Measurement of cardiac function
Trans-thoracic echocardiographic analysis was performed with an animal-specific instrument (VisualSonics® Vevo770®; VisualSonicsInc, Toronto, Canada), as previously described 16. The animals were anaesthetized with isoflurane before M-mode images of the left ventricle were recorded when the heart rate was near 400 bpm. To evaluate the function of their hearts, the following parameters were measured: the left ventricular end-diastolic diameter (LVIDd), left ventricular volume (LV Volume), left ventricular ejection fraction (LVEF), left ventricular short-axis fractional shortening (LVFS), left ventricular stroke volume (SV).
Transmission electron microscopy
From each group the cardiac tissues were minced into small pieces (≤1 mm3) and fixed in 2.5% glutaraldehyde in 0.1 mol/l sodium cacodylate buffer (pH 7.3) for 2 hrs. The specimens were rinsed in buffer, postfixed in cacodylate-buffered 2% OsO4, stained en bloc in uranyl acetate, dehydrated gradient in ethanol and embedded in epoxy resin. Finally, 50–70 nm super thin slices were prepared, stained with uranyl acetate and lead citrate and examined under an electron microscope (Philips TECNA10; Philips, Amsterdam, The Netherlands).
Measurement of mitochondria cytochrome c oxidase activity
Mitochondria were isolated from left ventricle as described previously 17. The final crude mitochondrial pellet was resuspended in sucrose-histidine-EDTA buffer, and the protein concentrations were determined via bicinchoninic acid method. The cytochrome c oxidase (CCO) activity was measured as described by Subbuswamy 18.
Quantitative RT-PCR
RNA was isolated and its concentrations were determined; quantitative real-time PCR analyses were performed as previously described 15. The primers for BNP, SERCA2a, PLB, RyR2 type 1, RyR2 type 2, PKCα, PKCβ and glyceraldehydes 3-phosphate dehydrogenase (GAPDH) were designed from Takara: For BNP gene, sense, 5′-CAGTCAGTCGCTTGGGCTGT -3′ and antisense, 5′-GCAGAGTCAGAAGCCGGAGT -3′. For SERCA2a gene, sense, 5′-TGAGGCCACCTCACAGCAAC-3′ and antisense, 5′-CAATGCCGTGGTCTTGGATG-3′. For PLB gene, sense, 5′-AACTAAACAGTCTGCATTGTGACGA-3′ and antisense, 5′-GCCGAGCGAGTAAGGTATTGGA-3′. For RyR2 type 1 gene, sense, 5′-GGCCATCCTTGTCCAGCATTAC-3′ and antisense, 5′- CTGCTCCGTAATGTAAAGCCCATC-3′. For RyR2 type 2 gene, sense, 5′-GAGAGCCCGGAAGCTCTGAA-3′ and antisense, 5′-GGCAACTCCATGGCACACAC-3′. For PKCα gene, sense, 5′-TCGGATCCTTACGTGAAGCTGA-3′ and antisense, 5′-AGTCGCCGGTCTTTGTCTGAA-3′. For PKCβ gene, sense, 5′- CTTGCAGAGCAAGGGCATCA-3′ and antisense, 5′-TGCCACAGAAGTCTTGGT TGTC-3′. The relative expression levels of the genes were normalized to those of GAPDH using 2−ΔΔCt method.
Measurement of SERCA-2 activity
The protein extraction of the cardiac tissues was prepared, as previously described 19; the activity of SERCA-2 was measured according to the operating instructions of Ca2+-ATPase kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Western blot
The ventricle tissues were removed rapidly from the rats to be stored at −80°C. The expressions of phosphorylated PKC and calcium-handling proteins were measured via Western blot and normalized to the protein level of β-actin or GAPDH. From the frozen ventricle tissues were extracted the total proteins, whose concentration was determined with a BCA protein assay. SERCA2a, PLB, PPI-1, NCX1 (1:1000; ABCAM, Cambridge, UK), CaMKII, RyR, PKCα, PKCβ1, PKCβ2, phospho-PKCα, phospho-PKCβ1 and phospho-PKCβ2 (1:300; Santa Cruz Biotechnology, Santa Cruz, CA, USA) were examined by Western blot as previously described 15, and the optic densities were analysed using ImagePro 5.0 (Media Cybernetics, Inc., Silver Spring, MD, USA).
Statistical analysis
The results were presented as mean ± S.E.M. and analysed using one-way anova followed by Fisher's LSD test for multiple comparisons using the SPSS software package, version 16.0 (SPSS Inc., Chicago, IL, USA), P < 0.05 considered as statistically significant.
Results
Lowering effect of Rosuvastatin on blood pressure
The average SBP was found to be higher in SHR controls than in WKYs by 59.4% (P < 0.01). However, no significant difference was observed among SHR+LD, SHR+HD and SHR controls after 4 and 8 weeks, respectively (P > 0.05; Table1).
Table 1.
Changes in systolic blood pressure (SBP)
| Group | SBP (mmHg) | ||
|---|---|---|---|
| Baseline | In the fourth week | In the eighth week | |
| WKY | 129.14 ± 7.76 | 124.22 ± 4.49 | 128.65 ± 4.44 |
| SHR | 205.90 ± 8.56* | 206.07 ± 6.96* | 201.80 ± 2.95* |
| SHR+LD | 200.35 ± 7.04* | 203.17 ± 7.51* | 206.07 ± 4.57* |
| SHR+HD | 200.31 ± 7.89* | 198.07 ± 6.69* | 202.81 ± 6.39* |
Values are mean ± S.E.M., n = 5, *P < 0.01 versus WKYs.
Effect of Rosuvastatin on cardiac structure and function of SHRs
Echocardiographic measurements were conducted in vitro to prove whether myocardium of SHR underwent heart malfunction (Fig.1A). It was found that LVEF and LVFS were lower in SHR controls than in WKYs by 37.5% and 45.5% respectively (P < 0.05). In SHR+HD, LVEF and LVFS increased by 52.9% and 66.3%, respectively, when compared with SHR controls (P < 0.05). However, SV showed no significant difference among these four groups (P > 0.05) (Fig.1B and C).
Fig 1.
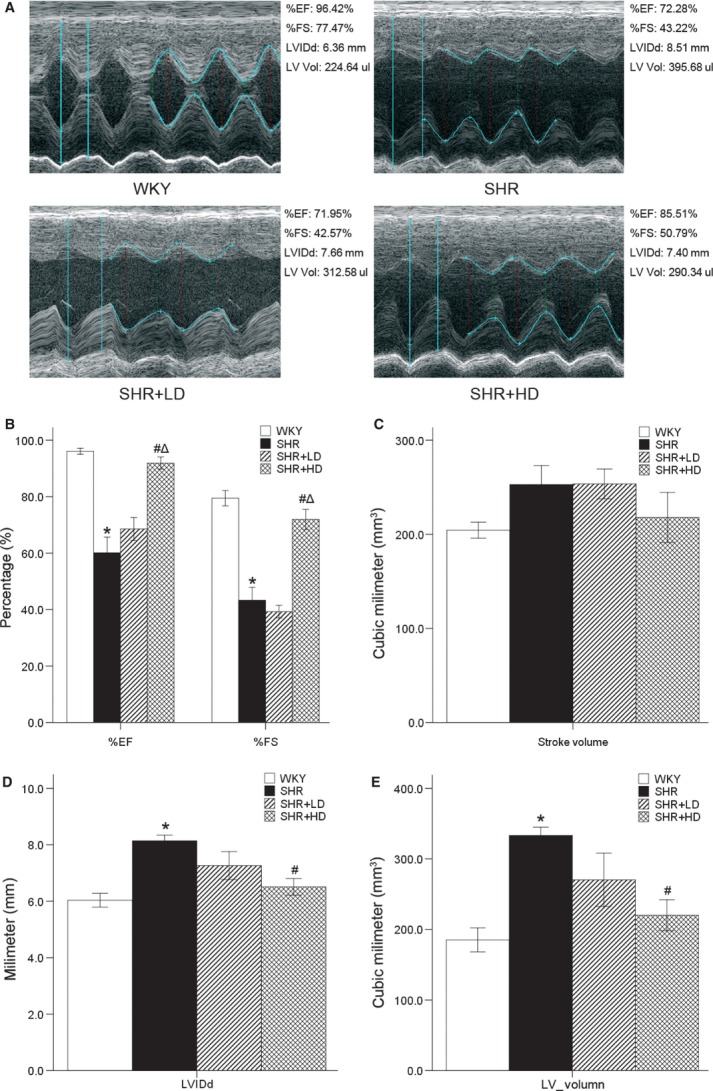
Echocardiographic data of %EF, %FS, SV, LVIDd and LV volume. (A) M-type echocardiographic images of WKYs, SHR controls, SHR+LD and SHR+HD. (B) Cardiac function decreased significantly in SHR controls compared with WKYs as indicated by both %EF and %FS (P < 0.05); %EF and %FS were significantly enhanced in SHR+HD (P < 0.05). (C) Stroke volume with no variety in all groups. (D) and (E) Left ventricule dilated significantly in SHR controls compared with WKYs as indicated by LVIDd and LV volume (P < 0.05), but reversed in SHR+HD (P < 0.05). Values are mean ± S.E.D., n = 4, *P < 0.05 versus WKYs; #P < 0.05 versus SHR controls. LVIDd: left ventricular end-diastolic diameter, LV volume: left ventricular volume, SV: left ventricular stroke volume, %EF: left ventricular ejection fraction, %FS: left ventricular short-axis fractional shortening.
The results also indicated an increase in LVIDd and LV volume in SHR controls by 34.8% and 80%, respectively, when compared with WKYs (P < 0.05). In SHR+HD, significant reductions developed in LVIDd and LV volume by 20.1% and 33.9%, respectively (P < 0.05; Fig.1D and E).
According to the general changes, the results of the transmission electron microscopy showed the losses and damages of myofibrils in SHR controls when compared with WKYs (Fig.2A). Swollen, fragmented and vacuolated mitochondria were observed to be evident in SHRs, and the cristae in the mitochondria appeared distorted and in some cases were completely lysed. Furthermore, there appeared dilated and swollen sarcoplasmic reticulum in SHRs (Fig.2B). Treated with Rosuvastatin, SHRs showed improved cardiac structure as indicated by the reversed losses and damages of myofibrils, and increased volume of myofibrils. Moreover, the swollen, fragmented and vacuolated mitochondria were alleviated, whereas the sarcoplasmic reticulum was not as dilated and swollen as that of SHR controls. The improvement of the cardiac structure was more apparent in SHR+HD than in SHR+LD (Fig.2C and D).
Fig 2.

Ultrastructure changes. (A) Normal cardiac ultrastructure in control (WKYs). (B) Losses and damages of myofibrils in SHR controls; swollen, fragmented and vacuolated mitochondria are observed in SHR controls; dilated and swollen sarcoplasmic reticulum elements in SHR controls. (C) and (D) Volume of myofibrils increased; swollen, fragmented and distended mitochondria alleviated; dilated and swollen sarcoplasmic reticulum ameliorated in SHR+LD and SHR+HD, respectively, especially in SHR+HD. SR: sarcoplasmic reticulum; mito: mitochondria; mf: myofibril.
Furthermore, compared with WKY, the CCO activity of SHR controls was decreased by 31.2% (P < 0.05), but increased in SHR+LD and SHR+HD by 12.8% (P > 0.05) and 36.0% (P < 0.05) respectively (Table2).
Table 2.
Effect of Rosuvastatin on cytochrome c oxidase (CCO) activity (k/min./mg of protein)
Values are mean ± S.E.M., n = 7, *P < 0.01 versus WKYs; #P < 0.01 versus SHR controls.
In addition to the morphologic alterations, plasma NT-proBNP levels were examined to be significantly increased in SHR controls in comparison with WKYs, and to be decreased in SHR+LD and SHR+HD by 12.1% and 19.5% respectively (P < 0.01; Table3). Meanwhile, the mRNA level of BNP examined by real-time RT-qPCR showed a 5.35-fold increase in SHR controls, and a decrease in SHR+LD and SHR+HD by 47.4% and 72.3% respectively (P < 0.01; Fig.3).
Table 3.
Effect of Rosuvastatin on plasma NT-proBNP (ng/ml)
| Group | WKY | SHR | SHR+LD | SHR+HD |
|---|---|---|---|---|
| NT-proBNP concentration | 158.27 ± 13.65 | 209.47 ± 20.91* | 184.18 ± 17.55# | 168.62 ± 13.76# |
Values are mean ± S.E.M., n = 7, BNP: B-type natriuretic peptide, *P < 0.01 versus WKYs; #P < 0.01 versus SHR controls.
Fig 3.
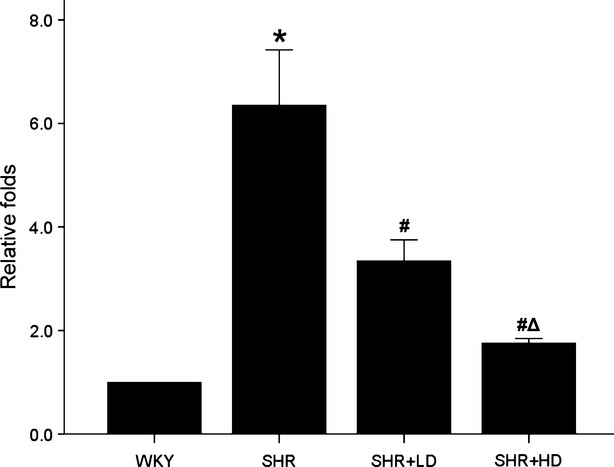
BNP (Nppb) gene expression by RT-qPCR. Real-time RT-qPCR analysis of BNP gene expression showing a significantly higher BNP expression in SHRs than WKYs (P < 0.01), but dose dependently down-regulated in SHR+LD and SHR+HD (P < 0.01). Values: mean ± S.E.M., n = 3, *P < 0.05 versus WKYs; #P < 0.05 versus SHR controls; ΔP < 0.01 versus SHR+LD. BNP: B-type natriuretic peptide.
Regulation of the mRNA and protein expressions of Ca2+-cycling protein in SHRs
To investigate the mechanism of Rosuvastatin in the attenuated cardiac function of SHRs, the expressions of myocardium Ca2+-handling proteins were evaluated.
PPI-1, dephosphorylation of the PLB and thus incapable of activating SERCA2a, was significantly up-regulated by 85.9% in SHR controls (P < 0.01), and significantly down-regulated in SHR+LD and SHR+HD by 56.4% and 77.3% respectively (P < 0.01). Meanwhile, the expression of CaMKII up-regulated significantly by 40.9% in SHR+HD (P < 0.01) but not SHR+LD (P > 0.05), when compared with SHR controls (Fig.4A).
Fig 4.
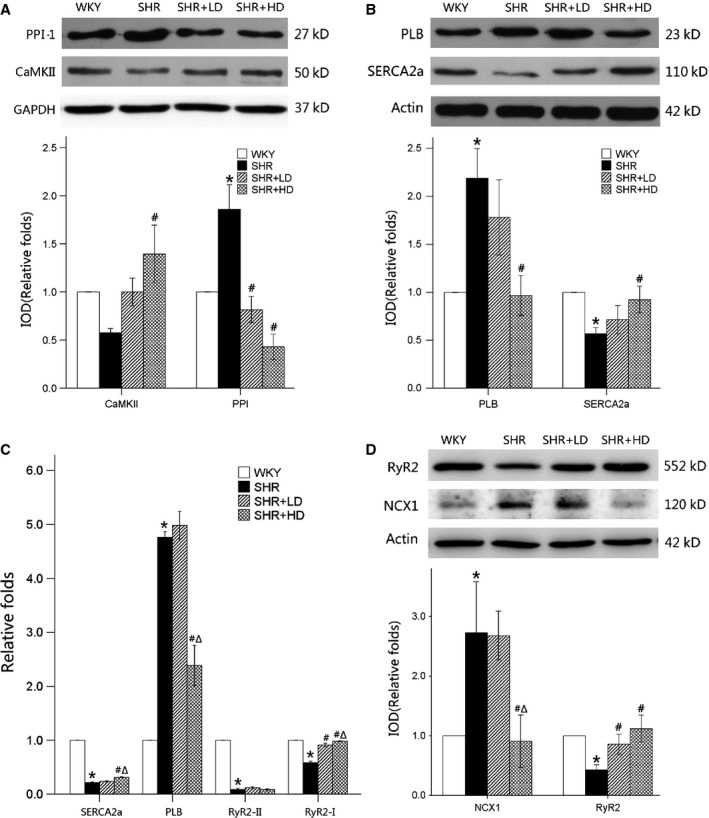
Ca2+-cycling protein and mRNA expression by Western blot and Real-time RT-qPCR. (A) Protein expressions of PPI-1 are up-regulated significantly in SHR controls (P < 0.01), and reversed in SHR+LD and SHR+HD (P < 0.01); the expression of CaMKII is up-regulated significantly in SHR+HD. (B) Protein expressions of SERCA2a are significantly suppressed, whereas PLB is significantly augmented in SHR controls compared with WKYs (P < 0.05), and reversed in SHR+HD (P < 0.05). (C) mRNA expression of SERCA2a, RyR2 type 2 and RyR2 type 1 are significantly suppressed, whereas PLB is elevated in SHR controls compared with WKYs (P < 0.05); the mRNA levels of SERCA2a and RyR2 type 1 are significantly elevated in SHR+HD (P < 0.05), and the mRNA level of PLB significantly reduced in SHR+HD (P < 0.05). (D) Protein expressions of RyR2 are significantly suppressed, whereas NCX1 is significantly augmented in SHR controls compared with WKYs (P < 0.05), and reversed in SHR+LD and SHR+HD, respectively (P < 0.05). Values are mean ± S.E.M., n = 3, *P < 0.05 versus WKYs; #P < 0.05 versus SHR controls; ΔP < 0.01 versus SHR+LD. PPI-1: protein phosphatase inhibitor-1; CaMKII: calcium/calmodulin-dependent protein kinase II, SERCA2a: sarcoplasmic reticulum calcium pump, PLB: phospholamban, RyR2: ryanodine receptor2, NCX1: sodium–calcium exchanger 1.
The results indicated that SERCA2a, RyR2 type 1 and RyR2 type 2 mRNA were significantly decreased by 78.4%, 41.3% and 91.1%, respectively (P < 0.05), whereas PLB expressions increased 4.8-fold in SHR controls when compared with WKYs. Furthermore, the expressions of SERCA2a, RyR2 type 1 mRNA were significantly up-regulated in SHR+HD (P < 0.05), whereas those of PLB were down-regulated (P < 0.05). However, RyR2 type 2 mRNA showed no significant changes among the three groups (Fig.4C).
The expression of NCX1 was significantly up-regulated in SHR controls, but reversed in SHR+HD. Meanwhile, the protein expressions of SERCA2a, PLB and RyR2 were highly consistent with those of RT-qPCR, as indicated by Western blot (Fig.4B and D).
The SERCA2 activity was significantly decreased by 20.6% in SHR when compared with WKY (P < 0.05), but increased by 11.7% and 19.7% in SHR+LD and SHR+HD (P < 0.05) respectively (Table4).
Table 4.
Effect of Rosuvastatin on SERCA-2 activity [μmol/(mg/prot/hr)]
Values are mean ± S.E.M., n = 7, SERCA2a: sarcoplasmic reticulum calcium pump, *P < 0.01 versus WKYs; #P < 0.01 versus SHR controls.
Decreased expressions and phosphorylation of PKCα and PKCβ2 in SHRs
PKC signalling pathway has been identified as a given pathway in the Ca2+-regulating process. It was found that Rosuvastatin down-regulated significantly the mRNA expression of PKCα and PKCβ by 64.5% and 67.7% in SHR+LD, respectively, and by 7.29% and 66.4% in SHR+HD respectively (P < 0.01). Furthermore, the ratios of phosphorylation to the total PKCα and PKCβ2 were significantly inhibited, but it was not true of PKCβ1 in SHR+LD and SHR+HD when compared with SHR controls (Fig.5).
Fig 5.
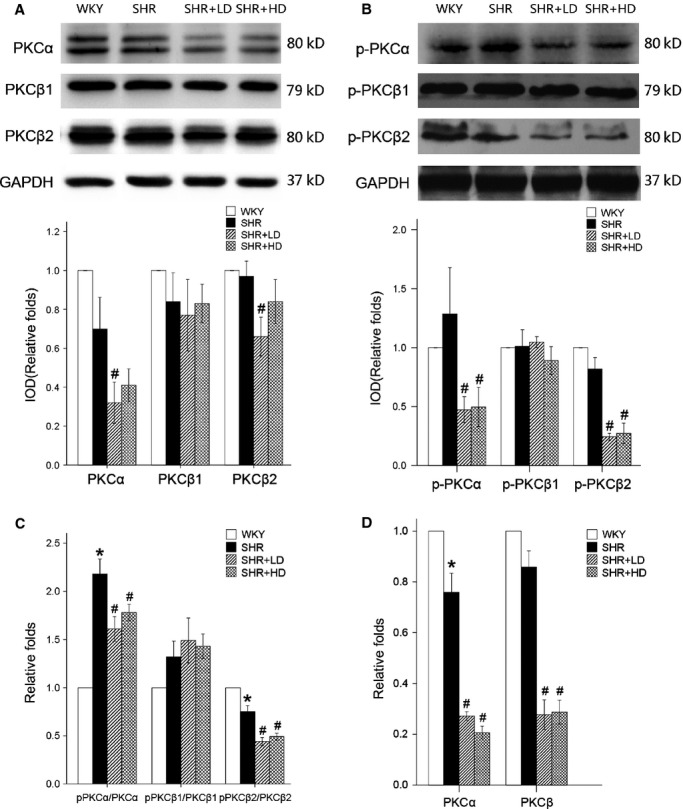
mRNA and protein expression of PKCα and PKCβ by real-time RT-qPCR and Western blot. (A) Expressions of total PKCα and PKCβ2 are significantly down-regulated in SHR+LD (P < 0.05), but not the same with the expression of total PKCβ1 (P > 0.05). (B) Expressions of phospho-PKCα and phospho-PKCβ2 are significantly down-regulated in SHR+LD and SHR+HD (P < 0.05), but no significant changes are found in the expression of phospho-PKCβ1 (P > 0.05). (C) Ratio of phosphor PKCα to total PKCα and the ratio of phosphor PKCβ2 to total PKCβ2 are down-regulated significantly in SHR+LD and SHR+HD (P < 0.01). (D) mRNA level of the whole PKCα (P < 0.05) and PKCβ (P > 0.05) gene expressions is down-regulated in SHR controls and lowered considerably (P < 0.05) in SHR+LD and SHR+HD. Values are mean ± S.E.M., n = 3, *P < 0.05 versus WKYs; #P < 0.05 versus SHR controls; ΔP < 0.01 versus SHR+LD. p-PKCα: phosphorylation of protein kinase Cα;p-PKCβ1: phosphorylation of protein kinase Cβ1;p-PKCβ2: phosphorylation of protein kinase Cβ2.
Discussion
Most of the hypertensives tend to develop cardiac hypertrophy and even heart failure, although they receive the administration of anti-hypertensive medications. Rosuvastatin has been reported to be controversial in attenuating cardiac function 9, improving survival rate in those with diastolic heart failure 10 and decreasing all-cause mortality following a diagnosis of heart failure 11. However, the mechanisms of these protective effects remain unknown.
In the present study, we used 52-week-old SHR as the model of the end-stage hypertensive heart disease, finding that the aged SHRs developed systolic heart failure characterized by the abnormal parameters of echocardiography and the increased NT-proBNP. We also found that the mitochondria were swollen, fragmented and vacuolated and the sarcoplasmic reticulum (SR) were dilated and swollen in SHRs. The mitochondria CCO and SERCA2 activity were down-regulated. In addition, the decreased expressions of SERCA2a, RyR2 and CaMKII were accompanied by an increase in PLB, NCX1 and PPI-1, suggesting a possible mechanism that the failure of SR can induce the impairments of Ca2+-handling proteins, thus causing heart failure.
Rosuvastatin treatment can ameliorate heart failure of aged SHRs independent of blood pressure lowering, via dephosphorylation of PKCα/β2, followed by dephosphorylation of PPI-1 and increased expression of CaMKII, thus promoting phosphorylation of PLB and up-regulating SERCA2 activity.
It is well recognized that high blood pressure increases ventricular wall tension, stimulates cardiomyocytes to express cytokines 20, causes ventricular remodelling and consequently develops heart failure, the pathological changes of which can be attenuated with a reduction in SBP, thereby improving cardiac function. Statins have been reported on lowering blood pressure in both SHR and WKY/L-NAME rats by reducing total peripheral vascular resistance 21. However, there have been controversies on their anti-hypertensive effects. Loch and Saka reported that statin treatment failed to alter blood pressure in Dahl salt-sensitive rats 22,23.
In the present study, no significant changes in SBP were observed among the three SHR groups. The main reason responsible for this could be that 52-week-old SHRs had developed severe endothelial compromise, perivascular fibrosis and arterial stiffness. In this case, Rosuvastatin with weak anti-hypertensive effect could not alter significantly the peripheral resistance and the resultant SBP. This result was consistent with the report by Prandin MG that the confounding factors, such as age and baseline SBP, might influence the blood pressure of each individual 24.
Previous studies have shown that statins can ameliorate BNP up-regulation 25 and restore heart function in hypertensives 9 in addition to their lipid-lowering effects. This is also the case for the results from this study that a high concentration of Rosuvastatin could reverse the expressions of SERCA-2a, RyR2, PLB, NCX1, PPI-1 and CaMKII, thus ameliorating the cardiac malfunction at the end stage of hypertension.
However, there have been controversies around the therapeutic effect of statins on cardiac malfunction. In John Kjekshus’ trial, even Rosuvastatin could reduce the total number of hospitalizations for heart failure, but produce no effect on heart failure mortality 26. The UNIVERSE trials also failed to demonstrate a therapeutic effect of statin on LV remodelling in the patients with chronic heart failure 27. It could be that those enrolled had been well treated for heart failure with a standard therapy before Rosuvastatin, which reduced significantly the population with potentially fatal heart failure enrolled in the trial. This could also be because the patients with the heart failure of ischaemic or non-ischaemic aetiology were not differentially enrolled. Thus, the different inclusion and exclusion criteria could cause different pathophysiological and aetiological changes in heart failure, confusing the consistence of the results in the cohort.
The previous literature has shown that PKCα/β function as fundamental regulators of cardiac contractility and Ca2+-handling proteins in cardiomyocyte 28–31, and PKCβ2 inhibition attenuates myocardial infarction and hypertension-induced heart failure 32,33. Moreover, the enhancement in cardiac contractility associated with PKCα gene deletion protected the myocardium against pressure overload-induced heart failure and dilated cardiomyopathy 34.
Our previous study 15 indicated that Rosuvastatin could decrease the expression of angiotensin II type 1 receptor (AT1R), one type of G-protein coupling receptor, in the ventricle, and resultantly inhibit mitogen-activated protein kinase (MAPK) signal pathway, thus ameliorate cardiac hypertrophy in hypertensive heart disease. To investigate whether PKC is involved in the inhibitory effect of AT1R by Rosuvastatin, we examined the mRNA and protein expression of PKCα/β in the myocardium, and found down-regulation of PKCα/β as well as decreased ratios of phosphorylation to total PKCα and PKCβ2 after Rosuvastatin treatment. On the basis of the data presented, we concluded that Rosuvastatin could decrease the expression of AT1R, and consequently suppress the activation of Gα subunit 35–37, restrain G-protein coupling signal pathway, thus inhibit the phosphorylation of PKCα/β2 and ameliorate the impairment of calcium-handling proteins.
It was reported that the decreased expression of PKC resulted in inactivation of PPI-1, activating the phosphorylation of protein phosphatase-1, which in turn regulated PLB phosphorylation 38,39, thereby promoting the function of SERCA-2a responsible for Ca2+ loading and the magnitude of the Ca2+ transients 40,41. In this study, therefore, a decrease in SERCA2a expression along with an increase in PLB and NCX1 protein level in SHRs could lead to a decreased SR function causing a decreased SR Ca2+ store and rate of Ca2+ releasing from the SR. These changes were found to be capable of affecting the contractile function of the heart, which were reversed following the administration of Rosuvastatin, suggesting that Rosuvastatin could ameliorate the changes in Ca2+-handling protein via PKC signalling pathway.
It can be concluded from this study that Rosuvastatin therapy was proved to exert a protective effect on the end stage of hypertension with dephosphorylation of PKCα/β2, causing the consequent dephosphorylation of PPI-1 and increased expression of CaMKII, which could result in the decreased expression of PLB and increased SERCA2a, RyR2 in the aged SHRs.
Beyond lipid lowering, statins have many pleiotropic effects, such as stimulating stem cells 42,43, activating immune system 44, activating heme oxygenase-1 at the stress response elements 45 and having synergistic effects with telmisartan on endothelial progenitor cells 46. Those we discovered above can be one of the statins’ pleiotropic effects, which can make statins potentially beneficial to patients with heart failure, for whom hypertension is the main risk factor.
Acknowledgments
This work was supported by the Outstanding Youth Grant from National Natural Science Foundation of China (30725036). The authors are thankful to Dr. Xinxing Xie from the Department of Cardiology of Qianfoshan Hospital, Shandong Province, for his animal dissections. We are also grateful to Dr. Jianguo Jia and Jian Wu from Shanghai Institute of Cardiovascular Diseases of Zhongshan Hospital, Fudan University, for their technical assistance in echocardiographic evaluation.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- Brown MJ, McInnes GT, Papst CC, et al. Aliskiren and the calcium channel blocker amlodipine combination as an initial treatment strategy for hypertension control (ACCELERATE): a randomised, parallel-group trial. Lancet. 2011;377:312–20. doi: 10.1016/S0140-6736(10)62003-X. [DOI] [PubMed] [Google Scholar]
- Calhoun DA, White WB, Krum H, et al. Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension: results of a randomized, double-blind, placebo- and active-controlled phase 2 trial. Circulation. 2011;124:1945–55. doi: 10.1161/CIRCULATIONAHA.111.029892. [DOI] [PubMed] [Google Scholar]
- Sciarretta S, Palano F, Tocci G, et al. Antihypertensive treatment and development of heart failure in hypertension: a Bayesian network meta-analysis of studies in patients with hypertension and high cardiovascular risk. Arch Intern Med. 2011;171:384–94. doi: 10.1001/archinternmed.2010.427. [DOI] [PubMed] [Google Scholar]
- Marzoll A, Melchior-Becker A, Cipollone F, et al. Small leucine-rich proteoglycans in atherosclerotic lesions: novel targets of chronic statin treatment. J Cell Mol Med. 2011;15:232–43. doi: 10.1111/j.1582-4934.2009.00986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibi K, Kimura T, Kimura K, et al. Clinically evident polyvascular disease and regression of coronary atherosclerosis after intensive statin therapy in patients with acute coronary syndrome: serial intravascular ultrasound from the Japanese assessment of pitavastatin and atorvastatin in acute coronary syndrome (JAPAN-ACS) trial. Atherosclerosis. 2011;219:743–9. doi: 10.1016/j.atherosclerosis.2011.08.024. [DOI] [PubMed] [Google Scholar]
- Domanski M, Coady S, Fleg J, et al. Effect of statin therapy on survival in patients with nonischemic dilated cardiomyopathy (from the Beta-blocker Evaluation of Survival Trial [BEST]) Am J Cardiol. 2007;99:1448–50. doi: 10.1016/j.amjcard.2006.12.080. [DOI] [PubMed] [Google Scholar]
- Sicard P, Zeller M, Dentan G, et al. Beneficial effects of statin therapy on survival in hypertensive patients with acute myocardial infarction: data from the RICO survey. Am J Hypertens. 2007;20:1133–9. doi: 10.1016/j.amjhyper.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Murphy SA, Cannon CP, Wiviott SD, et al. Reduction in recurrent cardiovascular events with intensive lipid-lowering statin therapy compared with moderate lipid-lowering statin therapy after acute coronary syndromes from the PROVE IT-TIMI 22 (Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis In Myocardial Infarction 22) trial. J Am Coll Cardiol. 2009;54:2358–62. doi: 10.1016/j.jacc.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Horwich TB, MacLellan WR, Fonarow GC. Statin therapy is associated with improved survival in ischemic and non-ischemic heart failure. J Am Coll Cardiol. 2004;43:642–8. doi: 10.1016/j.jacc.2003.07.049. [DOI] [PubMed] [Google Scholar]
- Tehrani F, Morrissey R, Phan A, et al. Statin therapy in patients with diastolic heart failure. Clin Cardiol. 2010;33:E1–5. doi: 10.1002/clc.20615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thambidorai SK, Deshmukh AR, Walters RW, et al. Impact of statin use on heart failure mortality. Int J Cardiol. 2011;147:438–43. doi: 10.1016/j.ijcard.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Danielson E, Fonseca FA, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373:1175–82. doi: 10.1016/S0140-6736(09)60447-5. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–8. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
- Braunwald E. Creating controversy where none exists: the important role of C-reactive protein in the CARE, AFCAPS/TexCAPS, PROVE IT, REVERSAL, A to Z, JUPITER, HEART PROTECTION, and ASCOT trials. Eur Heart J. 2012;33:430–2. doi: 10.1093/eurheartj/ehr310. [DOI] [PubMed] [Google Scholar]
- Zhang WB, Du QJ, Li H, et al. The therapeutic effect of Rosuvastatin on cardiac remodeling from hypertrophy to fibrosis during the end-stage hypertension in rats. J Cell Mol Med. 2012;16:2227–37. doi: 10.1111/j.1582-4934.2012.01536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Minamino T, Toko H, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–8. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- Shiva S, Brookes PS, Patel RP, et al. Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc Natl Acad Sci USA. 2001;98:7212–7. doi: 10.1073/pnas.131128898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabu SK, Anandatheerthavarada HK, Raza H, et al. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J Biol Chem. 2006;281:2061–70. doi: 10.1074/jbc.M507741200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson J, Poitevin P, Chillon JM, et al. Vascular Ca overload produced by vitamin D3 plus nicotine diminishes arterial distensibility in rats. Am J Physiol. 1994;266:H540–7. doi: 10.1152/ajpheart.1994.266.2.H540. [DOI] [PubMed] [Google Scholar]
- Hu X, Li T, Zhang C, et al. GATA4 regulates ANF expression synergistically with Sp1 in a cardiac hypertrophy model. J Cell Mol Med. 2011;15:1865–77. doi: 10.1111/j.1582-4934.2010.01182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susic D, Varagic J, Ahn J, et al. Beneficial pleiotropic vascular effects of rosuvastatin in two hypertensive models. J Am Coll Cardiol. 2003;42:1091–7. doi: 10.1016/s0735-1097(03)00926-4. [DOI] [PubMed] [Google Scholar]
- Loch D, Levick S, Hoey A, et al. Rosuvastatin attenuates hypertension-induced cardiovascular remodeling without affecting blood pressure in DOCA-salt hypertensive rats. J Cardiovasc Pharmacol. 2006;47:396–404. doi: 10.1097/01.fjc.0000210072.48991.f6. [DOI] [PubMed] [Google Scholar]
- Saka M, Obata K, Ichihara S, et al. Pitavastatin improves cardiac function and survival in association with suppression of the myocardial endothelin system in a rat model of hypertensive heart failure. J Cardiovasc Pharmacol. 2006;47:770–9. doi: 10.1097/01.fjc.0000211791.22411.0d. [DOI] [PubMed] [Google Scholar]
- Prandin MG, Cicero AF, Dormi A, et al. Prospective evaluation of the effect of statins on blood pressure control in hypertensive patients in clinical practice. Nutr Metab Cardiovasc Dis. 2010;20:512–8. doi: 10.1016/j.numecd.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Jarai R, Kaun C, Weiss TW, et al. Human cardiac fibroblasts express B-type natriuretic peptide: fluvastatin ameliorates its up-regulation by interleukin-1alpha, tumour necrosis factor-alpha and transforming growth factor-beta. J Cell Mol Med. 2009;13:4415–21. doi: 10.1111/j.1582-4934.2009.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florkowski CM, Molyneux SL, George PM. Rosuvastatin in older patients with systolic heart failure. N Engl J Med. 2008;358:1301. doi: 10.1056/NEJMc073536. [DOI] [PubMed] [Google Scholar]
- Chang SA, Kim YJ, Lee HW, et al. Effect of rosuvastatin on cardiac remodeling, function, and progression to heart failure in hypertensive heart with established left ventricular hypertrophy. Hypertension. 2009;54:591–7. doi: 10.1161/HYPERTENSIONAHA.109.131243. [DOI] [PubMed] [Google Scholar]
- Hambleton M, Hahn H, Pleger ST, et al. Pharmacological- and gene therapy-based inhibition of protein kinase Calpha/beta enhances cardiac contractility and attenuates heart failure. Circulation. 2006;114:574–82. doi: 10.1161/CIRCULATIONAHA.105.592550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte SL, Blaxall BC. PKC-ing is believing: targeting protein kinase C in heart failure. Circ Res. 2011;109:1320–2. doi: 10.1161/CIRCRESAHA.111.259358. [DOI] [PubMed] [Google Scholar]
- Palaniyandi SS, Sun L, Ferreira JC, et al. Protein kinase C in heart failure: a therapeutic target. Cardiovasc Res. 2009;82:229–39. doi: 10.1093/cvr/cvp001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qvit N, Mochly-Rosen D. Highly specific modulators of protein kinase C localization: applications to heart failure. Drug Discov Today Dis Mech. 2010;7:e87–87e93. doi: 10.1016/j.ddmec.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaniyandi SS, Ferreira JC, Brum PC, et al. PKCbetaII inhibition attenuates myocardial infarction induced heart failure and is associated with a reduction of fibrosis and pro-inflammatory responses. J Cell Mol Med. 2011;15:1769–77. doi: 10.1111/j.1582-4934.2010.01174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki K, Koyanagi T, Berry NC, et al. Pharmacological inhibition of epsilon-protein kinase C attenuates cardiac fibrosis and dysfunction in hypertension-induced heart failure. Hypertension. 2008;51:1565–9. doi: 10.1161/HYPERTENSIONAHA.107.109637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Chen X, Macdonnell SM, et al. Protein kinase C{alpha}, but not PKC{beta} or PKC{gamma}, regulates contractility and heart failure susceptibility: implications for ruboxistaurin as a novel therapeutic approach. Circ Res. 2009;105:194–200. doi: 10.1161/CIRCRESAHA.109.195313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrez N, Morel JL, Kalkbrenner F, et al. A betagamma dimer derived from G13 transduces the angiotensin AT1 receptor signal to stimulation of Ca2 + channels in rat portal vein myocytes. J Biol Chem. 1997;272:23180–5. doi: 10.1074/jbc.272.37.23180. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Griendling KK, Akers M, et al. Temporal dispersion of activation of phospholipase C-beta1 and -gamma isoforms by angiotensin II in vascular smooth muscle cells. Role of alphaq/11, alpha12, and beta gamma G protein subunits. J Biol Chem. 1998;273:19772–7. doi: 10.1074/jbc.273.31.19772. [DOI] [PubMed] [Google Scholar]
- Hansen JL, Theilade J, Haunso S, et al. Oligomerization of wild type and nonfunctional mutant angiotensin II type I receptors inhibits galphaq protein signaling but not ERK activation. J Biol Chem. 2004;279:24108–15. doi: 10.1074/jbc.M400092200. [DOI] [PubMed] [Google Scholar]
- Rodriguez P, Mitton B, Waggoner JR, et al. Identification of a novel phosphorylation site in protein phosphatase inhibitor-1 as a negative regulator of cardiac function. J Biol Chem. 2006;281:38599–608. doi: 10.1074/jbc.M604139200. [DOI] [PubMed] [Google Scholar]
- Sahin B, Shu H, Fernandez J, et al. Phosphorylation of protein phosphatase inhibitor-1 by protein kinase C. J Biol Chem. 2006;281:24322–35. doi: 10.1074/jbc.M603282200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louch WE, Hougen K, Mork HK, et al. Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J Physiol. 2010;588:465–78. doi: 10.1113/jphysiol.2009.183517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwano K, Arai M, Koitabashi N, et al. Lentiviral vector-mediated SERCA2 gene transfer protects against heart failure and left ventricular remodeling after myocardial infarction in rats. Mol Ther. 2008;16:1026–32. doi: 10.1038/mt.2008.61. [DOI] [PubMed] [Google Scholar]
- Revenco D, Morgan JP. Metabolic modulation and cellular therapy of cardiac dysfunction and failure. J Cell Mol Med. 2009;13:811–25. doi: 10.1111/j.1582-4934.2009.00759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi K, Takehara N, Matsubara H, et al. Stemming heart failure with cardiac- or reprogrammed-stem cells. J Cell Mol Med. 2008;12:2217–32. doi: 10.1111/j.1582-4934.2008.00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenbacher G, Gander H, Nussbaumer O, et al. IL-2 costimulation enables statin-mediated activation of human NK cells, preferentially through a mechanism involving CD56 + dendritic cells. Cancer Res. 2010;70:9611–20. doi: 10.1158/0008-5472.CAN-10-1968. [DOI] [PubMed] [Google Scholar]
- Kwok SC, Samuel SP, Handal J. Atorvastatin activates heme oxygenase-1 at the stress response elements. J Cell Mol Med. 2012;16:394–400. doi: 10.1111/j.1582-4934.2011.01324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz M, Brouwers C, Nickenig G, et al. Synergistic effects of telmisartan and simvastatin on endothelial progenitor cells. J Cell Mol Med. 2010;14:1645–56. doi: 10.1111/j.1582-4934.2009.00829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]