

Abstract
The lymphatic system is essential for the maintenance of tissue homeostasis and immunity. Its dysfunction in disease (such as lymphangioleiomyomatosis) can lead to chylous effusions, oedema or dissemination of malignant cells. Collagen IV has six α chains, of which some of the non-collagenous-1 domains have endogenous anti-angiogenic properties, however, little is known about specific endogenous anti-lymphangiogenic characteristics. In this study we sought to investigate the expression levels of collagen IV non-collagenous-1 domains in lung tissue of patients with and without lymphangioleiomyomatosis to explore the hypothesis that a member of the collagen IV family, specifically the non-collagenous domain-1 of α5, which we named lamstatin, has anti-lymphangiogenic properties. Levels of lamstatin detected by immunohistochemistry were decreased in lungs of lymphangioleiomyomatosis patients. We produced recombinant lamstatin in an E.coli expression system and synthesized a 17-amino acid peptide from a theoretically identified, active region (CP17) and tested their effects in vitro and in vivo. Recombinant lamstatin and CP17 inhibited proliferation, migration and cord formation of human microvascular lung lymphatic endothelial cells, in vitro. Furthermore, lamstatin and CP17 decreased complexity and dysplasia of the tumour-associated lymphatic network in a lung adenocarcinoma xenograft mouse model. In this study we identified a novel, direct inhibitor of lymphangiogenesis, derived from collagen IV. This may prove useful for exploring new avenues of treatment for lymphangioleiomyomatosis and metastasis via the lymphatic system in general.
Keywords: Lymphangiogenesis, Lymphangioleiomyomatosis, collagen, type IV collagen alpha5 chain
Introduction
Lymphangiogenesis, the formation of new lymphatic vessels from pre-existing ones, has an essential role in the maintenance of tissue homeostasis and immunity. The recent elucidation of the role of the lymphatic network in the dissemination of malignant tumour cells 1 has highlighted the need for a greater understanding of the regulatory mechanisms involved. It is suggested that lymphangiogenesis is regulated by pro- and anti-lymphangiogenic factors maintaining a steady-state balance under normal conditions, however, in the presence of inflammation or disease, this balance is shifted towards pro-lymphangiogenic factors. Vascular endothelial growth factor (VEGF)-C and -D are the most prominent members of this group, but others such as basic fibroblast growth factor (bFGF) and angiopoietin (Ang)-1 have also been shown to induce lymphangiogenesis 2–5. Vascular endothelial growth factor-C and -D bind to the tyrosine kinase receptor VEGFR-3 or Flt-4, which is highly expressed on lymphatic endothelial cells 6–10. However, direct, tissue-derived inhibitors of lymphangiogenesis have received very little attention to date.
Collagen IV (Col IV) is an important component of the extracellular matrix (ECM), deposited as heterotrimers formed when any three chains of the six genetically different α chains (α1–α6) combine. Each α chain consists of a helical and a non-collagenous (NC)1 domain and all are present in the healthy lung 11. Interestingly, the absence or decreased detection of members of the Col IV family, especially α5, is associated with higher levels of tumour cell invasion in adjacent tissue in breast, prostate, bronchoalveolar and colorectal cancer 12–16. Some NC1 domains have been reported to have anti-angiogenic properties (e.g. α1, α2, α3) but others, such as α4, α5 and α6 have not yet been ascribed a function, especially in terms of lymphangiogenesis (reviewed in 17,18).
Lymphangioleiomyomatosis (LAM) is a rare (up to 5 per million) but progressive disease affecting predominantly women of childbearing age 19. Lymphangioleiomyomatosis manifests itself either as part of the genetic disorder tuberous sclerosis (TSC) or occurs sporadically (∽60% of cases), and demonstrates characteristics of metastatic disease 20–23. The pulmonary component of LAM features alterations in the ECM, cystic lesions, as well as nodules consisting of and generated by abnormally proliferating and metastasizing smooth muscle-like cells (LAM cells) 24–26. In addition, the number of lymphatic vessels is vastly increased in lungs from patients with LAM compared with lungs from individuals without LAM, and the increase of lymphangiogenesis in LAM patients is correlated with decreased survival rates 27. Pro-lymphangiogenic factors, including VEGF-C (produced by LAM cells in vivo 27), VEGF-D (elevated in the serum of LAM patients 28) and bFGF (produced by tryptase+ mast cells in the vicinity of hyperplastic smooth muscle-like cells 29), are increased in LAM. However, the possibility that an endogenous anti-lymphangiogenic factor is dysregulated in the lungs of women with LAM has not been considered previously.
In this study we investigated the expression levels of Col IV NC1 domains in patients with and without LAM, and explored the hypothesis that a member of the Col IV family, specifically the NC1 domain of α5, which we named lamstatin, or a 17 amino acid fragment we identified as a theoretical active site and called CP17, have hitherto uncharacterized anti-lymphangiogenic properties.
Material and methods
Immunohistochemistry
We studied paraffin embedded tissue sections of bronchial rings from patients with LAM and sections derived from macroscopically normal tissue distant from tumours following cancer resections which served as controls, in addition to tissues from patients free of respiratory disease. All tissues were stained for the six collagen IV isoforms (NC1 domain-specific antibodies). An additional group of LAM patients was stained for collagen IV α1, α3 and α5. Bronchial rings from people with cystic fibrosis and bronchiectasis were also stained for collagen IV α3 and α5. We have previously described the staining of the controls used in this study 11. The controls for staining collagen IV α1–α6 were derived from macroscopically normal tissue taken from a location distant from the tumour following cancer resections. In addition, tissue was also harvested from endobronchial biopsies or lungs removed at transplantation from patients free of respiratory disease. We have also previously described the staining of tissues from patients with cystic fibrosis and those with bronchiectasis for collagen IV α3 11. Sections were deparaffinized and rehydrated through graded alcohol. Blocking serum (10% non-immune horse serum) was then added to the sections for 20 min. at room temperature. Without rinsing, either primary antibodies (collagen IV α1–6 NC1 a kind gift from Dr Sado at Shigei Medical Research Institute, Okayama, Japan (1 ng/ml) 16) or isotype control antibody (Rat IgG, Jackson ImmunoResearch, West Grove, PA, USA (1 ng/ml)) was added to the sections and incubated for 1 hr at room temperature. Sections were then washed with PBS and a goat anti-rat fluorescein isothiocyanate (FITC) [MP Biomedicals, Solon, OH, USA (1 ng/ml)] conjugated secondary antibody was added, and incubated for 30 min. at room temperature. Following a rinse with PBS, slides were mounted using Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA). Images were taken on an Olympus BX51 (Olympus Australia, Mt Waverley, Australia) fluorescence microscope and captured using Leica imaging software IM1000 (Leica, Wetzlar, Germany) as described previously 11. The presence and intensity of staining for the collagen IV α1–6 chain NC1 domains in the images from four controls and eight LAM patients was scored by three independent observers, who were blinded to the diagnosis of the participants. Images were scored as follows; 0: Absence of stain; 1: Weak and discontinuous staining; 2: Weak and continuous staining; 3: Strong and continuous staining. Scores from the three observers were averaged and the standard error of the mean calculated.
Gene cloning, expression and purification of recombinant protein
The DNA for the NC1 domain of human Col4α5 (lamstatin, chromosome Xq22.3) was extracted from primary human lung endothelial cells. Briefly, cells were extracted from blood vessels dissected from human donor lungs as described previously 11, expanded in tissue culture medium containing 10% FBS, 10 μg/ml endothelial cell growth supplement, 20 U/ml heparin and 2% antibiotics, and total RNA was extracted using the NucleoSpin RNA II kit according to the manufacturer's instructions (Macherey Nagel, Düren, Germany). Total RNA was transcribed to cDNA using hexameric primers (New England Biolabs, Ipswich, MA, USA) and Superscript III (Invitrogen, Carlsbad, CA, USA).
The MMP cleavage site prediction tool (http://www.dmbr.ugent.be/prx/bioit2-public/SitePrediction/index.php) was used to identify the MMP2 cleavage site at the beginning of the NC1 domain in the collagen IV α5 aa sequence, and primers that recognized the corresponding gene sequence were designed. The cDNA was then amplified with the following primers: 5′-TTCCATATGGGATTTCTTATTACA-3′ (forward), 5′-CGGGATCCTTATGTCCTCTTCATGCA-3′ (reverse) with restriction sites for NdeI (forward) and BamHI (reverse). PCR amplification was undertaken for 35 cycles with the following conditions: denaturation at 95°C for 15 sec., annealing at 60°C for 30 sec. and elongation at 72°C for 60 sec. The amplicon (675 bp) was eluted from a 1.5% Agarose gel (Amresco, Cochran Solon, OH, USA) using a QIAEX II gel extraction kit (Qiagen, Doncaster, VIC, Australia) and cloned into pcDNΑ5/FRT/TO-TOPO (Invitrogen) according to the manufacturer's recommendations. The vector was transformed into TOP10 E.coli (Invitrogen) and streaked on agar plates with ampicillin (100 μg/ml) (Sigma-Aldrich, St. Louis, MO, USA). Colonies were picked, expanded and the inserts within the isolated plasmids were subject to sequencing (Supamac, Sydney, Australia). Positive clones were selected and archived for later use. Lamstatin was then subcloned into pET15b (via BamHI and NdeI) and transformed into BL21 (DE3) (Bioline, Sydney, NSW, Australia) for expression. E.coli were grown overnight, and then expansion cultures were started with an innoculum of OD 0.1 and grown until they reached OD 0.5. Expression was then induced with 119.2 mg/l of isopropyl 1-thio-β-D-galactopyranoside (IPTG; Sigma-Aldrich) for 4 hrs and cells were pelleted thereafter at 4°C at 4000 × g for 20 min. Pellets were collected and washed twice with buffer A and then resuspended in buffer A (7.9 g/l Tris–HCl, 1.46 g/l EDTA, pH 7.5). Cells were then sonicated on ice for 50 cycles (4 sec. at 60% of max. amplitude and 6 sec. pause). The suspension was pelleted at 15,000 × g for 20 min. before washing with solubilization buffer 1 (1% Triton X-100 and 180.2 g/l urea). The supernatant (15,000 × g, 20 min.) was removed and inclusion bodies were incubated with solubilization buffer 2 (354.4 g/l guanidine, 10.3 g/l NaHPO3 and 1.58 g/l Tris–HCl, pH 5.5) for 2 hrs at RT. Insoluble debris was spun down and the lysate was either purified via a Nickel-sepharose column (AmershamPharmacia, GE Healthcare, Rydalmere, NSW, Australia) or directly processed by dilution and ultra filtration (Amicon Ultra15, 10 kD; Millipore, Billerica, MA, USA). Purified protein was analysed on PAGE for purity (Coomassie Blue staining) and stored at −80°C for later use. The protein concentration was measured by UV (280 nm; NanoDrop, Wilmington, DE, USA) and bicinchoninic acid assay (Sigma, Sydney, Australia). CP17 was obtained from AusPep (Tullamarine, Victoria, Australia) in HPLC grade purity.
Cells and media
Human lung lymphatic endothelial cells (HMVEC-LLy) were purchased from Lonza (Basel, Switzerland) together with the EGM-2 MV BulletKit [composition: hEGF, Hydrocortisone, GA-1000 (gentamicin, Amphotericin-B), FBS (Foetal Bovine Serum), VEGF, hFGF-B, R3-IGF-1, Ascorbic Acid (Lonza)] for expansion. Human umbilical vein endothelial cells (HUVECs) were a kind gift from Dr Anthony Ashton at the Kolling Institute and Prof Jenny Gamble at the Centenary Institute, The University of Sydney. Human umbilical vein endothelial cells were cultured on gelatin-coated flasks in medium M199 containing sodium bicarbonate, non-essential amino acids, sodium pyruvate, 20% foetal bovine serum (FBS), 1% antibiotic–antimycotic mix, 50 μg/ml endothelial cell growth supplement (BD Bioscience, San Jose, CA, USA) and 50 μg/ml heparin (Sigma-Aldrich). Cells were used at passage 3–5. Α549 cells originated from the ATCC (Manassas, Virginia, USA) and were grown in DMEM (Gibco, Invitrogen) with 10% FBS, 2% Penicillin-Streptomycin. Primary human lung fibroblasts and airway smooth muscle cells were isolated and grown as previously described 11,30–37. The study was approved by the Ethics Review Committee of the South West Sydney Area Health Service, Royal Prince Alfred Hospital, St. Vincent Hospital and The University of Sydney human research ethics committee. Written informed consent was obtained from all participants.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl Tetrazoliumbromide (MTT) cell viability assay
Cells in medium were seeded at a density of 5000/well in a 96-well plate (Nunc, Thermo Fisher Scientific, Rockford, IL, USA) and 24 hrs later treated with lamstatin or CP17 as indicated. To ensure specificity of CP17, another collagen IV derived, blood vascular endothelium anti-proliferative peptide (T3) identified by Maeshima et al. 38,39 was also tested. Cells were grown for 66 hrs after treatment, before MTT was added. After 6 hrs of incubation, MTT-formazan development was stopped with 10% SDS and 3.7 g/l HCl. OD was read at 570 nm (690 nm background reference reading) in a Spectramax M2 (Molecular Devices, Sunnyvale, CA, USA) spectrometer. Results were expressed as percentage of vehicle control. The vehicle for lamstatin was 292.2 ng/l EDTA/pH 3.5; that for the CP17 peptide was MilliQ water and for the T3 peptide we used 40% Acetonitrile with 0.1% trifluoracetate.
Trypan blue exclusion cell viability assay
Cells were seeded and stimulated as described above. Instead of addition of MTT, cells were harvested by detaching them with 0.05% trypsin in 198 mg/l disodium EDTA for 5 min. and resuspended in fresh Hanks buffered saline solution. Twenty microlitres of cell suspension was then diluted 1:1 with trypan blue and loaded onto a counting chamber of a Nexcelom Cellometer™ AutoT4 (Nexcelom Bioscience, Lawrence, MA, USA). Samples were automatically counted and visually inspected for viable cells.
Manual cell counts
Human umbilical vein endothelial cells were seeded (1 × 104 cells/cm2) in 12-well plates for 24 hrs before they were treated with lamstatin (2.5–10 μg/ml), vehicle (1 nM EDTA, pH3.5) or CP17 (2.5–10 μg/ml) for 72 hrs. Cells were then rinsed with PBS, detached using trypsin and stained with trypan blue. Cells were then counted manually using a haemocytometer.
Cord formation assay
Matrigel (BD Bioscience) was prepared in 48-well plates according to the manufacturer's recommendations. HMVEC-LLys were seeded at a concentration of 2 × 104 cells/well and allowed to attach for 1 hr. The cells were then grown in complete EBM-2 MV in the presence or absence of lamstatin or CP17 (both 1.25, 2.5, 5, 10 μg/ml) or vehicle (292.2 ng/l EDTA/pH 3.5). Effects were observed 24 hrs after addition of lamstatin or CP17. Human umbilical vein endothelial cells were seeded on to geltrex (Invitrogen) coated 24-well plates for 2 hrs before being treated with lamstatin (10 μg/ml), vehicle or CP17 (10 μg/ml) for up to 18 hrs at 37°C, 5% CO2. Images were taken using a Ti Elipse microscope (Nikon) with a constant magnification factor of 100× and the panorama setting on an Olympus CAMEDIA C-4000 camera (Olympus, Hamburg, Germany). The number of cords was counted from a 416 px × 333 px (=138.5 px2) area divided into nine segments of equal size, the cords per field were counted and the average number of cords per 1000 px2 was calculated.
Migration assay
Subconfluent HMVEC-LLy was harvested after a 4 hrs starving period in EBM-2 MV (no bullet kit supplement, but 0.1% BSA) using non-enzyme detachment solution (Trevigen, Gaithersburg, MD, USA), washed and the concentration adjusted to 4.0 × 105 cells/ml in EBM-2 MV (0.1% BSA). A 24-well insert (BD Falcon™ FluoroBlok, BioCoat for Endothelial Migration, BD Bioscience) was brought to room temperature and the top well was filled with 250 μl of cell suspension with either vehicle control or 2.5, 5 or 10 μg/ml lamstatin or CP17. Cells were incubated for 1 hr at 37°C, before EBM-2 MV (complete), with or without treatment (see above), was added to the bottom well. The presence of the inhibitor in the top and bottom wells ensured a constant concentration over the course of the experiment. Cells were allowed 22 hrs to migrate before the filter was washed with HBSS twice and stained with Calcein (4 μg/ml in HBSS, Invitrogen Molecular Probes) for 90 min. Fluorescence was read in a Spectramax M2 (Molecular Devices, Sunnyvale, CA, USA) at 488 nm excitation and 520 nm emission. Experiments were repeated three times.
Mouse model of angio- and lymphangiogenesis
All animal experiments were approved by the provincial state office of southern Finland and carried out in accordance with institutional guidelines. NOD/SCID/gamma mice were injected in the ears with LNM35 AΑV-EGFP-expressing tumour cells suspended in growth factor reduced Matrigel® with or without 10 or 100 μg/ml lamstatin, 10 or 100 μg/ml CP17 or respective vehicle (292.2 ng/l EDTA pH 3.5). After 12 days of tumour growth, animals were killed and ears were collected. Whole mount staining of the ears was performed as described earlier 40. Lymphatic vessels were stained with LYVE-1 (AlexaFluor 647; Invitrogen, Carlsbad, CA, USA), blood vessels were stained with PECAM-1 (BD Bioscience clone MEC13.3) and AlexaFluor 594. LNM35 tumour cells were detected using their green fluorescent protein (EGFP) expression. Confocal imaging (constant thickness and scanning intervals) enabled visualization of tumour-associated lymphatic networks. Analysis of the mean intensity (this value represents the mean intensity of the pixels inside the image boundaries) of LYVE-1 staining was performed with QuantityOne (Bio-Rad, Hercules, CA, USA). The number of branching points or loops was assessed as described by Shayan et al. 41. In brief, confocal images of lymphatic vessels were loaded into Image J (www.sbweb.nih.gov/ij/) and a 5 × 5 grid was overlaid. Branches were defined as two clearly distinguishable vessels that separate out without rejoining. Loops were defined as small, circular vessel structures, which had to be in the same focal plane. Branches or loops were counted separately in each image. One image, created from one stack of 64 confocal images from a single location in the experimental ear was analysed per animal and mean values were calculated for each treatment group. For the lamstatin experiments we studied three animals in which no tumour cells were present, 11 which received vehicle alone, eight which received 10 μg/ml lamstatin and nine which received 100 μg/ml lamstatin. Similarly, for experiments in which the effect of CP17 was examined we studied three animals in which no tumour cells were present, 11 which received vehicle alone, eight which received 10 μg/ml CP17 and six which received 100 μg/ml CP17.
After 12 days tumour sizes were measured in all animals treated with either vehicle (292.2 ng/l EDTA, pH 3.5), 10 or 100 μg/ml lamstatin. Tumour was assumed to be ellipsoid in shape and thickness, and width and length were measured using a micrometre calliper. Volume was calculated according to the formula for ellipsoid bodies and plotted in mm3.
Statistical analysis
Values were considered to be significantly different if P < 0.05. All calculations were performed with GraphPad Prism 5 (Macintosh version, GraphPad Software, San Diego, CA, USA). Non-parametric analysis of variance (anova) with repeated measures was used (Kruskal–Wallis) with Dunn's or Bonferroni's post-tests or paired Student's t-tests where appropriate.
Results
Collagen IV α3 and α5 NC1 domains are reduced in LAM lung sections
Paraffin embedded lung sections from LAM (n = 8) and control (n = 10) patients were immunohistochemically stained for all six isoforms of Col IV. An additional 2 LAM patients were stained for collagen IV α1, α3 and α5. In Figure1A, representative examples of LAM lung sections (top) are compared with well-defined control patients (which have been previously analysed for the expression of collagen IV α1–6 and published elsewhere in a separate study 11) (middle). The isotype control is shown below the specific stains. Detection of tumstatin (the NC1 domain of collagen IV α3) or lamstatin (NC1 of collagen IV α5) staining was significantly reduced in the tissue from all LAM patients examined. However, these isoforms were present in the control patients. We did not identify any other Col IV isoforms which were differentially stained.
Fig 1.
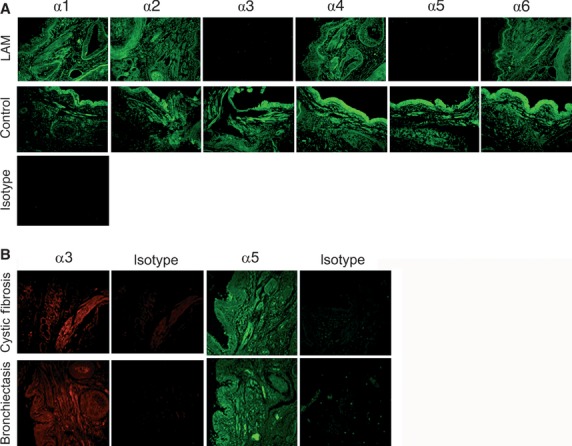
Immunohistochemical staining for Col IV NC1 isoforms. (A) Col IV NC1 domains α1–α6 were stained in human lung tissue sections using specific antibodies and detected using a secondary antibody conjugated to fluorescein isothiocyanate (FITC) (green staining). Eight lymphangioleiomyomatosis (LAM) patients and 10 controls were tested. Scale bar represents 50 μm. L: lumen; B: basal lamina; V: blood vessel. (B) Col IV α3 (red staining) and α5 (green staining) NC1 domains were detected in human lung tissue sections from four cystic fibrosis patients and one bronchiectasis patient. The isotype control was exposed to rat IgG and the secondary antibody.
Lymphangioleiomyomatosis (n = 8) and control (n = 4) sections were scored for the level of staining observed (Table1). All LAM sections had reduced scores for the level of tumstatin observed (0.5 ± 0.16) as well as for lamstatin (0.56 ± 0.12) compared with the control sections. We have previously explored the role of the collagen IV α3 NCI domain (tumstatin) in asthma 11, therefore in this study we chose to focus on the effects of the collagen IV α5 NCI domain (lamstatin).
Table 1.
Levels of the six collagen IV α chain NC1 domains observed in stained airway sections from lymphangioleiomyomatosis (LAM) and control individuals
| n | α1 | α2 | α3 | α4 | α5 | α6 | |
|---|---|---|---|---|---|---|---|
| LAM (SEM) | 8 | 2.16 (±0.26) | 2.28 (±0.18) | 0.5 (±0.16) | 2.63 (±0.17) | 0.56 (±0.12) | 2.33 (±0.12) |
| Control (SEM) | 4 | 1.72 (±0.3) | 2.21 (±0.17) | 2.29 (±0.20) | 2.72 (±0.20) | 2.47 (±0.22) | 2.5 (±0.20) |
Scoring results obtained from three independent observers blinded to the patient diagnosis using the following scale: 0: Absence of stain; 1: Weak and discontinuous staining; 2: Weak and continuous staining; 3: Strong and continuous staining.
To test whether the absence of lamstatin was a feature of other chronic respiratory disorders, tissue sections from patients with cystic fibrosis (n = 4) and bronchiectasis (n = 1) were stained for col IV α3 and α5 and all were positive for tumstatin and lamstatin as shown in Figure1B.
Identification of the potentially active peptide in lamstatin
We initially observed anti-proliferative activity of recombinant tumstatin (produced in our laboratory using a similar approach to that described here for lamstatin) and lamstatin on HMVEC-LLy (data not shown). To identify potentially active regions in lamstatin, we analysed the sequences of tumstatin and lamstatin for homology (using an approach similar to but not identical to that of Karagiannis et al. 17) and a single and unique stretch of 17 aa with a molecular weight of 2022 Da was identified, which we termed as CP17. The aa sequence of CP17 is VCNFASRNDYSYWLSTP and it runs between aa 66 and 82 of the lamstatin sequence.
Lamstatin and the consensus peptide CP17 reduce viability of proliferating HMVEC-LLy
We treated adherent, subconfluent (approx 50%) and proliferating HMVEC-LLy cells from two healthy donors for 72 hrs with increasing concentrations of lamstatin or CP17. Lamstatin significantly reduced cell viability at concentrations of 5 and 10 μg/ml (maximal reduction of ∽25% for lamstatin) (n = 5 experimental repeats) (Fig.2A). Conversely, lamstatin had no effect on the viability of primary lung fibroblasts (n = 3 non-LAM cell lines), primary airway smooth muscle cells (n = 6 non-LAM cell lines) and Α549 epithelial cells (n = 3 experimental repeats) (Fig.2A). CP17 produced a similar significant reduction in cell viability to lamstatin, at concentrations of 5 and 10 μg/ml (n = 3 experimental repeats) (Fig.2B). However, treatment with lamstatin had no effect on cell viability in growth factor-starved confluent HMVEC-LLys (n = 6 experimental repeats) (Fig.2C). We also compared the effect of the previously published peptide T3 (one of the active regions of tumstatin) 38 and found it had no effect on lymphatic endothelial cell proliferation (n = 3 experimental repeats) (data not shown).
Fig 2.
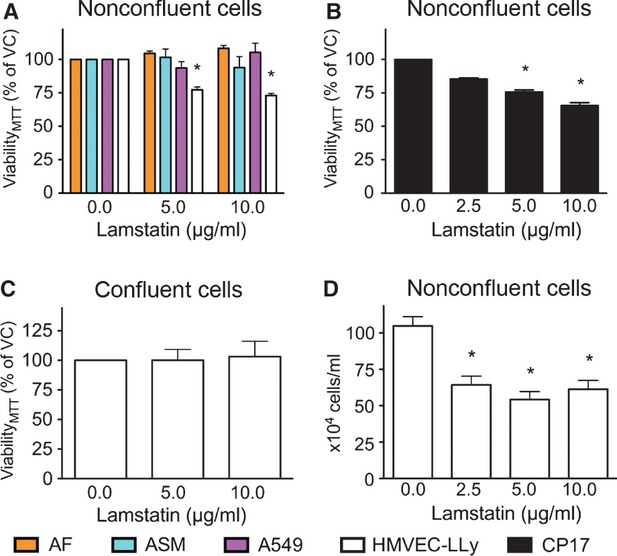
Lamstatin and CP17 reduce cell viability of proliferating human lung lymphatic endothelial cells (HMVEC-LLys). Subconfluent cells were treated for 72 hrs with lamstatin or CP17 and cell viability, as measured with MTT, expressed as percentage of vehicle control (% VC). (A) Primary lung fibroblasts [n = 3, non-lymphangioleiomyomatosis (LAM) cell lines] (orange), primary airway smooth muscle cells (n = 6 non-LAM cell lines) (light blue), Α549 cells (n = 3 experimental repeats) (pink) or HMVEC-LLYs (white) (n = 5 experimental repeats) treated with lamstatin; (B) HMVEC-LLys (n = 3 experimental repeats) treated with CP17; (C) Confluent HMVEC-LLys (n = 6 experimental repeats) were treated for 72 hrs with lamstatin. (D) Subconfluent HMVEC-LLys (n = 4 experimental repeats) were treated for 72 hrs with lamstatin and manually counted using trypan blue exclusion (data expressed as average number of cells/ml). All data are expressed as mean ± SE of the mean (SEM) and one-way repeated measures anova for comparisons (Bonferroni correction for multiple comparisons) of various concentrations versus no treatment *P < 0.05.
To confirm that MTT cleavage is reduced as a consequence of decreased cell numbers, HMVEC-LLys were treated with vehicle or increasing concentrations of lamstatin and a manual trypan blue exclusion cell count was carried out. Lamstatin significantly reduced the total number of trypan blue negative cells over a period of 72 hrs (Fig.2D) at concentrations of 2.5, 5 and 10 μg/ml (n = 4 experimental repeats).
Lamstatin and CP17 reduce cord formation
To further characterize lamstatin's anti-lymphangiogenic properties, we seeded HMVEC-LLy on Matrigel ® and induced cord formation (an in vitro equivalent of tube formation and a prerequisite for HMVEC-LLy neo-vascularization) with EBM-2 MV growth medium (Fig.3A). Lamstatin significantly reduced the number of cords from 0.059 per 1000 px2 to 0.028 per 1000 px2 at a concentration of 5 μg/ml (∽52% reduction, P < 0.05) and to 0.024 cords per 1000 px2 at 10 μg/ml (∽59% reduction, P < 0.05) (n = 3 experimental repeats) (Fig.3B). To support the notion that CP17 is the active region, the cord formation assay was repeated with concentrations of 1.25, 2.5, 5 and 10 μg/ml CP17. There were significant, comparable reductions in the number of cords after 24 hrs treatment with CP17 at concentrations of 5 (from 0.062 per 1000 px2 to 0.024 cords per 1000 px2, ∽61% reduction, P < 0.05) and 10 μg/ml (to 0.019 cords per 1000 px2, ∽69% reduction, P < 0.01) (n = 3 experimental repeats) consistent with our findings with lamstatin (Fig.3B).
Fig 3.
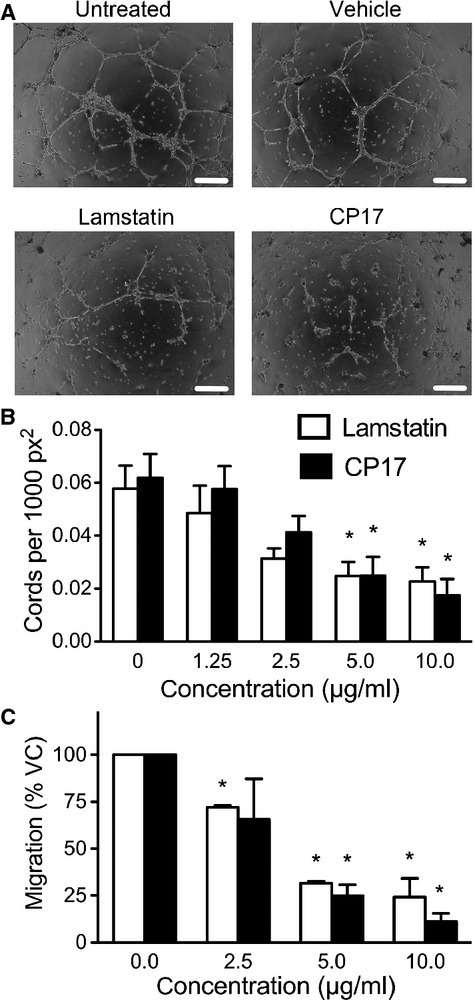
Lamstatin and CP17 inhibit cord formation and migration of human lung lymphatic endothelial cells (HMVEC-LLys) in vitro. HMVEC-LLys were seeded on Matrigel (BD Bioscience) for 1 hr to allow attachment before EBM-2 MV medium (complete) with increasing concentration of lamstatin, CP17 or vehicle was added. Cord formation was assessed after 24 hrs. (A) Cord formation images from a representative experiment. Lamstatin (10 μg/ml) compared with vehicle control (1 nM EDTA, pH 3.5) and CP17 (10 μg/ml) compared with vehicle control (sterile MilliQ water). Black scale bar represents 500 μm. (B) Cord formation in the presence of lamstatin (white bars) (n = 3 experimental repeats) or CP17 (black bars) (n = 3 experimental repeats). Data are expressed as mean ± SE of the mean (SEM) of cords per 1000 square pixel (px2) and one-way repeated measures anova for comparisons of various concentrations versus no treatment, *P < 0.05. (C) HMVEC-LLys were seeded at 4.0 × 105 cells/ml per transwell and serum starved (EBM-2 MV, no supplements, 0.1% BSA) before EBM-2 MV medium (complete) with lamstatin (white bars) (n = 3 experimental repeats) or CP17 (black bars) (n = 3 experimental repeats) was added. Cells were allowed to migrate for 24 hrs and then stained with Calcein-AM for detection. Data were normalized to vehicle controls and presented as mean ± SEM per cent migration (100% VC: vehicle control). One-way repeated measures anova treatment versus no treatment, *P < 0.05.
Lamstatin and CP17 are inhibitors of HMVEC-LLy migration
Effective induction of tissue lymphangiogenesis requires HMVEC-LLys to be able to migrate towards pro-lymphangiogenic gradients (e.g. of VEGF-C, VEGF-D, FGF-2 and other factors). We used full growth media (EBM-2 MV) as a chemoattractant, which is supplemented, with FGF-2 (see methods) amongst other agents. Human lung lymphatic endothelial cells exposed to increasing concentrations of either lamstatin or CP17 exhibited significantly reduced migration from 100% with no treatment to 25% with 10 μg/ml of lamstatin (P < 0.01) and from 100% to 13% for 10 μg/ml CP17 (P < 0.01) (n = 3 and three experimental repeats respectively) (Fig.3C).
Lamstatin and CP17 inhibit HUVEC proliferation and cord formation in vitro
We treated proliferating HUVECs for 72 hrs with increasing concentrations of lamstatin or CP17. Lamstatin significantly reduced cell number (manual cell counts) at concentrations of 5 and 10 μg/ml (maximal reduction of ∽45% for lamstatin) (n = 6 experimental repeats) (Fig.4A). MTT cleavage was slightly reduced in HUVECs treated with increasing concentrations of both lamstatin and CP17 (Fig.4 B and C) at concentrations of 5 and 10 μg/ml (n = 6 experimental repeats).
Fig 4.
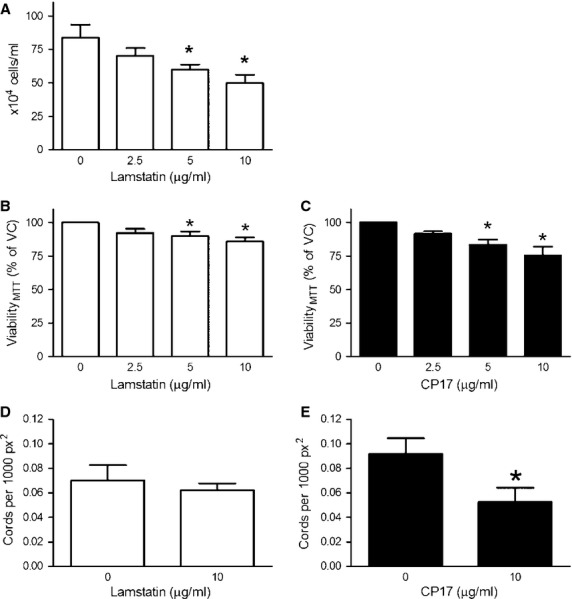
Lamstatin and CP17 reduce cell viability and inhibit cord formation of proliferating human umbilical vein endothelial cells (HUVECs). Subconfluent HUVECs were treated for 72 hrs with (A) lamstatin and manually counted using trypan blue exclusion (n = 6 experimental repeats) (data expressed as average number of cells/ml). Subconfluent HUVECs were treated for 72 hrs with (B) lamstatin or (C) CP17 and cell viability, measured with MTT, was expressed as percentage of vehicle control (% VC) (n = 6 and n = 6 experimental repeats). All data are expressed as mean ± SE of the mean (SEM) and compared using one-way repeated measures anova (Bonferroni correction for multiple comparisons) of various concentrations versus no treatment *P < 0.05. HUVECs were seeded on geltrex-coated plates for 2 hrs before being treated with lamstatin (10 μg/ml), vehicle or CP17 (10 μg/ml) for up to 18 hrs. (D) Cord formation in the presence of lamstatin (n = 4 experimental repeats) or (E) CP17 (n = 4 experimental repeats). Data are expressed as mean ± SE of the mean (SEM) of cords per 1000 square pixel (px2) and compared using paired student's t-test, *P < 0.05.
We repeated the cord formation assay with HUVECs in the presence and absence of lamstatin and CP17. Lamstatin did not reduce the number of cords per 1000 px2 at 10 μg/ml (n = 4 experimental repeats) (Fig.4D). However, there were significant reductions in the number of cords after 24 hrs treatment with CP17 10 μg/ml (from 0.092 cords per 1000 px2 to 0.052 cords per 1000 px2, ∽50% reduction, P < 0.05) (n = 4 experimental repeats) (Fig.4E).
Lamstatin and CP17 are potent inhibitors of tumour-induced lymphangiogenesis in a murine model of lymphatic dysplasia
To address the hypothesis that lamstatin and its anti-lymphangiogenic component CP17 are inhibitors of lymphangiogenesis in vivo, we used a LNM35 adenocarcinoma xenograft model which is known to have tumour-associated angiogenesis and lymphangiogenesis 40. Animals were given a single administration of lamstatin or CP17 together with the LNM35 AAV-EGFP tumour cells and subsequent tumour and associated lymphatic and blood vessel network development were observed by confocal microscopy (lamstatin experiment no tumour n = 3, vehicle n = 11, 10 and 100 μg/ml lamstatin n = 8 and n = 9, CP17 experiment no tumour n = 3, vehicle n = 11, 10 and 100 μg/ml n = 8 and n = 6 mice respectively) (Fig.5). Lamstatin produced an overall significant reduction in the degree of lymphatic vascularization, as indicated by the decrease in overall LYVE-1 staining by ∽60% (P < 0.05) (Fig.6A). Lamstatin at a concentration of 10 μg/ml reduced the mean intensity of LYVE-1 staining from 11.3 intensity units (IU) (vehicle) to 4.3 IU (P < 0.05) and at 100 μg/ml to 4.6 IU (P < 0.05). More specifically, in the presence of lamstatin the lymphatic network dysplasia was reduced and the vessels appearance was less disorganized and more like that seen in the non-tumour tissue (Fig.5, top row: lamstatin 10 compared with lamstatin 100 or vehicle and no tumour control). The tumour cells (LNM35 AAV-EGFP) (Fig.5, second row), visualized in green with EGFP expression, clustered together to form a mass, whereas those of the lymphatic or blood lineages were more organized, lining themselves up into vessel structures. In the matrigel plugs containing tumour cells, treatment with lamstatin or CP17 had no effect on the tumour cells. However, lamstatin at the highest concentration (100 μg/ml) significantly reduced both the number of loops (P < 0.05) (Fig.6B) and the number of branches (P < 0.05) (Fig.6C) (indications of tumour-induced lymphangiogenesis) to levels observed in no-tumour, control animals. Consistent with our hypotheses, CP17 also proved to be a potent inhibitor in this model (Fig.5). CP17 at the highest concentration (100 μg/ml) significantly reversed the increased numbers of loops (P < 0.05; Fig.6B) and branches (P < 0.05; Fig.6C) to levels observed in healthy animals.
Fig 5.
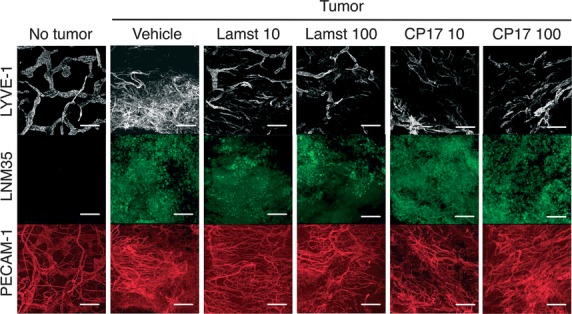
Lamstatin and CP17 inhibit lymphangiogenesis in a murine model of tumour-induced lymphangiogenesis. LNM35 tumour cells, expressing EGFP, were injected intradermally into ears of NOD/SCID/gamma mice with or without lamstatin or CP17 and Matrigel. No tumour = 200 μl Matrigel bolus only. Vehicle = tumour cells, Matrigel and vehicle (292.2 ng/l EDTA, pH 3.5). Lamst = Lamstatin either 10 or 100 μg/ml in a 200 μl bolus of LNM35 and Matrigel. CP17 = CP17 10 or 100 μg/ml in a 200 μl bolus of LNM35 and Matrigel. Images of representative staining for treatment of tumour-induced lymphangiogenesis with lamstatin and CP17 (lamstatin: no tumour n = 3, vehicle n = 11, 10 and 100 μg/ml lamstatin, n = 8 and n = 9 mice; CP17: no tumour n = 3, vehicle n = 11, 10 and 100 μg/ml CP17, n = 8 and n = 6 mice respectively). White layer = LYVE-1, lymphatics; Green layer = LNM35 tumour; Red layer = PECAM-1 (CD31), blood vasculature. Scale bar represents 200 μm.
Fig 6.
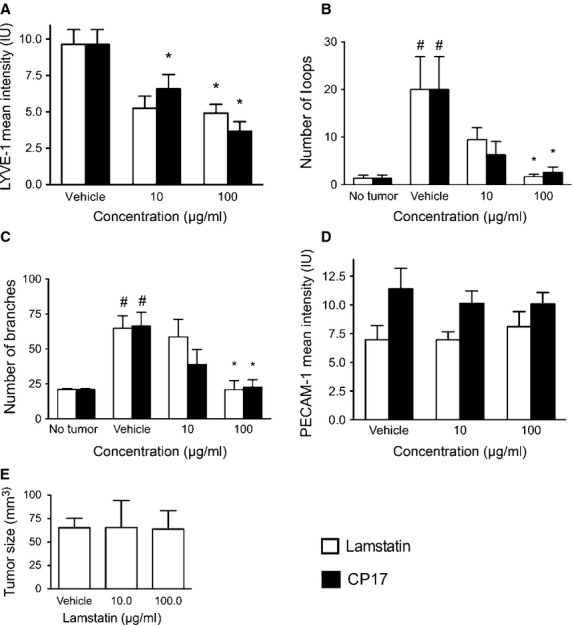
Lamstatin and CP17 inhibit lymphangiogenesis in a murine model of tumour-induced lymphangiogenesis. LNM35 tumour cells, expressing EGFP, were injected intradermally into the ears of NOD/SCID/gamma mice with or without lamstatin (white bars) or CP17 (black bars) and Matrigel. (A) Mean intensity values of LYVE-1 fluorescence staining in lamstatin and CP17 treated animals (Lamstatin vehicle n = 11, 10 and 100 μg/ml lamstatin n = 8 and n = 9, CP17 vehicle n = 11, 10 and 100 μg/ml CP17 n = 8 and n = 6, mice respectively). *P < 0.05 one-way anova (Kruskal–Wallis, with Dunn's multiple comparisons corrections) treatment versus vehicle. *P < 0.05. (B) Assessment of loops in tumour ear model of lamstatin-treated animals (no tumour n = 3, vehicle n = 11, 10 and 100 μg/ml lamstatin, n = 8 and n = 9 mice respectively) and CP17-treated animals (no tumour n = 3, vehicle n = 11, 10 and 100 μg/ml CP17, n = 8 and n = 6 mice respectively). (C) Morphological analysis of branching points of lymphatic vessels in whole mount staining of LNM35 tumours with lamstatin (no tumour n = 3, vehicle n = 11, 10 and 100 μg/ml lamstatin, n = 8 and n = 9 mice respectively) or CP17 (no tumour n = 3, vehicle n = 11, 10 and 100 μg/ml CP17, n = 8 and n = 6 mice respectively) treatment; (D) Percentage of mean PECAM-1-positive blood vessel area (% mean PECAM-1 PVA) of animals treated with vehicle, lamstatin (vehicle (292.2 ng/l EDTA, pH 3.5) n = 11, 10 and 100 μg/ml lamstatin n = 8 and n = 9 mice respectively) or CP17 (vehicle (MilliQ water) n = 11, 10 and 100 μg/ml CP17 n = 8 and n = 6 mice respectively). All data presented as mean ± SEM, one-way anova (Kruskal–Wallis, with Dunn's multiple comparisons corrections) treatment versus no treatment, *P < 0.05. (E) Lamstatin has no effect on tumour size after 12 days. After 12 days tumour sizes were measured in animals treated with either vehicle (292.2 ng/l EDTA, pH 3.5), 10 or 100 μg/ml lamstatin. Data are expressed as mean ± SEM. Vehicle n = 11, 10 and 100 μg/ml lamstatin n = 8 and n = 9 mice.
In contrast, lamstatin had no significant effect on the fluorescence intensity (IU) of the blood vessels at either 10 or 100 μg/ml (PECAM-1, red staining in Fig.5 and analysis in Fig.6D). CP17 also did not change the mean fluorescence intensity (IU, PECAM-1, red staining in Fig.5) of the blood vessels at either 10 and 100 μg/ml, compared with the vehicle control (Fig.6D). To exclude the possibility that lamstatin indirectly inhibited lymphangiogenesis by reducing the initial tumour mass, cell viability of LNM35 tumour cells in vitro (via the MTT assay) was measured. No significant effect of lamstatin on tumour cell viability was detected (data not shown). To support this result, we measured the in vivo tumour sizes of treated animals and no effect of either 10 or 100 μg/ml lamstatin treatment was detected when we compared matrigel plugs with tumour + vehicle and those with tumour + lamstatin (Fig.6E).
Discussion
In this study we report that the Col IV α5 NC1 (which we have named lamstatin) is significantly reduced in lung tissue sections of LAM patients and has novel anti-lymphangiogenic properties. In vitro, lamstatin and the 17 aa functional peptide (CP17) reduced migration of lymphatic endothelial cells. Furthermore, in vivo, in a murine model of tumour-induced lymphangiogenesis, lamstatin and CP17 dramatically reduced lymphangiogenesis. Thus, we identify lamstatin as a direct inhibitor of lymphangiogenesis, the absence of which may profoundly influence lymphangiogenesis and lymphatic endothelial cell survival.
Reports of endogenous lymphangiogenic inhibitors are few in number. Recently, Albuquerque and colleagues identified a soluble VEGFR-2 monomer, able to capture and deplete VEGF-C, which is responsible for the lack of lymph vessels observed in the cornea of mice and possibly human beings 42. The angiostatic proteins 16K human prolactin and collagen IV α2 non-collagenous-1 (NC1) domain (canstatin) have also now been described as inhibitors of lymphangiogenesis 43,44. Shao and Xie showed that endostatin, a proteolytic fragment of the NC1 domain of Col XVIII directly reduced the viability, in vitro, of pig thoracic duct lymphatic endothelial cells. In addition, endostatin directly interfered with the migration of lymphatic endothelial cells in an in vitro assay 45 and inhibited lymphatic tumour growth and lymphangiogenesis in a Lewis lung carcinoma xenograft 46. In contrast, Brideau and colleagues showed an indirect anti-lymphangiogenic effect of endostatin in vivo 47. Endostatin decreased in vitro and in vivo migration of tumour-associated mast cells, a substantial source of pro-lymphangiogenic VEGF-C. The effects of endostatin, 16K human prolactin and canstatin were not limited to the lymphatic system as they also decreased new blood vessel formation 47. We did not identify significant effects of lamstatin on new blood vessel formation in vivo, however, in vitro inhibition of HUVEC proliferation and cord formation was evident to some extent. The reason for the disparity in our in vivo and in vitro findings may reflect the differences in endothelial cell types examined—HUVECS versus murine tumour-associated endothelial cells. Tumour endothelial cells are abnormal 48,49, compared with non-diseased cells, and thus their response to lamstatin and CP17 may be different.
The absence or decreased levels of histochemical detection for members of the Col IV family, especially Col IV α5 as we have reported here in LAM, are associated with higher levels of invasion in breast, prostate, bronchoalveolar and colorectal cancer 12,13,50–52. In addition, increased lymphangiogenesis is commonly found in those cancers (explicitly breast and prostate) and results in lymph node metastasis and invasion 14,53–56. It is therefore conceivable that lamstatin functions as a general inhibitor of lymphangiogenesis in the human body, and depletion could lead to the formation of new lymphatic vessels constituting a pro-lymphangiogenic environment and facilitating invasiveness and metastasis.
Lamstatin, and the consensus peptide CP17, had inhibitory effects on the individual mechanisms underlying lymphangiogenesis in vitro. Both molecules reduced the viability of proliferating but not quiescent HMVEC-LLy cells. This reduced cell viability was the result of a reduction in the number of viable cells, potentially due to the induction of cell death or through the inhibition of cell progression through the cell cycle. Conversely, lamstatin had no effect on the viability of primary lung fibroblasts, primary airway smooth muscle cells and Α549 epithelial cells, indicating that, within the cell types we tested, the effect is specific for HMVEC-LLy cells. Lamstatin and CP17 reduced HMVEC-LLy cord formation, an in vitro equivalent of tube formation, and a prerequisite for HMVEC-LLy neo-vascularization. In addition, lamstatin and CP17 inhibited the capacity of HMVEC-LLys to migrate towards a pro-lymphangiogenic gradient. This suggests that lamstatin interferes with several crucial lymphangiogenic properties of HMVEC-LLy. Furthermore, CP17 harbours a functional site of 17 or fewer amino acids which may be responsible for the effects seen with lamstatin, as these properties are also observed with the peptide.
Lamstatin and CP17 were effective at reducing lymphangiogenesis in our in vivo model. We found that lamstatin inhibited lymphangiogenesis, but had little or no effect on blood vascularization (angiogenesis) or tumour cell proliferation. The observed effects of lamstatin in this model were restricted to the dysplastic lymphatic network. We can exclude the possibility that lamstatin indirectly inhibited lymphangiogenesis via reducing the initial tumour mass because lamstatin did not alter the viability of the LNM35 tumour cells in vitro nor was tumour size in vivo related to the treatment in vivo. CP17 also proved to be a potent inhibitor of all of the indices of lymphangiogenesis measured in this model. These findings confirm our in vitro data and strongly suggest that lamstatin and its functional component CP17 are inhibitors of lymphangiogenesis in vitro and in vivo, even in the highly pro-lymphangiogenic environment such as that exists in the vicinity of a tumour.
Although LAM is not yet considered to be a malignant disease, LAM cells are highly mobile and metastasize, and it is suggested that dissemination of LAM cell clusters occurs via the lymphatic system 57 which is increased in patients with LAM 27, 57, 58. In this study, lamstatin was undetectable in the airway tissue sections of all eight LAM patients we tested. Therefore, the absence of lamstatin in LAM lung tissue may facilitate the dissemination process because lymphatic vessels can easily be formed in the absence of an endogenous anti-lymphangiogenic factor.
In summary, we identified a novel inhibitor of lymphangiogenesis absent in lung tissue sections from patients with LAM. Lamstatin and CP17 decrease proliferation and migration of HMVEC-LLy in vitro and decrease tumour-induced lymphatic neo-vascularization in an in vivo model. Lamstatin may offer insights into the pathophysiology of LAM and may also provide a novel avenue for the treatment of diseases which are associated with excessive lymphangiogenesis, such as metastatic cancer.
Conflict of interest
The authors confirm that there are no conflicts of interests.
Acknowledgments
We would like to thank foremost all LAM and non-LAM patients, who kindly donated their explanted lungs. We acknowledge the collaborative effort of the cardiopulmonary transplant team and the pathologists at St Vincent's Hospital, Sydney and the thoracic surgeons and pathologists at Royal Prince Alfred Hospital Sydney, The Alfred Hospital, Melbourne, Strathfield Private Hospital and Rhodes Pathology, Sydney. We would like to thank Maree Svolos, Beray Kazak and Ho Yin Ng for excellent technical assistance and Professor Kari Alitalo for his support. Special thanks to Sarah Boustany for her contributions. This work was supported in part by the Cooperative Research Centre for Asthma and Airways, and the National Health and Medical Research Council, Australia grant #1009156. J.K. Burgess is supported by a NH&MRC Career Development Fellowship #1032695 and J.L. Black by a NH&MRC Senior Principal Research Fellowship #571098. C.A. Heckman is supported by an American Thoracic Society Research Grant.
References
- Albrecht I, Christofori G. Molecular mechanisms of lymphangiogenesis in development and cancer. Int J Dev Biol. 2011;55:483–94. doi: 10.1387/ijdb.103226ia. [DOI] [PubMed] [Google Scholar]
- Chang LK, Garcia-Cardena G, Farnebo F, et al. Dose-dependent response of FGF-2 for lymphangiogenesis. Proc Natl Acad Sci U S A. 2004;101:11658–63. doi: 10.1073/pnas.0404272101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Aldrich TH, Jones PF, et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87:1161–9. doi: 10.1016/s0092-8674(00)81812-7. [DOI] [PubMed] [Google Scholar]
- Kubo H, Cao R, Brakenhielm E, et al. Blockade of vascular endothelial growth factor receptor-3 signaling inhibits fibroblast growth factor-2-induced lymphangiogenesis in mouse cornea. Proc Nat Acad Sci USA. 2002;99:8868–73. doi: 10.1073/pnas.062040199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JW, Min M, Larrieu-Lahargue F, et al. Prox1 promotes lineage-specific expression of fibroblast growth factor (FGF) receptor-3 in lymphatic endothelium: a role for FGF signaling in lymphangiogenesis. Mol Biol Cell. 2006;17:576–84. doi: 10.1091/mbc.E05-04-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achen MG, Jeltsch M, Kukk E, et al. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4) Proc Nat Acad Sci USA. 1998;95:548–53. doi: 10.1073/pnas.95.2.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont DJ, Jussila L, Taipale J, et al. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science. 1998;282:946–9. doi: 10.1126/science.282.5390.946. [DOI] [PubMed] [Google Scholar]
- Joukov V, Sorsa T, Kumar V, et al. Proteolytic processing regulates receptor specificity and activity of VEGF-C. EMBO J. 1997;16:3898–911. doi: 10.1093/emboj/16.13.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaipainen A, Korhonen J, Mustonen T, et al. Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Nat Acad Sci USA. 1995;92:3566–70. doi: 10.1073/pnas.92.8.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen TA, Arola J, Saaristo A, et al. VEGF-C and VEGF-D expression in neuroendocrine cells and their receptor, VEGFR-3, in fenestrated blood vessels in human tissues. FASEB J. 2000;14:2087–96. doi: 10.1096/fj.99-1049com. [DOI] [PubMed] [Google Scholar]
- Burgess JK, Boustany S, Moir LM, et al. Reduction of tumstatin in asthmatic airways contributes to angiogenesis, inflammation, and hyperresponsiveness. Am J Respir Crit Care Med. 2010;181:106–15. doi: 10.1164/rccm.200904-0631OC. [DOI] [PubMed] [Google Scholar]
- Dehan P, Waltregny D, Beschin A, et al. Loss of type IV collagen alpha 5 and alpha 6 chains in human invasive prostate carcinomas. Am J Pathol. 1997;151:1097–104. [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Iyama K, Ishikawa N, et al. Loss of expression of type IV collagen alpha5 and alpha6 chains in colorectal cancer associated with the hypermethylation of their promoter region. Am J Pathol. 2006;168:856–65. doi: 10.2353/ajpath.2006.050384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Yasuoka H, Tsujimoto M, et al. Lymph vessel density correlates with nodal status, VEGF-C expression, and prognosis in breast cancer. Breast Cancer Res Treat. 2005;91:125–32. doi: 10.1007/s10549-004-5783-x. [DOI] [PubMed] [Google Scholar]
- Nakano KY, Iyama KI, Mori T, et al. Loss of alveolar basement membrane type IV collagen alpha3, alpha4, and alpha5 chains in bronchioloalveolar carcinoma of the lung. J Pathol. 2001;194:420–7. doi: 10.1002/path.928. [DOI] [PubMed] [Google Scholar]
- Ninomiya Y, Kagawa M, Iyama K, et al. Differential expression of two basement membrane collagen genes, COL4A6 and COL4A5, demonstrated by immunofluorescence staining using peptide-specific monoclonal antibodies. J Cell Biol. 1995;130:1219–29. doi: 10.1083/jcb.130.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis ED, Popel AS. A systematic methodology for proteome-wide identification of peptides inhibiting the proliferation and migration of endothelial cells. Proc Nat Acad Sci USA. 2008;105:13775–80. doi: 10.1073/pnas.0803241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega N, Werb Z. New functional roles for non-collagenous domains of basement membrane collagens. J Cell Sci. 2002;115:4201–14. doi: 10.1242/jcs.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohman DW, Noghrehkar D, Ratnayake S. Lymphangioleiomyomatosis: A review. Eur J Intern Med. 2008;19:319–24. doi: 10.1016/j.ejim.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Henske EP. Metastasis of benign tumor cells in tuberous sclerosis complex. Genes Chromosom Cancer. 2003;38:376–81. doi: 10.1002/gcc.10252. [DOI] [PubMed] [Google Scholar]
- Pacheco-Rodriguez G, Kumaki F, Steagall WK, et al. Chemokine-enhanced chemotaxis of lymphangioleiomyomatosis cells with mutations in the tumor suppressor TSC2 gene. J Immunol. 2009;182:1270–7. doi: 10.4049/jimmunol.182.3.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts S, Oberstein EM, Glassberg MK. Benign metastasizing leiomyoma and lymphangioleiomyomatosis: sex-specific diseases? Clin Chest Med. 2004;25:343–60. doi: 10.1016/j.ccm.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Yu J, Henske EP. mTOR activation, lymphangiogenesis, and estrogen-mediated cell survival: the “perfect storm” of pro-metastatic factors in LAM pathogenesis. Lymphat Res Biol. 2010;8:43–9. doi: 10.1089/lrb.2009.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JL, Ge Q, Boustany S, et al. In vitro studies of lymphangioleiomyomatosis. Eur Res J. 2005;26:569–76. doi: 10.1183/09031936.05.00016905. [DOI] [PubMed] [Google Scholar]
- Goncharova EA, Goncharov DA, Spaits M, et al. Abnormal growth of smooth muscle-like cells in lymphangioleiomyomatosis: Role for tumor suppressor TSC2. Am J Respir Cell Mol Biol. 2006;34:561–72. doi: 10.1165/rcmb.2005-0300OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrilees MJ, Hankin EJ, Black JL, et al. Matrix proteoglycans and remodelling of interstitial lung tissue in lymphangioleiomyomatosis. J Pathol. 2004;203:653–60. doi: 10.1002/path.1577. [DOI] [PubMed] [Google Scholar]
- Kumasaka T, Seyama K, Mitani K, et al. Lymphangiogenesis in lymphangioleiomyomatosis: its implication in the progression of lymphangioleiomyomatosis. Am J Surg Pathol. 2004;28:1007–16. doi: 10.1097/01.pas.0000126859.70814.6d. [DOI] [PubMed] [Google Scholar]
- Seyama K, Kumasaka T, Souma S, et al. Vascular endothelial growth factor-D is increased in serum of patients with lymphangioleiomyomatosis. Lymphat Res Biol. 2006;4:143–52. doi: 10.1089/lrb.2006.4.143. [DOI] [PubMed] [Google Scholar]
- Inoue Y, King TE, Barker E, et al. Basic fibroblast growth factor and its receptors in idiopathic pulmonary fibrosis and lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2002;166:765–73. doi: 10.1164/rccm.2010014. [DOI] [PubMed] [Google Scholar]
- Moir LM, Trian T, Ge Q, et al. PI 3-Kinase isoform specific effects in airway mesenchymal cell function. J Pharmacol Exp Ther. 2011;337:557–66. doi: 10.1124/jpet.110.173583. [DOI] [PubMed] [Google Scholar]
- Burgess JK, Carlin S, Pack RA, et al. Detection and characterization of OX40 ligand expression in human airway smooth muscle cells: a possible role in asthma? J Allergy Clin Immunol. 2004;113:683–9. doi: 10.1016/j.jaci.2003.12.311. [DOI] [PubMed] [Google Scholar]
- Burgess JK, Ge Q, Boustany S, et al. Increased sensitivity of asthmatic airway smooth muscle cells to prostaglandin E2 might be mediated by increased numbers of E-prostanoid receptors. J Allergy Clin Immunol. 2004;113:876–81. doi: 10.1016/j.jaci.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Burgess JK, Oliver BGG, Poniris MH, et al. A phosphodiesterase 4 inhibitor inhibits matrix protein deposition in airways in vitro. J Allergy Clin Immunol. 2006;118:649–57. doi: 10.1016/j.jaci.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Hostettler KE, Roth M, Burgess JK, et al. Cyclosporine A mediates fibroproliferation through epithelial cells. Transplantation. 2004;77:1886–93. doi: 10.1097/01.tp.0000131149.78168.dd. [DOI] [PubMed] [Google Scholar]
- Johnson PR, Black JL, Carlin S, et al. The production of extracellular matrix proteins by human passively sensitized airway smooth-muscle cells in culture: the effect of beclomethasone. Am J Respir Crit Care Med. 2000;162:2145–51. doi: 10.1164/ajrccm.162.6.9909111. [DOI] [PubMed] [Google Scholar]
- Johnson PRA, Roth M, Tamm M, et al. Airway smooth muscle cell proliferation is increased in asthma. Am J Respir Crit Care Med. 2001;164:474–7. doi: 10.1164/ajrccm.164.3.2010109. [DOI] [PubMed] [Google Scholar]
- Moir L, Ng H, Poniris M, et al. Doxycycline inhibits matrix metalloproteinase-2 secretion from TSC2-null mouse embryonic fibroblasts and lymphangioleiomyomatosis cells. Br J Pharmacol. 2011;164:83–92. doi: 10.1111/j.1476-5381.2011.01344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima Y, Manfredi M, Reimer C, et al. Identification of the anti-angiogenic site within vascular basement membrane-derived tumstatin. J Biol Chem. 2001;276:15240–8. doi: 10.1074/jbc.M007764200. [DOI] [PubMed] [Google Scholar]
- Maeshima Y, Yerramalla UL, Dhanabal M, et al. Extracellular matrix-derived peptide binds to alpha(v)beta(3) integrin and inhibits angiogenesis. J Biol Chem. 2001;276:31959–68. doi: 10.1074/jbc.M103024200. [DOI] [PubMed] [Google Scholar]
- Heckman CA, Holopainen T, Wirzenius M, et al. The tyrosine kinase inhibitor cediranib blocks ligand-induced vascular endothelial growth factor receptor-3 activity and lymphangiogenesis. Cancer Res. 2008;68:4754–62. doi: 10.1158/0008-5472.CAN-07-5809. [DOI] [PubMed] [Google Scholar]
- Shayan R, Karnezis T, Tsantikos E, et al. A system for quantifying the patterning of the lymphatic vasculature. Growth Factors. 2007;25:417–25. doi: 10.1080/08977190801932550. [DOI] [PubMed] [Google Scholar]
- Albuquerque RJ, Hayashi T, Cho WG, et al. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat Med. 2009;15:1023–30. doi: 10.1038/nm.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinet V, Castermans K, Herkenne S, et al. The angiostatic protein 16K human prolactin significantly prevents tumor-induced lymphangiogenesis by affecting lymphatic endothelial cells. Endocrinology. 2011;152:4062–71. doi: 10.1210/en.2011-1081. [DOI] [PubMed] [Google Scholar]
- Hwang-Bo J, Yoo KH, Park JH, et al. Recombinant canstatin inhibits angiopoietin-1-induced angiogenesis and lymphangiogenesis. Int J Cancer. 2012;131:298–309. doi: 10.1002/ijc.26353. [DOI] [PubMed] [Google Scholar]
- Shao XJ, Xie FM. Influence of angiogenesis inhibitors, endostatin and PF-4, on lymphangiogenesis. Lymphology. 2005;38:1–8. [PubMed] [Google Scholar]
- Dong X, Zhao X, Xiao T, et al. Endostar, a recombined humanized endostatin, inhibits lymphangiogenesis and lymphatic metastasis of Lewis lung carcinoma xenograft in mice. Thorac Cardiovasc Surg. 2011;59:133–6. doi: 10.1055/s-0030-1250152. [DOI] [PubMed] [Google Scholar]
- Brideau G, Makinen MJ, Elamaa H, et al. Endostatin overexpression inhibits lymphangiogenesis and lymph node metastasis in mice. Cancer Res. 2007;67:11528–35. doi: 10.1158/0008-5472.CAN-07-1458. [DOI] [PubMed] [Google Scholar]
- Hashizume H, Baluk P, Morikawa S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156:1363–80. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley AC. Tumor endothelial cells. Cold Spring Harb Perspect Med. 2012;2:a006536. doi: 10.1101/cshperspect.a006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiki Y, Iyama K, Tsuruta J, et al. Differential distribution of basement membrane type IV collagen alpha1(IV), alpha2(IV), alpha5(IV) and alpha6(IV) chains in colorectal epithelial tumors. Pathol Int. 2002;52:224–33. doi: 10.1046/j.1440-1827.2002.01341.x. [DOI] [PubMed] [Google Scholar]
- Hinenoya N, Naito I, Momota R, et al. Type IV collagen alpha chains of the basement membrane in the rat bronchioalveolar transitional segment. Arch Histol Cytol. 2008;71:185–94. doi: 10.1679/aohc.71.185. [DOI] [PubMed] [Google Scholar]
- Nakano S, Iyama K, Ogawa M, et al. Differential tissular expression and localization of type IV collagen alpha1(IV), alpha2(IV), alpha5(IV), and alpha6(IV) chains and their mRNA in normal breast and in benign and malignant breast tumors. Lab Invest. 1999;79:281–92. [PubMed] [Google Scholar]
- Skobe M, Hawighorst T, Jackson DG, et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7:192–8. doi: 10.1038/84643. [DOI] [PubMed] [Google Scholar]
- Stacker SA, Caesar C, Baldwin ME, et al. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001;7:186–91. doi: 10.1038/84635. [DOI] [PubMed] [Google Scholar]
- Yano A, Fujii Y, Iwai A, et al. Glucocorticoids suppress tumor lymphangiogenesis of prostate cancer cells. Clin Cancer Res. 2006;12:6012–7. doi: 10.1158/1078-0432.CCR-06-0749. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Opeskin K, Horvath LG, et al. Lymphatic vessel density and lymph node metastasis in prostate cancer. Prostate. 2005;65:222–30. doi: 10.1002/pros.20288. [DOI] [PubMed] [Google Scholar]
- Kumasaka T, Seyama K, Mitani K, et al. Lymphangiogenesis-mediated shedding of LAM cell clusters as a mechanism for dissemination in lymphangioleiomyomatosis. Am J Surg Pathol. 2005;29:1356–66. doi: 10.1097/01.pas.0000172192.25295.45. [DOI] [PubMed] [Google Scholar]
- Hirama M, Atsuta R, Mitani K, et al. Lymphangioleiomyomatosis diagnosed by immunocytochemical and genetic analysis of lymphangioleiomyomatosis cell clusters found in chylous pleural effusion. Intern Med. 2007;46:1593–6. doi: 10.2169/internalmedicine.46.0225. [DOI] [PubMed] [Google Scholar]