

Abstract
Infection of the CNS with HIV-1 occurs rapidly after primary peripheral infection. HIV-1 can induce a wide range of neurological deficits, collectively known as HIV-1-associated neurocognitive disorders. Our previous work has shown that the selected neurotoxic effects induced by individual viral proteins, Tat and gp120, and by HIV+ supernatant are enhanced by co-exposure to morphine. This mimics co-morbid neurological effects observed in opiate-abusing HIV+ patients. Although there is a correlation between opiate drug abuse and progression of HIV-1-associated neurocognitive disorders, the mechanisms underlie interactions between HIV-1 and opiates remain obscure. Previous studies have shown that HIV-1 induces neurotoxic effects through abnormal activation of GSK3β. Interestingly, expression of GSK3β has shown to be elevated in brains of young opiate abusers indicating that GSK3β is also linked to neuropathology seen with opiate abusing patients. Thus, we hypothesize that GSK3β activation is a point of convergence for HIV- and opiate-mediated interactive neurotoxic effects. Neuronal cultures were treated with supernatant from HIV-1SF162-infected THP-1 cells, in the presence or absence of morphine and GSK3β inhibitors. Our results show that GSK3β inhibitors, including valproate and small molecule inhibitors, significantly reduce HIV-1-mediated neurotoxic outcomes, and also negate interactions with morphine that result in cell death, suggesting that GSK3β-activation is an important point of convergence and a potential therapeutic target for HIV- and opiate-mediated neurocognitive deficits.
Keywords: NeuroAIDS, HIV-1, morphine, neurodegeneration, synaptodendritic injury, GSK3β
INTRODUCTION
Infection of the central nervous system (CNS) with human immunodeficiency virus-1 (HIV-1) occurs rapidly after primary peripheral infection (An et al., 1996; An et al., 1999b; Davis et al., 1992; Kramer-Hammerle et al., 2005). HIV-1 can induce a wide range of CNS deficits, collectively known as HIV-1-associated neurocognitive disorders (HAND); nearly 50% of HIV-1-infected individuals suffer from HAND (Fischer-Smith and Rappaport, 2005; McArthur, 2004; McArthur et al., 2005; McArthur et al., 2003). Infected and/or activated glial and immune cells in the CNS release various viral and cellular factors that drive direct and indirect neuronal toxicity, leading to HAND (Buescher et al., 2007; Epstein and Gendelman, 1993; Gonzalez-Scarano and Martin-Garcia, 2005; Kaul et al., 2005; Kramer-Hammerle et al., 2005). The advent of combination antiretroviral therapy (cART) has reduced the severity of HAND, but the disease prevalence remains the same (Cysique and Brew, 2009; Cysique et al., 2004; Ellis et al., 2007; Robertson et al., 2007; Robinson-Papp et al., 2009; Sacktor, 2002; Sacktor et al., 2002), likely due to longer patient survival (Dore et al., 2003) and the relatively poor CNS penetrance of many antiretroviral drugs (Ellis et al., 2007; Kerza-Kwiatecki and Amini, 1999).
Drug abuse is a major risk factor for HIV infection; nearly 30% of HIV+ patients have a history of drug abuse involving opiates (Bokhari et al., 2009; Robertson et al., 1994). A large fraction of HIV+ patients are also exposed to opiates for treatment of AIDS-related chronic pain syndromes. Opiate drugs of abuse modulate immune function (Novick et al., 1989; Rogers and Peterson, 2003; Sheng et al., 1997), and also promote HIV-1 replication in vitro (Peterson et al., 2004; Peterson et al., 1990). Since opiates by themselves promote outcomes involved in CNS dysfunction, such as blood-brain barrier breakdown, immune and glial cell activation, and neuronal damage (Bell et al., 2006; Hauser et al., 2005; Hu et al., 2002; Sheng et al., 1997), it can be predicted that opiate drug abuse might exacerbate HIV-1 pathogenesis in the CNS. Our previous work has shown that certain neurotoxic effects induced by the individual HIV-1 proteins, trans-activator of transcription (Tat) and glycoprotein 120 (gp120) (Fitting et al., 2010a; Gurwell et al., 2001; Podhaizer et al., 2012; Suzuki et al., 2011; Zou et al., 2011b), and by HIV+ supernatant (HIV+sup) (Masvekar et al., 2014), are enhanced by co-exposure to morphine, the major metabolite of heroin in the CNS (Sawynok, 1986). This mimics co-morbid neurological effects observed in opiate-abusing HIV+ patients (Anthony et al., 2008; Bell et al., 2002; Byrd et al., 2011; Meijerink et al., 2014; Robinson-Papp et al., 2012; Smith et al., 2014). Although there is a correlation between opiate drug abuse and HAND progression, the mechanisms that underlie interactions between HIV-1 and opiates remain obscure; the main aim of this work was to identify point(s) of convergence for HIV-1 and morphine signaling in neurons.
Identified originally as a regulator of glycogen metabolism, glycogen synthase kinase-3β (GSK3β) is a central component of various signaling pathways in neurons, including those affecting neuronal plasticity, gene expression, and cell survival (Frame and Cohen, 2001; Grimes and Jope, 2001; Jacobs et al., 2012). GSK3β activity appears to be dysregulated in multiple neuropathological conditions, including Alzheimer’s disease, Parkinson’s disease, schizophrenia, autism, and bipolar mood disorder, and pharmacological inhibition of GSK3β is effective against some symptomatology in these diseases (Emamian et al., 2004; Frame and Cohen, 2001; Haenisch et al., 2014; Jacobs et al., 2012; Kaytor and Orr, 2002; Koistinaho et al., 2011; Kozikowski et al., 2006; Leroy et al., 2007; Schaffer et al., 2008). In previous studies, HIV-1 neurotoxicity was linked to abnormal activation of GSK3β (Crews et al., 2009; Dou et al., 2003; Dou et al., 2005; Everall et al., 2002; Maggirwar et al., 1999; Sui et al., 2006), GSK3β has also been linked to neuropathology seen in opiate-abusing patients (Anthony et al., 2010; Ramage et al., 2005). We therefore tested GSK3β activation as a point of convergent signaling for interaction between HIV-1 and morphine.
Both lethal and sublethal effects of HIV+sup ± morphine treatments were assessed on neuron populations, and also by time-lapse imaging of individual cells over 72 h. Valproic acid and small molecule GSK3β inhibitors significantly reduced HIV+sup-mediated neurotoxic outcomes. Interactions between HIV and morphine that resulted in neuronal death were also abrogated, implicating GSK3β as a mediator of specific neurotoxic events initiated by combined exposure to HIV-1 and opiates.
MATERIALS and METHODS
All experimental procedures were reviewed and approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee.
Neuron cultures
As the striatum is a principal, sub-cortical target of HIV-1, and as levels of opioid receptors in the striatum are relatively high (Arvidsson et al., 1995; Berger and Arendt, 2000; Berger and Nath, 1997), it is a region in which HIV-opiate interactions are likely to occur. Thus, our culture model system primarily used murine striatal neurons. We also tested effects of HIV+sup and morphine on cortical and hippocampal neurons. Neurons were cultured as previously described (Masvekar et al., 2014; Podhaizer et al., 2012; Zou et al., 2011a; Zou et al., 2011b). In brief, striata or cortices from E15–E16 or hippocampi from P0–P1 ICR (CD-1) mice (Charles River Laboratories International, Inc., Wilmington, MA) were dissected, minced and incubated with trypsin (2.5 mg/ml; Sigma-Aldrich, St. Louis, MO) and deoxyribonuclease (DNase; 0.015 mg/ml; Sigma-Aldrich) in neurobasal medium (Gibco, Grand Island, NY) for 30 min at 37°C. Tissue was resuspended in neurobasal medium supplemented with B-27 additives (Gibco), L-glutamine (0.5 mM; Gibco), glutamate (25 μM; Sigma-Aldrich) and Antibiotic-Antimycotic (Gibco), triturated, and filtered twice through 70 μm pore, nylon mesh filters (BD Biosciences, San Jose, CA). Cells were seeded into culture plates (Corning Inc., Corning, NY) pre-coated with poly-L-lysine (0.5 mg/ml; Sigma-Aldrich), in supplemented neurobasal medium. Purity was determined by immunostaining for microtubule-associated protein 2 (MAP-2, a dendrite marker; Abcam, Cambridge, MA; ab32454), and cultures were found to be > 80% neurons.
HIV-1 supernatant
Cells of the acute monocytic leukemia cell line, THP1 (ATCC, Manassas, VA), were cultured at 0.5 × 105 cells/ml in RPMI-1640 medium (Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco) and 100 U/ml Penicillin-Streptomycin (Gibco), and stimulated with interleukin-2 (IL-2; 100 ng/ml; Sigma-Aldrich) and phytohaemagglutinin (PHA; 5 μg/ml; Sigma-Aldrich), for 48 h. Stimulated cells were treated with polybrene (2 μg/ml; Sigma-Aldrich) for 30 min at 37°C, resuspended in fresh medium, and exposed to HIV-1SF162 (p24 = 50 pg/ml; a R5-tropic HIV-1 strain; from Dr. Jay Levy (Cheng-Mayer and Levy, 1988), through NIH AIDS Research and Reference Reagent Program, Germantown, MD). After 5 d, supernatants (HIV+sup) were collected by filtering through a 0.20-μm filter, and viral infection was confirmed by quantifying HIV-p24 levels using ELISA (Advanced Bioscience Laboratories, Rockville, MD), typically in 5 d a 3–5 fold increase in HIV-p24 levels were observed. Supernatant from untreated/uninfected THP1 cells (Controlsup) was used as a control. Cell culture supernatants were aliquoted and stored at −80°C. To minimize variability, the same supernatant was used for all the experiments.
Treatments
To avoid the potential confound of morphine increasing HIV replication (Peterson et al., 2004; Peterson et al., 1993; Peterson et al., 1990), morphine was only added to cell-free supernatants. Our previous studies showed that supernatants from HIV-infected cells induce neurotoxic effects in a concentration-dependent manner over a range of p24 levels (10–500 pg/ml), but the interactions with morphine were significant only at lower HIV-exposure levels (p24 = 10 and 25 pg/ml) (Masvekar et al., 2014). Thus, current studies used an HIV+sup exposure level of p24 = 25 pg/ml. HIV+sup and Controlsup were diluted similarly and added to neuronal cultures in the presence or absence of morphine (morphine sulfate; Sigma-Aldrich), at a dose that maximally stimulates neuronal and glial μ-opioid receptors (MORs) in vitro (500 nM) (Bruce-Keller et al., 2008; El-Hage et al., 2005; Gurwell et al., 2001; Ikeda et al., 2010; Masvekar et al., 2014; Miyatake et al., 2009; Podhaizer et al., 2012; Zou et al., 2011b). To determine the role of GSK3β in HIV- and opiate-mediated neurotoxicity, HIV+sup ± morphine treatments were conducted in the presence of a standard GSK3β inhibitor, valproate (valproic acid, VPA; 1 mM; Sigma-Aldrich), or in the presence of two very selective, small molecule inhibitors, SB415286 (10 μM; Sigma-Aldrich; a competitive inhibitor of the ATP binding site on GSK3β and GSK3α with an IC50 value of 78 nM (Coghlan et al., 2000)) or GSK3β inhibitor XXVI (XXVI; 1 μM; EMD Millipore, Billerica, MA; a cell-permeable, pyrazolone GSK3β inhibitor with an IC50 value of 34 nM).
TUNEL assay
Cells were fixed in 4% paraformaldehyde (Sigma-Aldrich) overnight at 4°C, permeabilized in 0.1% Triton-X 100 (Molecular Probes) and 0.1% BSA (Invitrogen, Grand Island, NY) for 15 min, blocked in 0.1% BSA (Invitrogen, Grand Island, NY) and 1% horse serum (Invitrogen) for 30 min. Cells were subsequently labeled with Hoechst 33342 (Sigma-Aldrich) and TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling; Roche Applied Sciences, Mannheim, Germany). Cells were visualized and digital images were acquired using the Axio Observer Z.1 microscope and Zen 2010 software (Zeiss Inc., Thornwood, NY). Neuronal apoptosis was assessed by manually counting the percentage of TUNEL(+) cells.
Assessment of neuron viability
Time-lapse digital images of pre-selected neurons were captured at 1 h intervals for 72 h after treatments using a microscope with a computer-regulated stage (Axio Vision 4.6; Carl Zeiss Inc.) and in a controlled environmental chamber (37°C, 95% humidity, 5% CO2). In digital images, neurons were assessed for viability at 6 h intervals using rigorous morphological criteria confirming cell death, including cytoplasmic swelling, nuclear destruction, cell body fragmentation, excessive neurite degeneration, and loss of phase brightness (Masvekar et al., 2014; Podhaizer et al., 2012; Singh et al., 2004; Zou et al., 2011a; Zou et al., 2011b). Findings were reported as the average percentage of neuronal survival, with respect to pre-treatment neuron count ± the standard error of the mean (SEM). The effect of each treatment on neuronal survival was analyzed using a repeated-measure analysis of variance (ANOVA) followed by Duncan’s post hoc test (Statistica 8.0; StatSoft, Tulsa, OK) from n = 3–5 experiments in which at least 50 neurons per treatment group were followed per experiment (150–250 neurons total per treatment group).
Assessment of neuritic arborization
Cells were fixed, permeabilized, blocked, and subsequently labeled for MAP-2 (Abcam), TUNEL (Roche Applied Sciences), and Hoechst 33342 (Sigma-Aldrich). Cells were visualized and digital images were acquired. Neuritic arborization was evaluated using Sholl analysis; a ‘Sholl score’ was measured by counting the number of intersections of MAP-2-positive neurites with equidistant concentric circles of increasing radius, centered on the cell body (Sholl, 1953). To avoid the inclusion of dead cells, we specifically evaluated changes in the arborization only of TUNEL(−) cells.
Immunoblotting
Whole cell extracts were prepared using radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich) with protease and phosphatase inhibitors (Roche Applied Sciences), and total protein concentrations were determined by bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, Rockford, IL). Cell lysates containing equal amounts of total protein (~5–10 μg) were heated at 100° C for 5 minutes in laemmli buffer (Sigma-Aldrich), electrophoretically separated on 10% SDS-polyacrylamide gels (Bio-Rad, Hercules, CA), and transferred onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad). Membranes were incubated with primary antibodies to phospho-GSK3β-Ser9 (Cell Signaling Technology, Danvers, MA; 5558), GSK3β (Cell Signaling Technology; 9832), β-catenin (Cell Signaling Technology; 9562), glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Abcam; ab8245), MAP-2 (Abcam) and postsynaptic density protein 95 (PSD-95; UC Davis/NIH NeuroMab Facility, Davis, CA; 73-028). Appropriate horseradish peroxidase-conjugated secondary antibodies (SouthernBiotech, Birmingham, AL) were used. Membranes were detected using SuperSignal® West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific), and visualized using a Kodak Image Station 440CF.
Statistical Analyses
All data were expressed as average ± SEM. Unless otherwise indicated, data were analyzed statistically using a one-way ANOVA followed by Duncan’s post hoc test using Statistica 8.0 (StatSoft); an α level of p < 0.05 was considered significant.
RESULTS
HIV- and morphine-mediated GSK3β activation
GSK3β activation/inactivation was assessed at 4 h after treatments. Cell lysates were immunoblotted for phospho-GSK3β-Ser9 (p-GSK3β-S9; an inactive form of GSK3β) (Frame et al., 2001; Grimes and Jope, 2001; Jacobs et al., 2012; Kaytor and Orr, 2002), GSK3β (total GSK3β; t-GSK3β), β-catenin, and GAPDH (Fig. 1). HIV+sup, with or without morphine, significantly decreased p-GSK3β-S9 (normalized with t-GSK3β; p-GSK3β-S9/t-GSK3β; Fig. 1A), suggesting an increase in GSK3β activation. VPA significantly abrogated HIV+sup ± morphine-mediated effects. β-Catenin is a downstream target of GSK3β; active GSK3β phosphorylates β-catenin and initiates rapid ubiquitin-mediated degradation by the proteasome (Aberle et al., 1997; Jacobs et al., 2012; Kaytor and Orr, 2002). HIV+sup, with or without morphine, significantly reduced β-Catenin levels (Fig. 1B), suggesting an enhancement in GSK3β activity. VPA completely inhibited the effect of HIV+sup + morphine and partially inhibited the effect of HIV+sup alone, returning both to levels that were not different from control levels.
Figure 1. HIV+sup ± morphine-mediated GSK3β activation.
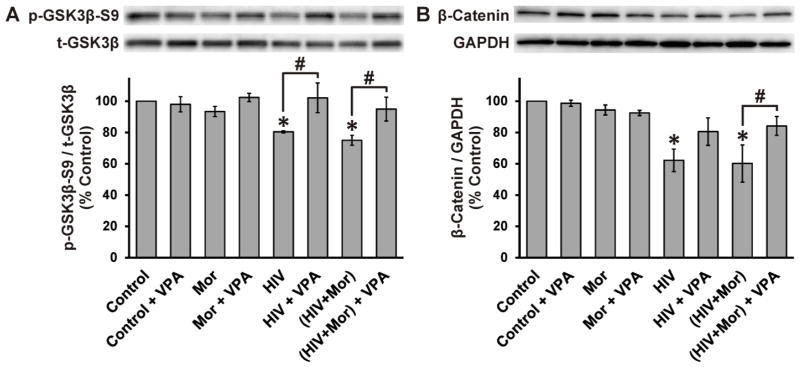
At 4 h after treatments, cells were lysed and protein levels were detected by immunoblotting. Findings were reported as average normalized protein levels (% control) ± SEM. Significance was analyzed by one-way ANOVA and Duncan’s post hoc test; n = 3 separate experiments. (A) Immunoblotting for phospho-GSK3β-Ser9 (p-GSKβ-S9) and total GSK3β (t-GSK3β); levels of p-GSKβ-S9 were normalized with t-GSK3β (p-GSKβ-S9/t-GSK3β). HIV+sup significantly reduced p-GSK3β-S9 (*p < 0.05 vs. Control), without any significant morphine interaction. VPA significantly abrogated HIV+sup ± morphine-mediated effects (#p < 0.05). (B) Immunoblotting for β-catenin and GAPDH; levels of β-catenin were normalized to GAPDH (β-catenin/GAPDH). HIV+sup significantly decreased β-catenin (*p < 0.05 vs. Control) without any morphine interaction. VPA restored β-catenin levels to control values (#p < 0.05), although in the case of neurons treated with HIV+sup alone the effect was less significant (HIV + VPA was not different from either HIV or control). Control = controlsup; HIV = HIV+sup; Mor = morphine; VPA = valproate.
Significant individual or interactive effects of morphine were not detected at 4 h. Thus, based on the time-dependent effects of opiates observed by others (Dobashi et al., 2010; Xie et al., 2010), we examined HIV+sup and morphine-mediated effects at longer time intervals (Fig. 2). At all assessed time points (12, 24, and 72 h), HIV+sup significantly reduced p-GSK3β-S9. At 24 and 72 h, morphine alone also induced significant loss of p-GSK3β-S9. A significant interaction between HIV+sup and morphine was seen only at 24 h.
Figure 2. HIV+sup- and morphine-mediated interactive effects on GSK3β-activation.

At 12, 24, and 72 h after treatments, cells were lysed and immunoblotted for p-GSKβ-S9 and t-GSK3β. Findings were reported as average levels of p-GSKβ-S9 normalized with t-GSK3β (p-GSKβ-S9/t-GSK3β) as a percentage of control values ± SEM. Significance was analyzed by one-way ANOVA and Duncan’s post hoc test; n = 4 separate experiments. At all time points, HIV+sup significantly reduced p-GSK3β-S9 (*p < 0.05 vs. Control). At 24 and 72 h, morphine alone also induced significant loss of p-GSK3β-S9, but a significant interaction with HIV+sup was seen only at 24 h ($p < 0.05). Control = controlsup; HIV = HIV+sup; Mor = morphine.
Role of GSK3β in HIV ± morphine-mediated cell death
Death of pre-selected neurons was observed using time-lapse imaging; cells were repeatedly imaged at 1 h intervals throughout the 72 h treatment period (Fig. 3A). Neurons exposed to morphine and/or VPA had survival values equivalent to control, while all groups treated with HIV+sup showed significantly reduced neuronal survival (Fig. 3B). Morphine significantly enhanced HIV+sup-mediated neuronal death. HIV+sup ± morphine-mediated effects were partially, but significantly, reversed by VPA co-treatment. The survival of neurons treated with ‘HIV+sup + VPA’ and ‘HIV+sup + morphine + VPA’ was not significantly different, showing that VPA effectively negated any HIV+sup-morphine interactions. Cell death was additionally confirmed using TUNEL staining (Fig. 3C). HIV+sup induced a significant increase in TUNEL(+) neurons, which was augmented by morphine co-treatment. HIV+sup ± morphine-mediated effects were partially, but significantly, reversed by VPA co-treatment. As seen for the time-lapse analyses, VPA also nullified the interactions between HIV+sup and morphine.
Figure 3. Role of GSK3β in HIV+sup ± morphine-mediated cell death.

(A) Cells were repeatedly imaged for 72 h after treatments. Digital images show the same cells/fields at 0, 24 and 72 h (white arrowheads indicate dead cells that were alive in previous image). (B) Cells were assessed for viability at 6 h intervals in digital images. Findings were reported as the average percentage of neuronal survival as a proportion of pre-treatment neuron count ± SEM. Significance was analyzed by repeated measures ANOVA and Duncan’s post hoc test; n = 3 separate experiments (at least 150 neurons per treatment group). HIV+sup significantly reduced neuronal survival (*p < 0.05 vs. Control), which was significantly augmented by morphine co-treatment ($p < 0.05). HIV+sup ± morphine-mediated effects were partially, but significantly, reversed by VPA (#p < 0.05). VPA also effectively negated HIV+sup-morphine interactions. (C) At 72 h after treatments, cells were fixed, permeabilized, and labeled for TUNEL and Hoechst 33342. The rate of neuronal apoptosis was reported as the average percentage of TUNEL(+) cells ± SEM. Significance was analyzed by one-way ANOVA and Duncan’s post hoc test; n = 3 separate experiments. HIV+sup significantly increased the percentage of TUNEL(+) cells (*p < 0.05 vs. Control), and morphine augmented the effect ($p < 0.05). HIV+sup ± morphine-mediated effects were partially, but significantly, reversed by VPA co-treatment (#p < 0.05), and VPA negated interactions between HIV+sup and morphine. Control = controlsup; HIV = HIV+sup; Mor = morphine; VPA = valproate.
Role of GSK3β in HIV ± morphine-mediated changes in neuritic arborization, MAP-2 and PSD-95
Since neurite degeneration and synapse losses are the major substrate for HAND (Bellizzi et al., 2006; Ellis et al., 2007; Everall et al., 1999; Kim et al., 2008; Masliah et al., 1997) we assessed changes in neuritic arborization using MAP-2 immunostaining (Fig. 4A) followed by Sholl analysis (Fig. 4B). At 72 h, HIV+sup induced a significant reduction in neuritic arborization. There was no significant morphine interaction, and the HIV+sup effect was partially, but significantly, reversed by VPA co-treatment. Neuritic/synaptic degeneration was additionally confirmed by immunoblotting for MAP-2 and PSD-95 (Fig. 4C and D). At 72 h, HIV+sup significantly decreased MAP-2. Similar to the Sholl analysis, there was no significant morphine interaction (Fig. 4C), and VPA significantly abrogated HIV+ sup-mediated loss of MAP-2. HIV+sup also induced a significant loss in PSD-95, but in this case, there was an increased loss with morphine co-treatment (Fig. 4D). VPA partially, but significantly, reversed HIV+sup ± morphine effects on PSD-95. The HIV-morphine interaction on PSD-95 levels was maintained in the presence of VPA.
Figure 4. Role of GSK3β in HIV+sup ± morphine-mediated changes in neuritic arborization, MAP-2 and PSD-95.

(A) At 72 h after treatments, cells were fixed, permeabilized, and labeled for MAP-2 (green), TUNEL (red) and Hoechst 33342 (blue) (B) Neurite arborization was measured by Sholl analysis in digital images of TUNEL(−) neurons. The findings were reported as average Sholl score ± SEM. HIV+sup significantly reduced the Sholl score (*p < 0.05 vs. Control), without any significant morphine interaction. HIV+sup ± morphine-mediated effects were partially, but significantly, reversed by VPA co-treatment (#p < 0.05). (C) At 72 h after treatments, cells were lysed and immunoblotted for MAP-2 and GAPDH. Findings were reported as average normalized MAP-2 (% control) ± SEM. HIV+sup, with or without morphine, significantly reduced MAP-2 (*p < 0.05 vs. Control). VPA significantly inhibited HIV+sup ± morphine-mediated effects (#p < 0.05). (D) Cell lysates were immunoblotted for PSD-95 and GAPDH. Findings were reported as average normalized PSD-95 (% control) ± SEM. HIV+sup induced a significant loss of PSD-95 (*p < 0.05 vs. Control), which was augmented by morphine co-treatment ($p < 0.05). HIV+sup ± morphine-mediated effects were partially, but significantly, reversed by VPA co-treatment (#p < 0.05). Even in the presence of VPA, morphine significantly augmented the effects of HIV+sup ($p < 0.05). In all studies, significance was analyzed by one-way ANOVA and Duncan’s post hoc test, from n = 3 separate experiments. Control = controlsup; HIV = HIV+sup; Mor = morphine; VPA = valproate.
Neuritic/synaptic degeneration was also assessed in cortical and hippocampal neurons (Fig. 5). In cortical neurons (Fig. 5A), HIV+sup significantly reduced MAP-2 and PSD-95, without significant morphine interactions. VPA significantly abrogated HIV+sup-mediated loss of MAP-2 and partially, but significantly, abrogated loss of PSD-95. In hippocampal neurons (Fig. 5B), HIV+sup induced a significant loss of MAP-2 and PSD-95, which was significantly augmented by morphine co-treatment. VPA significantly reversed HIV+sup ± morphine-mediated effects on MAP-2 and partially, but significantly, reversed effects on PSD-95.
Figure 5. Role of GSK3β in HIV+sup ± morphine-mediated changes in MAP-2 and PSD-95 in cortical and hippocampal neurons.

At 72 h after treatments, cells were lysed and immunoblotted for MAP-2, PSD-95 and GAPDH. Findings were reported as average normalized protein levels (% control) ± SEM. Significance was analyzed by one-way ANOVA and Duncan’s post hoc test; n = 3 separate experiments. (A) Cortical Neurons. MAP-2: HIV+sup ± morphine induced significant loss of MAP-2 (*p < 0.05 vs. Control), VPA significantly inhibited HIV+sup-mediated effects (#p < 0.05). PSD-95: HIV+sup, with or without morphine, significantly reduced PSD-95 (*p < 0.05 vs. Control). HIV+sup-mediated loss of PSD-95 was partially, but significantly, reversed by VPA co-treatment (#p < 0.05). (B) Hippocampal Neurons. MAP-2: HIV+sup induced a significant loss of MAP-2 (*p < 0.05 vs. Control), which was augmented by morphine co-treatment ($p < 0.05). HIV+sup ± morphine-mediated loss of MAP-2 was significantly reversed by VPA co-treatment (#p < 0.05). PSD-95: HIV+sup induced a significant loss of PSD-95 (*p < 0.05 vs. Control), which was augmented by morphine co-treatment ($p < 0.05). HIV+sup ± morphine-mediated effects were partially, but significantly, reversed by VPA co-treatment (#p < 0.05). Control = controlsup; HIV = HIV+sup; Mor = morphine; VPA = valproate.
Effects of small molecule GSK3β-inhibitors
The role of GSK3β activation in HIV+sup ± morphine-mediated neurotoxicity was confirmed additionally using the small molecule GSK3β-inhibitors, SB415286 or XXVI (Fig. 6), both of which have very high specificity for GSK3β compared to VPA. Both small molecule inhibitors partially, but significantly, reduced HIV+sup ± morphine-mediated neuronal death, and also abrogated HIV+sup-morphine interactions (Fig. 6A and B). Both inhibitors reversed the effect of HIV+sup ± morphine on neurite losses, although SB415286 only resulted in partial recovery (Fig. 6C).
Figure 6. Effects of small molecule GSK3β-inhibitors.

(A) Cell viability was assessed by time-lapse imaging analysis. Neurons exposed to ‘Control + SB’, ‘Control + XXVI’, ‘Mor’, ‘Mor + SB’ and ‘Mor + XXVI’ treatments had survival equivalent to ‘Control’, but are not shown here in order to highlight other groups. HIV+sup significantly reduced neuronal survival (*p < 0.05 vs. Control), with a significant morphine interaction ($p < 0.05). The effects of HIV+sup were partially, but significantly, reversed by both small molecule inhibitors (#p < 0.05 vs. respective HIV or HIV + Mor). Both SB415286 and XXVI also effectively negated interactions between HIV+sup and morphine. (B) Cell death was examined using TUNEL-staining. HIV+sup significantly increased the percentage of TUNEL(+) cells (*p < 0.05 vs. Control), with a significant morphine interaction ($p < 0.05). Both small molecule inhibitors partially reversed HIV+sup ± morphine-mediated effects (#p < 0.05) and also eradicated the HIV+sup-morphine interaction. (C) Neuritic arborization was evaluated using MAP-2 immunostaining and Sholl analysis. HIV+sup significantly reduced the Sholl score (*p < 0.05 vs. Control), without a morphine interaction. All losses in arborization were partially, but significantly, reversed by both small molecule inhibitors (#p < 0.05), except that XXVI completely reversed the effect of HIV+sup. Control = controlsup; HIV = HIV+sup; Mor = morphine; SB = SB415286; XXVI = GSK3β inhibitor XXVI.
DISCUSSION
Multiple neurodegenerative processes, including HAND, involve dysregulation of GSK3β pathways (Crews et al., 2009; Dou et al., 2003; Dou et al., 2005; Everall et al., 2002; Maggirwar et al., 1999; Sui et al., 2006). The present work supports those findings, and importantly also demonstrates that GSK3β-activation is a point of convergence for specific interactions between HIV and opiates that lead to neurodegenerative outcomes. We used multiple GSK3β-inhibitors, and found that inhibition entirely negated the ability of morphine to enhance HIV+sup-mediated striatal neuron death. GSK3β-inhibitors did not entirely disrupt HIV+sup-morphine interactive effects on PSD95 in striatal neurons, and only partially reversed certain effects of HIV+sup ± morphine, indicating the likely involvement of other signaling pathways in various aspects of HIV ± opiate-related neurotoxicity.
Though HIV-1 induces profound changes in neuronal morphology, function, and survival, the virus itself rarely, if ever, infects neurons (An et al., 1999a; Bagasra et al., 1996; Kramer-Hammerle et al., 2005; Takahashi et al., 1996); instead, infected and activated glial cells produce cellular and viral products that drive secondary, “bystander” toxicity in neurons (Buescher et al., 2007; Epstein and Gendelman, 1993; Gonzalez-Scarano and Martin-Garcia, 2005; Kaul et al., 2005; Kramer-Hammerle et al., 2005). Our previous studies have shown that glia are crucial in determining the extent of neurotoxic interactions between HIV-1 proteins (Podhaizer et al., 2012; Zou et al., 2011b) or HIV-infective supernatant (Masvekar et al., 2014), and opiates. Since the present studies focused on understanding pathway(s) that underlie HIV-1 and opiate interactions within neurons, glia were excluded from our culture system. Still, the importance of glial-to-neuron signaling in HIV- and opiate-mediated neurotoxicity should be considered and not to be underestimated.
In accord with many previous studies (Crews et al., 2009; Dou et al., 2003; Dou et al., 2005; Everall et al., 2002; Maggirwar et al., 1999; Sui et al., 2006), our results show that HIV+sup induces GSK3β-activation, and that treatments to inhibit GSK3β activation ameliorate neuron death, and neurite and synaptic damage caused by HIV+sup. Previous studies have shown that the HIV-1 proteins Tat and gp120 abnormally activate GSK3β (Crews et al., 2009; Dewhurst et al., 2007; Everall et al., 2002; Maggirwar et al., 1999; Sui et al., 2006). But as our model system uses supernatant from HIV-infected cells, which includes various viral proteins and cytotoxic factors, it is hard to predict which proteins/factors play a role in GSK3β-activation. Presumably, our results may reflect the combined effects of multiple proteins and factors present in the supernatant. Some previous studies have shown that viral proteins induce abnormal GSK3β activation by reducing expression of neurotrophic factors such as fibroblast growth factors (FGFs), which otherwise inhibit GSK3β activity via phosphorylation at Ser 9 residue through activation of phosphatidylinositol-3-kinase (PI3K)/Akt (Crews et al., 2009; Everall et al., 2002).
Abnormally activated GSK3β can promote neuronal damage by dysregulating the function/stability of various structural, metabolic, and signaling proteins, including tau, β-catenin, MAP-2, activator protein 1 (AP-1), cyclic AMP response element binding protein (CREB), nuclear factor-kappa B (NF-κB), heat shock factor-1 (HSF-1), and others (Frame and Cohen, 2001; Grimes and Jope, 2001; Kaytor and Orr, 2002; Plyte et al., 1992). GSK3β inhibitors only partially reversed certain neurotoxic outcomes induced by HIV+sup, suggesting the involvement of signaling molecules/pathways other than GSK3β. Of interest in this regard are kinases such as mitogen-activated protein kinase 10 (MAPK10), double-stranded RNA-activated protein kinase (PKR), and cyclin-dependent kinase 5 (CDK5), all of which are known to play important role in HIV-mediated neurodegenerative processes (Alirezaei et al., 2007; Crews et al., 2009; Patrick et al., 2011).
Studies from the brains of opiate-abusing patients have indirectly implicated the role of GSK3β-activation in opiate-associated neuropathology (Anthony et al., 2010; Ramage et al., 2005), showing increased levels of phosphorylated tau, a target of GSK3β, in the brains of HIV−, opiate abusers at several ages. However, other studies have disagreed on whether opiates induce GSK3β activation (Dobashi et al., 2010; Xie et al., 2010) or inactivation (Li et al., 2010; Polakiewicz et al., 1998); this may be because the effects of opiates on GSK3β-activity occur in a dose- and time-dependent manner (Xie et al., 2010). Acute activation of MORs tends to inhibit GSK3β activity via PI3K/Akt signaling pathway (Polakiewicz et al., 1998), while chronic activation of MORs tends to elevate GSK3β activity (Dobashi et al., 2010; Xie et al., 2010), perhaps through increased intracellular calcium (Dobashi et al., 2010; Hartigan and Johnson, 1999; Quillan et al., 2002). In accord with these studies, our results also show that morphine exerts time-dependent effects on GSK3β activation (Figs. 1 and 2). At earlier time points (4 and 12 h) morphine effects were not observed, but at later time points (24 and 72 h) morphine significantly induced GSK3β activation.
At 24 h, morphine significantly augmented HIV+sup-mediated GSK3β activation (Fig. 2), but this transient interactive effect of morphine was not seen at 72 h. This loss in interaction may be due to selective loss of MOR-expressing neurons, as nearly 50% of cells have died by this time (Figs. 3 and 6). Even though there was no significant interaction between HIV and morphine at 72 h, both HIV and morphine alone induced significant GSK3B activation at that time.
Although there was no interactive effect of morphine on the relative expression of p-GSK3β at 72 h (Fig. 2), morphine did exacerbate the effects of HIV+sup on both PSD-95 and neuron death at that time (Figs. 3 and 4). This discordance may be due to temporal difference in the ‘cause’ and the ‘effect’. The interactive elevation in GSK3β activity seen prior to 72 h is the ‘cause’, which results in the aggravation of neurotoxic outcomes, an ‘effect’ seen later.
In terms of HIV+sup-morphine interactions, VPA co-treatment negated the interaction that augmented striatal neuron death (Fig. 3), but had no effect on interactive reduction in PSD-95 (Fig. 4D). This discrepancy likely reflects the role(s) of signaling molecules/pathways other than GSK3β, and is understandable given that multiple downstream pathways and events are triggered by opiates, HIV virions, and factors secreted from HIV-infected/activated cells. For example, CDK5, like GSK3β, is known to be involved in both HIV- and opiate-associated neuropathology (Anthony et al., 2010; Crews et al., 2009; Parkitna et al., 2006; Patrick et al., 2011; Wang et al., 2007), and might be a likely candidate in this regard.
The studies with striatal neurons used isolated cells lacking excitatory presynaptic inputs that would normally originate from trophic, glutamatergic corticostriatal afferents. Although isolated striatal neurons do not elaborate excitatory PSD-95-containing dendritic spines in normal numbers, they express relatively high levels of PSD-95. Interestingly, HIV+sup-morphine interactions were not observed to affect neurite length (Fig. 4B) or the expression of the neurite marker protein, MAP-2 (Fig. 4C); however, morphine significantly augmented the HIV-mediated loss of PSD-95 in striatal neurons (Fig. 4D). This suggests that HIV-opiate interactions induce adverse effects on synaptic stability instead on neurite outgrowth/pruning.
HIV and opiate neurotoxicity frequently show regional specificity in the brain (Fitting et al., 2010b; Pang et al., 2013). This is partly due to variability in expression of receptors important for HIV binding and entry, as well as opiate receptors (Arvidsson et al., 1995; Berger and Arendt, 2000; Berger and Nath, 1997; Mansour et al., 1995; Mansour et al., 1988; Nath, 2014; Podhaizer et al., 2012; Stiene-Martin et al., 1998; Turchan-Cholewo et al., 2008). However, studies presented here also showed different interactive effects of HIV and morphine in isolated neuron cultures derived from different brain regions. For example, in all neuron cultures (striatal, cortical and hippocampal; Figs. 4 and 5), HIV+sup induced loss of PSD-95, but only in striatal and hippocampal neurons were these effects augmented by morphine co-treatment. Thus, the regional specificity of HIV and morphine interactions in the brain likely has both neuronal and glial contribution.
In previous studies, VPA significantly ameliorated HIV-mediated neurotoxic outcomes both in experimental models (Crews et al., 2009; Dewhurst et al., 2007; Dou et al., 2003) and clinically (Schifitto et al., 2006). Though VPA is a potent GSK3β inhibitor, it is not a specific inhibitor; VPA targets additional molecules/pathways, including histone deacetylase (HDAC), Na+ channels, Ca2+ channels, voltage-gated K+ channels, GSK3α, and others (Chateauvieux et al., 2010). Therefore, we also tested the effects of the small molecule GSK3β-inhibitors, SB415286 and XXVI, which have much greater target specificity. Both small molecule inhibitors ameliorated the individual or combined neurotoxic outcomes due to HIV+sup and morphine (Fig. 6), providing confirmation for a role of GSK3β.
HIV+ patients who abuse opiates show more serious neuropathologies and behavioral and cognitive deficits even though they receive anti-retroviral therapy (Anthony et al., 2008; Bell et al., 2002; Byrd et al., 2011; Meijerink et al., 2014; Robinson-Papp et al., 2012; Smith et al., 2014). Patients with HIV-related pain syndromes, for whom opiates are usually prescribed, might also be at risk for such effects over chronic treatment times. And although recent studies suggest that certain aspects of HIV-related neuropathologies may be reversible (Bellizzi et al., 2006; Ellis et al., 2007; Kim et al., 2008), the situation may be less positive for opiate-exposed patients. Our recent study showed that neurite recovery from an HIV insult, normally robust after return to control conditions, was limited in the continued presence of morphine (Masvekar et al., 2014). This raises the prospect that neurologic symptoms may be more difficult to reverse in opiate-exposed patients even when viral titers are controlled. As the cellular basis of opiate-enhanced HIV-1 neuropathology is not understood, it is not clear whether a different, or additional, therapeutic course would be helpful for patients with dual exposure. The current findings predict that therapeutics specifically limiting GSK3β-activation might be particularly useful as an adjunctive therapy for reducing HAND symptomatology in HIV patients who abuse or are otherwise exposed to opiates.
HIGHLIGHTS.
We demonstrate that GSK3β-activation is a point of convergence for specific interactions between HIV and opiates that lead to neurodegenerative outcomes.
Multiple GSK3β-inhibitors, including valproic acid and small molecule inhibitors, were used to determine GSK3β pathway involvement.
GSK3β-inhibitors entirely negated the ability of morphine to enhance the HIV -mediated death of striatal neurons.
GSK3β-inhibitors partially reversed effects of HIV and HIV+morphine on PSD95 levels and neurite reduction, and did not negate interactive effects, implying the involvement of additional signaling pathways in these outcomes.
Acknowledgments
We gratefully acknowledge the following support from the NIH: R01 DA034231 (PEK/KFH); K02 DA027374, R01 DA033200 (KFH); R01 DA036154 (NE).
Abbreviations
- CNS
central nervous system
- HIV-1
human immunodeficiency virus-1
- HAND
HIV-1-associated neurocognitive disorders
- cART
combination antiretroviral therapy
- Tat
trans-activator of transcription
- gp120
glycoprotein 120
- GSK3β
glycogen synthase kinase-3β
- MAP-2
microtubule-associated protein 2
- MORs
μ-opioid receptors
- VPA
valproate
- p-GSK3β-S9
phospho-GSK3β-Ser9
- PSD-95
postsynaptic density protein 95
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- FGFs
fibroblast growth factors (FGFs)
- PI3K
phosphatidylinositol-3-kinase
- AP-1
activator protein 1
- CREB
cyclic AMP response element binding protein
- NF-κB
nuclear factor-kappa B
- HSF-1
heat shock factor-1
- MAPK10
mitogen-activated protein kinase 10
- PKR
double-stranded RNA-activated protein kinase
- CDK5
cyclin-dependent kinase 5
- HDAC
histone deacetylase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. The EMBO journal. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirezaei M, Watry DD, Flynn CF, Kiosses WB, Masliah E, Williams BR, Kaul M, Lipton SA, Fox HS. Human immunodeficiency virus-1/surface glycoprotein 120 induces apoptosis through RNA-activated protein kinase signaling in neurons. J Neurosci. 2007;27:11047–11055. doi: 10.1523/JNEUROSCI.2733-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An SF, Giometto B, Scaravilli F. HIV-1 DNA in brains in AIDS and pre-AIDS: correlation with the stage of disease. Ann Neurol. 1996;40:611–617. doi: 10.1002/ana.410400411. [DOI] [PubMed] [Google Scholar]
- An SF, Groves M, Giometto B, Beckett AA, Scaravilli F. Detection and localisation of HIV-1 DNA and RNA in fixed adult AIDS brain by polymerase chain reaction/in situ hybridisation technique. Acta Neuropathol. 1999a;98:481–487. doi: 10.1007/s004010051113. [DOI] [PubMed] [Google Scholar]
- An SF, Groves M, Gray F, Scaravilli F. Early entry and widespread cellular involvement of HIV-1 DNA in brains of HIV-1 positive asymptomatic individuals. J Neuropathol Exp Neurol. 1999b;58:1156–1162. doi: 10.1097/00005072-199911000-00005. [DOI] [PubMed] [Google Scholar]
- Anthony IC, Arango JC, Stephens B, Simmonds P, Bell JE. The effects of illicit drugs on the HIV infected brain. Front Biosci. 2008;13:1294–1307. doi: 10.2741/2762. [DOI] [PubMed] [Google Scholar]
- Anthony IC, Norrby KE, Dingwall T, Carnie FW, Millar T, Arango JC, Robertson R, Bell JE. Predisposition to accelerated Alzheimer-related changes in the brains of human immunodeficiency virus negative opiate abusers. Brain. 2010;133:3685–3698. doi: 10.1093/brain/awq263. [DOI] [PubMed] [Google Scholar]
- Arvidsson U, Riedl M, Chakrabarti S, Lee JH, Nakano AH, Dado RJ, Loh HH, Law PY, Wessendorf MW, Elde R. Distribution and targeting of a mu-opioid receptor (MOR1) in brain and spinal cord. J Neurosci. 1995;15:3328–3341. doi: 10.1523/JNEUROSCI.15-05-03328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagasra O, Lavi E, Bobroski L, Khalili K, Pestaner JP, Tawadros R, Pomerantz RJ. Cellular reservoirs of HIV-1 in the central nervous system of infected individuals: identification by the combination of in situ polymerase chain reaction and immunohistochemistry. AIDS. 1996;10:573–585. doi: 10.1097/00002030-199606000-00002. [DOI] [PubMed] [Google Scholar]
- Bell JE, Arango JC, Anthony IC. Neurobiology of multiple insults: HIV-1-associated brain disorders in those who use illicit drugs. J Neuroimmune Pharmacol. 2006;1:182–191. doi: 10.1007/s11481-006-9018-2. [DOI] [PubMed] [Google Scholar]
- Bell JE, Arango JC, Robertson R, Brettle RP, Leen C, Simmonds P. HIV and drug misuse in the Edinburgh cohort. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S35–42. doi: 10.1097/00126334-200210012-00003. [DOI] [PubMed] [Google Scholar]
- Bellizzi MJ, Lu SM, Gelbard HA. Protecting the synapse: evidence for a rational strategy to treat HIV-1 associated neurologic disease. J Neuroimmune Pharmacol. 2006;1:20–31. doi: 10.1007/s11481-005-9006-y. [DOI] [PubMed] [Google Scholar]
- Berger JR, Arendt G. HIV dementia: the role of the basal ganglia and dopaminergic systems. Journal of psychopharmacology. 2000;14:214–221. doi: 10.1177/026988110001400304. [DOI] [PubMed] [Google Scholar]
- Berger JR, Nath A. HIV dementia and the basal ganglia. Intervirology. 1997;40:122–131. doi: 10.1159/000150539. [DOI] [PubMed] [Google Scholar]
- Bokhari SM, Yao H, Bethel-Brown C, Fuwang P, Williams R, Dhillon NK, Hegde R, Kumar A, Buch SJ. Morphine enhances Tat-induced activation in murine microglia. J Neurovirol. 2009;15:219–228. doi: 10.1080/13550280902913628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Turchan-Cholewo J, Smart EJ, Geurin T, Chauhan A, Reid R, Xu R, Nath A, Knapp PE, Hauser KF. Morphine causes rapid increases in glial activation and neuronal injury in the striatum of inducible HIV-1 Tat transgenic mice. Glia. 2008;56:1414–1427. doi: 10.1002/glia.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buescher JL, Gross S, Gendelman HE, Ikezu T. The neuropathogenesis of HIV-1 infection. Handb Clin Neurol. 2007;85:45–67. doi: 10.1016/S0072-9752(07)85004-4. [DOI] [PubMed] [Google Scholar]
- Byrd DA, Fellows RP, Morgello S, Franklin D, Heaton RK, Deutsch R, Atkinson JH, Clifford DB, Collier AC, Marra CM, Gelman B, McCutchan JA, Duarte NA, Simpson DM, McArthur J, Grant I. Neurocognitive impact of substance use in HIV infection. J Acquir Immune Defic Syndr. 2011;58:154–162. doi: 10.1097/QAI.0b013e318229ba41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chateauvieux S, Morceau F, Dicato M, Diederich M. Molecular and therapeutic potential and toxicity of valproic acid. Journal of biomedicine & biotechnology. 2010;2010 doi: 10.1155/2010/479364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng-Mayer C, Levy JA. Distinct biological and serological properties of human immunodeficiency viruses from the brain. Ann Neurol. 1988;23(Suppl):S58–61. doi: 10.1002/ana.410230716. [DOI] [PubMed] [Google Scholar]
- Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, Mills D, Brown MJ, Haigh D, Ward RW, Smith DG, Murray KJ, Reith AD, Holder JC. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chemistry & biology. 2000;7:793–803. doi: 10.1016/s1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- Crews L, Patrick C, Achim CL, Everall IP, Masliah E. Molecular pathology of neuro-AIDS (CNS-HIV) Int J Mol Sci. 2009;10:1045–1063. doi: 10.3390/ijms10031045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cysique LA, Brew BJ. Neuropsychological functioning and antiretroviral treatment in HIV/AIDS: a review. Neuropsychol Rev. 2009;19:169–185. doi: 10.1007/s11065-009-9092-3. [DOI] [PubMed] [Google Scholar]
- Cysique LA, Maruff P, Brew BJ. Prevalence and pattern of neuropsychological impairment in human immunodeficiency virus-infected/acquired immunodeficiency syndrome (HIV/AIDS) patients across pre- and post-highly active antiretroviral therapy eras: a combined study of two cohorts. J Neurovirol. 2004;10:350–357. doi: 10.1080/13550280490521078. [DOI] [PubMed] [Google Scholar]
- Davis LE, Hjelle BL, Miller VE, Palmer DL, Llewellyn AL, Merlin TL, Young SA, Mills RG, Wachsman W, Wiley CA. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology. 1992;42:1736–1739. doi: 10.1212/wnl.42.9.1736. [DOI] [PubMed] [Google Scholar]
- Dewhurst S, Maggirwar SB, Schifitto G, Gendelman HE, Gelbard HA. Glycogen synthase kinase 3 beta (GSK-3 beta) as a therapeutic target in neuroAIDS. J Neuroimmune Pharmacol. 2007;2:93–96. doi: 10.1007/s11481-006-9051-1. [DOI] [PubMed] [Google Scholar]
- Dobashi T, Tanabe S, Jin H, Nishino T, Aoe T. Valproate attenuates the development of morphine antinociceptive tolerance. Neurosci Lett. 2010;485:125–128. doi: 10.1016/j.neulet.2010.08.084. [DOI] [PubMed] [Google Scholar]
- Dore GJ, McDonald A, Li Y, Kaldor JM, Brew BJ. Marked improvement in survival following AIDS dementia complex in the era of highly active antiretroviral therapy. AIDS. 2003;17:1539–1545. doi: 10.1097/00002030-200307040-00015. [DOI] [PubMed] [Google Scholar]
- Dou H, Birusingh K, Faraci J, Gorantla S, Poluektova LY, Maggirwar SB, Dewhurst S, Gelbard HA, Gendelman HE. Neuroprotective activities of sodium valproate in a murine model of human immunodeficiency virus-1 encephalitis. J Neurosci. 2003;23:9162–9170. doi: 10.1523/JNEUROSCI.23-27-09162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H, Ellison B, Bradley J, Kasiyanov A, Poluektova LY, Xiong H, Maggirwar S, Dewhurst S, Gelbard HA, Gendelman HE. Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis. J Neurosci. 2005;25:8375–8385. doi: 10.1523/JNEUROSCI.2164-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF. Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia. 2005;50:91–106. doi: 10.1002/glia.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8:33–44. doi: 10.1038/nrn2040. [DOI] [PubMed] [Google Scholar]
- Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nature genetics. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- Epstein LG, Gendelman HE. Human immunodeficiency virus type 1 infection of the nervous system: pathogenetic mechanisms. Ann Neurol. 1993;33:429–436. doi: 10.1002/ana.410330502. [DOI] [PubMed] [Google Scholar]
- Everall IP, Bell C, Mallory M, Langford D, Adame A, Rockestein E, Masliah E. Lithium ameliorates HIV-gp120-mediated neurotoxicity. Mol Cell Neurosci. 2002;21:493–501. doi: 10.1006/mcne.2002.1196. [DOI] [PubMed] [Google Scholar]
- Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, Grant I, Mallory M, Masliah E. Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC Group. HIV Neurobehavioral Research Center. Brain Pathol. 1999;9:209–217. doi: 10.1111/j.1750-3639.1999.tb00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer-Smith T, Rappaport J. Evolving paradigms in the pathogenesis of HIV-1-associated dementia. Expert reviews in molecular medicine. 2005;7:1–26. doi: 10.1017/S1462399405010239. [DOI] [PubMed] [Google Scholar]
- Fitting S, Xu R, Bull C, Buch SK, El-Hage N, Nath A, Knapp PE, Hauser KF. Interactive comorbidity between opioid drug abuse and HIV-1 Tat: chronic exposure augments spine loss and sublethal dendritic pathology in striatal neurons. Am J Pathol. 2010a;177:1397–1410. doi: 10.2353/ajpath.2010.090945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Zou S, Chen W, Vo P, Hauser KF, Knapp PE. Regional heterogeneity and diversity in cytokine and chemokine production by astroglia: differential responses to HIV-1 Tat, gp120, and morphine revealed by multiplex analysis. Journal of proteome research. 2010b;9:1795–1804. doi: 10.1021/pr900926n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. The Biochemical journal. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Molecular cell. 2001;7:1321–1327. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Gurwell JA, Nath A, Sun Q, Zhang J, Martin KM, Chen Y, Hauser KF. Synergistic neurotoxicity of opioids and human immunodeficiency virus-1 Tat protein in striatal neurons in vitro. Neuroscience. 2001;102:555–563. doi: 10.1016/s0306-4522(00)00461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haenisch F, Alsaif M, Guest PC, Rahmoune H, Dickerson F, Yolken R, Bahn S. Multiplex immunoassay analysis of plasma shows prominent upregulation of growth factor activity pathways linked to GSK3beta signaling in bipolar patients. Journal of affective disorders. 2014;156:139–143. doi: 10.1016/j.jad.2013.12.008. [DOI] [PubMed] [Google Scholar]
- Hartigan JA, Johnson GV. Transient increases in intracellular calcium result in prolonged site-selective increases in Tau phosphorylation through a glycogen synthase kinase 3beta-dependent pathway. J Biol Chem. 1999;274:21395–21401. doi: 10.1074/jbc.274.30.21395. [DOI] [PubMed] [Google Scholar]
- Hauser KF, El-Hage N, Buch S, Berger JR, Tyor WR, Nath A, Bruce-Keller AJ, Knapp PE. Molecular targets of opiate drug abuse in neuroAIDS. Neurotox Res. 2005;8:63–80. doi: 10.1007/BF03033820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Sheng WS, Lokensgard JR, Peterson PK. Morphine induces apoptosis of human microglia and neurons. Neuropharmacology. 2002;42:829–836. doi: 10.1016/s0028-3908(02)00030-8. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Miyatake M, Koshikawa N, Ochiai K, Yamada K, Kiss A, Donlin MJ, Panneton WM, Churchill JD, Green M, Siddiqui AM, Leinweber AL, Crews NR, Ezerskiy LA, Rendell VR, Belcheva MM, Coscia CJ. Morphine modulation of thrombospondin levels in astrocytes and its implications for neurite outgrowth and synapse formation. J Biol Chem. 2010;285:38415–38427. doi: 10.1074/jbc.M110.109827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs KM, Bhave SR, Ferraro DJ, Jaboin JJ, Hallahan DE, Thotala D. GSK-3beta: A Bifunctional Role in Cell Death Pathways. International journal of cell biology. 2012;2012:930710. doi: 10.1155/2012/930710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA. HIV-1 infection and AIDS: consequences for the central nervous system. Cell Death Differ. 2005;12(Suppl 1):878–892. doi: 10.1038/sj.cdd.4401623. [DOI] [PubMed] [Google Scholar]
- Kaytor MD, Orr HT. The GSK3 beta signaling cascade and neurodegenerative disease. Current opinion in neurobiology. 2002;12:275–278. doi: 10.1016/s0959-4388(02)00320-3. [DOI] [PubMed] [Google Scholar]
- Kerza-Kwiatecki AP, Amini S. CNS as an HIV-1 reservoir; BBB and drug delivery. J Neurovirol. 1999;5:113–114. doi: 10.3109/13550289909021992. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Martemyanov KA, Thayer SA. Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci. 2008;28:12604–12613. doi: 10.1523/JNEUROSCI.2958-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho J, Malm T, Goldsteins G. Glycogen synthase kinase-3beta: a mediator of inflammation in Alzheimer’s disease? International journal of Alzheimer’s disease. 2011;2011:129753. doi: 10.4061/2011/129753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozikowski AP, Gaisina IN, Petukhov PA, Sridhar J, King LT, Blond SY, Duka T, Rusnak M, Sidhu A. Highly potent and specific GSK-3beta inhibitors that block tau phosphorylation and decrease alpha-synuclein protein expression in a cellular model of Parkinson’s disease. ChemMedChem. 2006;1:256–266. doi: 10.1002/cmdc.200500039. [DOI] [PubMed] [Google Scholar]
- Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005;111:194–213. doi: 10.1016/j.virusres.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Leroy K, Yilmaz Z, Brion JP. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol. 2007;33:43–55. doi: 10.1111/j.1365-2990.2006.00795.x. [DOI] [PubMed] [Google Scholar]
- Li Y, Li H, Zhang Y, Sun X, Hanley GA, LeSage G, Zhang Y, Sun S, Peng Y, Yin D. Toll-like receptor 2 is required for opioids-induced neuronal apoptosis. Biochem Biophys Res Commun. 2010;391:426–430. doi: 10.1016/j.bbrc.2009.11.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S. HIV-1 Tat-mediated activation of glycogen synthase kinase-3beta contributes to Tat-mediated neurotoxicity. J Neurochem. 1999;73:578–586. doi: 10.1046/j.1471-4159.1999.0730578.x. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Akil H, Watson SJ. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. 1995;18:22–29. doi: 10.1016/0166-2236(95)93946-u. [DOI] [PubMed] [Google Scholar]
- Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ. Anatomy of CNS opioid receptors. Trends Neurosci. 1988;11:308–314. doi: 10.1016/0166-2236(88)90093-8. [DOI] [PubMed] [Google Scholar]
- Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol. 1997;42:963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- Masvekar RR, El-Hage N, Hauser KF, Knapp PE. Morphine Enhances HIV-1SF162-Mediated Neuron Death and Delays Recovery of Injured Neurites. PLoS One. 2014;9:e100196. doi: 10.1371/journal.pone.0100196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur JC. HIV dementia: an evolving disease. J Neuroimmunol. 2004;157:3–10. doi: 10.1016/j.jneuroim.2004.08.042. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005;4:543–555. doi: 10.1016/S1474-4422(05)70165-4. [DOI] [PubMed] [Google Scholar]
- McArthur JC, Haughey N, Gartner S, Conant K, Pardo C, Nath A, Sacktor N. Human immunodeficiency virus-associated dementia: an evolving disease. J Neurovirol. 2003;9:205–221. doi: 10.1080/13550280390194109. [DOI] [PubMed] [Google Scholar]
- Meijerink H, Wisaksana R, Iskandar S, den Heijer M, van der Ven AJ, Alisjahbana B, van Crevel R. Injecting drug use is associated with a more rapid CD4 cell decline among treatment naive HIV-positive patients in Indonesia. Journal of the International AIDS Society. 2014;17:18844. doi: 10.7448/IAS.17.1.18844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatake M, Rubinstein TJ, McLennan GP, Belcheva MM, Coscia CJ. Inhibition of EGF-induced ERK/MAP kinase-mediated astrocyte proliferation by mu opioids: integration of G protein and beta-arrestin 2-dependent pathways. J Neurochem. 2009;110:662–674. doi: 10.1111/j.1471-4159.2009.06156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A. Eradication of human immunodeficiency virus from brain reservoirs. J Neurovirol. 2014 doi: 10.1007/s13365-014-0291-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick DM, Ochshorn M, Ghali V, Croxson TS, Mercer WD, Chiorazzi N, Kreek MJ. Natural killer cell activity and lymphocyte subsets in parenteral heroin abusers and long-term methadone maintenance patients. J Pharmacol Exp Ther. 1989;250:606–610. [PubMed] [Google Scholar]
- Pang X, Panee J, Liu X, Berry MJ, Chang SL, Chang L. Regional Variations of Antioxidant Capacity and Oxidative Stress Responses in HIV-1 Transgenic Rats With and Without Methamphetamine Administration. J Neuroimmune Pharmacol. 2013;8:691–704. doi: 10.1007/s11481-013-9454-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkitna JR, Obara I, Wawrzczak-Bargiela A, Makuch W, Przewlocka B, Przewlocki R. Effects of glycogen synthase kinase 3beta and cyclin-dependent kinase 5 inhibitors on morphine-induced analgesia and tolerance in rats. J Pharmacol Exp Ther. 2006;319:832–839. doi: 10.1124/jpet.106.107581. [DOI] [PubMed] [Google Scholar]
- Patrick C, Crews L, Desplats P, Dumaop W, Rockenstein E, Achim CL, Everall IP, Masliah E. Increased CDK5 expression in HIV encephalitis contributes to neurodegeneration via tau phosphorylation and is reversed with Roscovitine. Am J Pathol. 2011;178:1646–1661. doi: 10.1016/j.ajpath.2010.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson PK, Gekker G, Hu S, Cabral G, Lokensgard JR. Cannabinoids and morphine differentially affect HIV-1 expression in CD4(+) lymphocyte and microglial cell cultures. J Neuroimmunol. 2004;147:123–126. doi: 10.1016/j.jneuroim.2003.10.026. [DOI] [PubMed] [Google Scholar]
- Peterson PK, Gekker G, Schut R, Hu S, Balfour HH, Jr, Chao CC. Enhancement of HIV-1 replication by opiates and cocaine: the cytokine connection. Adv Exp Med Biol. 1993;335:181–188. doi: 10.1007/978-1-4615-2980-4_26. [DOI] [PubMed] [Google Scholar]
- Peterson PK, Sharp BM, Gekker G, Portoghese PS, Sannerud K, Balfour HH., Jr Morphine promotes the growth of HIV-1 in human peripheral blood mononuclear cell cocultures. AIDS. 1990;4:869–873. doi: 10.1097/00002030-199009000-00006. [DOI] [PubMed] [Google Scholar]
- Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, Woodgett JR. Glycogen synthase kinase-3: functions in oncogenesis and development. Biochimica et biophysica acta. 1992;1114:147–162. doi: 10.1016/0304-419x(92)90012-n. [DOI] [PubMed] [Google Scholar]
- Podhaizer EM, Zou S, Fitting S, Samano KL, El-Hage N, Knapp PE, Hauser KF. Morphine and gp120 Toxic Interactions in Striatal Neurons are Dependent on HIV-1 Strain. J Neuroimmune Pharmacol. 2012;7:877–891. doi: 10.1007/s11481-011-9326-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakiewicz RD, Schieferl SM, Gingras AC, Sonenberg N, Comb MJ. mu-Opioid receptor activates signaling pathways implicated in cell survival and translational control. J Biol Chem. 1998;273:23534–23541. doi: 10.1074/jbc.273.36.23534. [DOI] [PubMed] [Google Scholar]
- Quillan JM, Carlson KW, Song C, Wang D, Sadee W. Differential effects of mu-opioid receptor ligands on Ca(2+) signaling. J Pharmacol Exp Ther. 2002;302:1002–1012. doi: 10.1124/jpet.302.3.1002. [DOI] [PubMed] [Google Scholar]
- Ramage SN, Anthony IC, Carnie FW, Busuttil A, Robertson R, Bell JE. Hyperphosphorylated tau and amyloid precursor protein deposition is increased in the brains of young drug abusers. Neuropathol Appl Neurobiol. 2005;31:439–448. doi: 10.1111/j.1365-2990.2005.00670.x. [DOI] [PubMed] [Google Scholar]
- Robertson JR, Ronald PJ, Raab GM, Ross AJ, Parpia T. Deaths, HIV infection, abstinence, and other outcomes in a cohort of injecting drug users followed up for 10 years. BMJ. 1994;309:369–372. doi: 10.1136/bmj.309.6951.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KR, Smurzynski M, Parsons TD, Wu K, Bosch RJ, Wu J, McArthur JC, Collier AC, Evans SR, Ellis RJ. The prevalence and incidence of neurocognitive impairment in the HAART era. AIDS. 2007;21:1915–1921. doi: 10.1097/QAD.0b013e32828e4e27. [DOI] [PubMed] [Google Scholar]
- Robinson-Papp J, Elliott KJ, Simpson DM. HIV-related neurocognitive impairment in the HAART era. Curr HIV/AIDS Rep. 2009;6:146–152. doi: 10.1007/s11904-009-0020-1. [DOI] [PubMed] [Google Scholar]
- Robinson-Papp J, Gelman BB, Grant I, Singer E, Gensler G, Morgello S National Neuro ATC. Substance abuse increases the risk of neuropathy in an HIV-infected cohort. Muscle & nerve. 2012;45:471–476. doi: 10.1002/mus.23231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers TJ, Peterson PK. Opioid G protein-coupled receptors: signals at the crossroads of inflammation. Trends Immunol. 2003;24:116–121. doi: 10.1016/s1471-4906(03)00003-6. [DOI] [PubMed] [Google Scholar]
- Sacktor N. The epidemiology of human immunodeficiency virus-associated neurological disease in the era of highly active antiretroviral therapy. J Neurovirol. 2002;8(Suppl 2):115–121. doi: 10.1080/13550280290101094. [DOI] [PubMed] [Google Scholar]
- Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, Stern Y, Albert S, Palumbo D, Kieburtz K, De Marcaida JA, Cohen B, Epstein L. HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol. 2002;8:136–142. doi: 10.1080/13550280290049615. [DOI] [PubMed] [Google Scholar]
- Sawynok J. The therapeutic use of heroin: a review of the pharmacological literature. Canadian journal of physiology and pharmacology. 1986;64:1–6. doi: 10.1139/y86-001. [DOI] [PubMed] [Google Scholar]
- Schaffer BA, Bertram L, Miller BL, Mullin K, Weintraub S, Johnson N, Bigio EH, Mesulam M, Wiedau-Pazos M, Jackson GR, Cummings JL, Cantor RM, Levey AI, Tanzi RE, Geschwind DH. Association of GSK3B with Alzheimer disease and frontotemporal dementia. Arch Neurol. 2008;65:1368–1374. doi: 10.1001/archneur.65.10.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schifitto G, Peterson DR, Zhong J, Ni H, Cruttenden K, Gaugh M, Gendelman HE, Boska M, Gelbard H. Valproic acid adjunctive therapy for HIV-associated cognitive impairment: a first report. Neurology. 2006;66:919–921. doi: 10.1212/01.wnl.0000204294.28189.03. [DOI] [PubMed] [Google Scholar]
- Sheng WS, Hu S, Gekker G, Zhu S, Peterson PK, Chao CC. Immunomodulatory role of opioids in the central nervous system. Arch Immunol Ther Exp (Warsz) 1997;45:359–366. [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. Journal of anatomy. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Singh IN, Goody RJ, Dean C, Ahmad NM, Lutz SE, Knapp PE, Nath A, Hauser KF. Apoptotic death of striatal neurons induced by human immunodeficiency virus-1 Tat and gp120: Differential involvement of caspase-3 and endonuclease G. J Neurovirol. 2004;10:141–151. doi: 10.1080/13550280490441103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DB, Simmonds P, Bell JE. Brain viral burden, neuroinflammation and neurodegeneration in HAART-treated HIV positive injecting drug users. J Neurovirol. 2014;20:28–38. doi: 10.1007/s13365-013-0225-3. [DOI] [PubMed] [Google Scholar]
- Stiene-Martin A, Zhou R, Hauser KF. Regional, developmental, and cell cycle-dependent differences in mu, delta, and kappa-opioid receptor expression among cultured mouse astrocytes. Glia. 1998;22:249–259. [PMC free article] [PubMed] [Google Scholar]
- Sui Z, Sniderhan LF, Fan S, Kazmierczak K, Reisinger E, Kovacs AD, Potash MJ, Dewhurst S, Gelbard HA, Maggirwar SB. Human immunodeficiency virus-encoded Tat activates glycogen synthase kinase-3beta to antagonize nuclear factor-kappaB survival pathway in neurons. Eur J Neurosci. 2006;23:2623–2634. doi: 10.1111/j.1460-9568.2006.04813.x. [DOI] [PubMed] [Google Scholar]
- Suzuki M, El-Hage N, Zou S, Hahn YK, Sorrell ME, Sturgill JL, Conrad DH, Knapp PE, Hauser KF. Fractalkine/CX3CL1 protects striatal neurons from synergistic morphine and HIV-1 Tat-induced dendritic losses and death. Molecular neurodegeneration. 2011;6:78–95. doi: 10.1186/1750-1326-6-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Wesselingh SL, Griffin DE, McArthur JC, Johnson RT, Glass JD. Localization of HIV-1 in human brain using polymerase chain reaction/in situ hybridization and immunocytochemistry. Ann Neurol. 1996;39:705–711. doi: 10.1002/ana.410390606. [DOI] [PubMed] [Google Scholar]
- Turchan-Cholewo J, Dimayuga FO, Ding Q, Keller JN, Hauser KF, Knapp PE, Bruce-Keller AJ. Cell-specific actions of HIV-Tat and morphine on opioid receptor expression in glia. J Neurosci Res. 2008;86:2100–2110. doi: 10.1002/jnr.21653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, White MG, Akay C, Chodroff RA, Robinson J, Lindl KA, Dichter MA, Qian Y, Mao Z, Kolson DL, Jordan-Sciutto KL. Activation of cyclin-dependent kinase 5 by calpains contributes to human immunodeficiency virus-induced neurotoxicity. J Neurochem. 2007;103:439–455. doi: 10.1111/j.1471-4159.2007.04746.x. [DOI] [PubMed] [Google Scholar]
- Xie N, Li H, Wei D, LeSage G, Chen L, Wang S, Zhang Y, Chi L, Ferslew K, He L, Chi Z, Yin D. Glycogen synthase kinase-3 and p38 MAPK are required for opioid-induced microglia apoptosis. Neuropharmacology. 2010;59:444–451. doi: 10.1016/j.neuropharm.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou S, El-Hage N, Podhaizer EM, Knapp PE, Hauser KF. PTEN gene silencing prevents HIV-1 gp120(IIIB)-induced degeneration of striatal neurons. J Neurovirol. 2011a;17:41–49. doi: 10.1007/s13365-010-0016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou S, Fitting S, Hahn YK, Welch SP, El-Hage N, Hauser KF, Knapp PE. Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at mu-opioid receptor-expressing glia. Brain. 2011b;134:3616–3631. doi: 10.1093/brain/awr281. [DOI] [PMC free article] [PubMed] [Google Scholar]