

Abstract
Aims/hypothesis
Type 2 diabetes mellitus in parents is a strong determinant of diabetes risk in their offspring. We hypothesise that offspring diabetes risk associated with parental diabetes is mediated by metabolic risk factors.
Methods
We studied initially non-diabetic participants of the Framingham Offspring Study. Metabolic risk was estimated using beta cell corrected insulin response (CIR), HOMA-IR or a count of metabolic syndrome components (metabolic syndrome score [MSS]). Dietary risk and physical activity were estimated using questionnaire responses. Genetic risk score (GRS) was estimated as the count of 62 type 2 diabetes risk alleles. The outcome of incident diabetes in offspring was examined across levels of parental diabetes exposure, accounting for sibling correlation and adjusting for age, sex and putative mediators. The proportion mediated was estimated by comparing regression coefficients for parental diabetes with (βadj) and without (βunadj) adjustments for CIR, HOMA-IR, MSS and GRS (percentage mediated = 1 – βadj / βunadj).
Results
Metabolic factors mediated 11% of offspring diabetes risk associated with parental diabetes, corresponding to a reduction in OR per diabetic parent from 2.13 to 1.96. GRS mediated 9% of risk, corresponding to a reduction in OR per diabetic parent from 2.13 to 1.99.
Conclusions/interpretation
Metabolic risk factors partially mediated offspring type 2 diabetes risk conferred by parental diabetes to a similar magnitude as genetic risk. However, a substantial proportion of offspring diabetes risk associated with parental diabetes remains unexplained by metabolic factors, genetic risk, diet and physical activity, suggesting that important familial influences on diabetes risk remain undiscovered.
Keywords: Diet, Family history, Genetic risk, Metabolic syndrome
Introduction
Rates of type 2 diabetes mellitus are rapidly increasing worldwide [1]. A number of risk factors have been clearly demonstrated to contribute to incident diabetes, including lifestyle factors such as diet and physical activity, metabolic factors such as increased central adiposity and triacylglycerol levels, and family history of diabetes [2-4]. Towards elucidating genetic contributions to familial diabetes risk, genome-wide and exome-wide association studies of ever-increasing size have revealed over 65 loci associated with type 2 diabetes and over 50 loci associated with glycaemic quantitative traits such as fasting glucose and fasting insulin [5-11]. Notably, diabetes in an individual's parents confers risk of incident diabetes even after adjusting for known common genetic risk alleles, suggesting that diabetes risk associated with family history is transduced through pathways in addition to simple genetic transmission [12, 13]. Health-related behaviours such as diet, physical activity and smoking, as well as aberrant glucose and insulin metabolism might be shared across generations and account for familial type 2 diabetes risk.
Recent work has attempted to address the uncertainty regarding the mechanism through which family history of diabetes confers incident diabetes risk on offspring. In a multicentre European cohort, 35 common genetic risk alleles, physical activity, adherence to a Mediterranean diet, smoking and education were examined as putative mediators of familial risk of type 2 diabetes [14]. In that study, although the risk of incident diabetes in offspring associated with having a parent with diabetes was comparable with that of previous studies, all of the examined factors together mediated only 15–20% of familial diabetes risk, with lifestyle factors contributing negligibly [14]. As studies in multiple longitudinal cohorts have shown an adverse metabolic profile in non-diabetic offspring of individuals with diabetes [15-22], we were interested in whether or not metabolic derangement might explain familial diabetes risk.
A clearer understanding of the mechanisms by which parental diabetes confers diabetes risk to offspring could be valuable. It is possible that diabetes risk in individuals with family history might be mitigated if mediating risk factors such as adverse diet or metabolic dysfunction are targeted for intervention. To this end, we conducted a mediation analysis that examines metabolic risk factors, diet, physical activity and 62 common genetic risk alleles as putative mediators of the association between parental diabetes and risk of incident diabetes in offspring. Given that non-diabetic offspring of diabetic individuals carry an adverse metabolic profile [19], we hypothesise that metabolic risk factors will be strongly correlated with parental diabetes and will substantively mediate type 2 diabetes risk associated with family history of diabetes.
Methods
Study population
We used data from the Framingham Offspring Study (FOS), which is a well-described prospective population cohort that began in 1971, comprised of the offspring of members of the original Framingham Heart Study (FHS) and their spouses [13]. As many of the exposure measurements occurred at the fifth follow-up exam of the FOS, we limited our analyses to FOS participants who were non-diabetic at exam five and who underwent whole-genome common variant genotyping, for whom anthropometric and laboratory exposures were measured at exam five, and who had directly assessed parental diabetes status. In all, 2631 individuals in the FOS had directly assessed parental diabetes status, the primary exposure, and diabetes status, the primary outcome, as well as metabolic mediators and genotyping (HOMA-IR and corrected insulin response [CIR] could not be calculated in 96 and 82 participants, respectively, and genotyping was unavailable in 282 participants). The Partners Human Research Committee approved these analyses and all participants in the FOS provided informed consent to their participation.
Parental history of diabetes
For the participants in this study, diabetes was defined in the parents as described in prior work examining type 2 diabetes in the original cohort of the FHS; specifically, casual glucose measurement ≥11.1 mmol/l, glucose level ≥11.1 mmol/l after a 50-g oral glucose challenge, or on insulin or oral hypoglycaemic medications [19]. Since only participants with directly assessed diabetes in both parents were included in the study, we then assigned a parental diabetes variable with value of 0, 1 or 2 to each participant, corresponding to the number of parents with diabetes.
Metabolic variables
Each FOS follow-up visit included a detailed medical history, physical examination, and fasting blood glucose and lipids, including triacylglycerol, HDL-cholesterol and total cholesterol; exam five also included fasting insulin and a 2-h OGTT with measurement of post-challenge glucose and insulin. The physical exam component included height and weight measurements, such that the BMI could be calculated as the weight (in kilograms) divided by the height (in metres) squared.
We estimated metabolic risk at exam five (mean age 54.2 years) with three metrics: CIR as a proxy for beta cell function, HOMA-IR, and a count of metabolic syndrome components to create a score, both proxies for insulin resistance. We calculated CIR using 120 min post-OGTT values of serum insulin and glucose, and HOMA-IR using fasting values of serum insulin and glucose as previously described [23]. We calculated a metabolic syndrome score (MSS) using a four-point scale that was modified from the definition of metabolic syndrome specified by the American Heart Association [24]. We assigned a point for the score for increased waist circumference >102 cm in men or >88 cm in women; for fasting serum triacylglycerol >1.7 mmol/l, or HDL-cholesterol <1.0 mmol/l in men or <1.3 mmol/l in women, or for those on lipid medication; for systolic blood pressure >130 mmHg or diastolic blood pressure >85 mmHg; or for fasting blood glucose >7.0 mmol/l or those on hyperglycaemia medication. Thus, we included a point for aberrant values in each of four domains: adiposity, lipidaemia, blood pressure and glycaemia. A higher score indicates greater metabolic abnormality.
Lifestyle exposures
Previous work has demonstrated that increased glycaemic load, decreased cereal fibre intake, decreased polyunsaturated fat intake, increased monounsaturated fat intake and increased trans fat intake are associated with increased risk of diabetes [4]. Each of these dietary components was estimated from a 126-item Food Frequency Questionnaire (FFQ) in FOS at exam five [4, 25, 26]. The FFQ consisted of a list of foods with standard serving sizes and a selection of nine frequency categories ranging from none or <1 serving per month to ≥6 servings per day. Nutrient intake was calculated by multiplying the frequency of consumption of a food item by the nutrient content per standard serving size for that food item. Dietary information was considered valid only if reported energy intake was as follows: ≥2.5 MJ/day (600 kcal/day) for both men and women; <16.7 MJ/day (4,000 kcal/day) for women; <17.5 MJ/day (4,200 kcal/day) for men; or if <13 food items were left blank on the FFQ. Diet data were available in 2,159 out of 2,361 participants and did not appear to be missing differentially across parental diabetes categories. We created a composite diabetes-related diet score as previously described by assigning each participant a score between one and five for cereal fibre intake, glycaemic load, trans fat intake and ratio of poly- to monounsaturated fat, corresponding to his/her quintile of intake for that component, and summing the four quintile scores to generate a single composite score [4]. For consistency with the other risk factors examined in this study for which a higher value corresponds with increased diabetes risk, we assigned the quintile scores such that a score of 1 corresponded to the lowest-risk quintile. That is, a low diabetogenic diet score corresponded to a diet low in trans fat and glycaemic load and high in cereal fibre with a high ratio of poly- to monounsaturated fat.
Physical activity was measured in FOS at exam five as described previously [27]. Briefly, participants were asked in a structured questionnaire to indicate the number of hours spent in each of five levels of activity—asleep, sedentary, light, moderate and heavy. Their responses contributed to a weighted sum, the physical activity index, with a score of 120 representing 24 h/day spent in strenuous (‘heavy’) activity and a score of 24 representing 24 h/day spent asleep. Physical activity data were available in 2,098 out of 2,361 participants but did not appear to be differentially missing across parental diabetes categories.
Genetic risk
Genotyping method and quality control in FOS, and the calculation of a genetic risk score (GRS) based on index or proxy single nucleotide polymorphisms (SNPs) at 62 loci associated with type 2 diabetes in the Diabetes Genetics Replication and Meta-Analysis (DIAGRAM) Consortium, have been described previously [13, 28]. Briefly, presence of 0, one or two diabetes-associated risk alleles was determined for each individual in the study at 62 out of 65 loci identified in DIAGRAMv3, with suitable genotype at the index or proxy SNP unavailable at three loci: rs11063069 (CCND2), rs11651052 (HNF1B) and rs8108269 (GIPR). The GRS was calculated as the weighted sum of the number of alleles at each of the 62 loci, weighted by the β coefficient (effect size) from DIAGRAMv3 [28].
Type 2 diabetes outcome
Included participants were followed through the most recent follow-up visit (exam eight) for a median follow-up of 13 years, with the primary outcome being incident type 2 diabetes in offspring during the follow-up period. Type 2 diabetes was defined as having a fasting glucose value >7.0 mmol/l or being on diabetes medications [13], and there were a total of 265 cases of incident diabetes over the follow-up period.
Statistical analysis
The association between incident type 2 diabetes in offspring and parental diabetes status was examined using generalised estimating equations with the logit link function, adjusting for age and sex and accounting for sibling correlation through clustering by family. Association between putative mediators—CIR, MSS, HOMA-IR, GRS, diabetogenic diet and physical activity—and parental diabetes status was examined using generalised estimating equations to account for sibling correlation. Pairwise correlations between putative mediators were examined using Pearson correlation. Diet and physical activity data were missing in approximately 10% of the study participants. We performed multiple imputation with chained equations using PROC MI in SAS 9.3 (SAS Institute, Cary, NC, USA) and analysed output from 25 datasets to impute values for the summary diet score and physical activity index, accounting for sibling correlation in the imputation procedure [29].
Mediation analysis was performed using established methods for dichotomous outcomes [30]. Figure 1 shows a schematic representation of mediation analysis in which the total effect of a predictor on an outcome is decomposed into direct and indirect effects. To provide a context for the results, we took an approach analogous to prior work examining diabetes and parental history [14]. Briefly, in logistic regression models, we compared the β coefficient for parental diabetes status associated with odds of incident diabetes, comparing models including only age, sex and parental diabetes with models including age, sex, parental diabetes and putative mediators. The ‘proportion mediated’ was calculated as the difference in the parental diabetes status β coefficients between the model with and without mediators divided by the parental diabetes status β coefficient from the model without mediators. Similarly, to examine metabolic mediators after adjusting for genetic risk, we compared the parental diabetes β coefficients from models with and without mediators after adjusting all models for GRS in addition to age and sex.
Fig. 1.
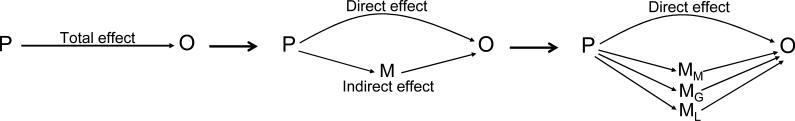
Schematic diagram of mediation analysis. An association (total effect) between a predictor (P) and outcome (O) can be decomposed to a direct effect and an indirect effect via a mediator (M). In this study, P is parental type 2 diabetes and O is incident offspring diabetes, and we examine three putative mediators—metabolic (MM), genetic (MG) and lifestyle (ML) risk factors
To be as inclusive of putative mediators as possible, we used a nominal p value threshold of 0.05 for association tests between mediators and either parental diabetes or incident offspring diabetes. Thus, all putative mediators had to at least be associated with parental diabetes at that nominal p value threshold. Given the correlations between metabolic mediators, evaluating each independently as a mediator would likely lead to biased mediation estimates. To avoid this bias, we performed mediation analysis with all three metabolic mediators (CIR, HOMA-IR and MSS) together; however, this modelling choice made it impossible to generate CIs and perform a formal statistical hypothesis test for estimates of proportion mediated by mediators examined together. To examine significance of putative mediators individually, we estimated the indirect or mediated effect as the product of the coefficients from parental history-mediator and mediator-diabetes regression; the approach has been previously described in detail [31]. All statistical analyses were performed using the SAS 9.3 statistical package.
Results
Participant characteristics
Table 1 shows relevant baseline characteristics of non-diabetic participants stratified by the number of parents with diabetes. Individuals with no, uni- or bi-parental diabetes were similar with respect to sex distribution and were comparably aged, and those with parental diabetes had a BMI approximately 1 kg/m2 higher than those without parental diabetes.
Table 1.
Characteristics of FOS participants stratified by parental diabetes status
| Characteristic | Number of parents with diabetes |
p value | ||
|---|---|---|---|---|
| None (n=1965) | One (n=370) | Two (n=26) | ||
| Age (years) | 54.5 ± 9.9 | 52.5 ± 9.7 | 54.9 ± 9.8 | 0.002 |
| Men | 889 (45%) | 181 (49%) | 11 (42%) | 0.40 |
| BMI (kg/m2) | 27.1 ± 4.6 | 28.0 ± 5.1 | 27.9 ± 5.4 | 0.001 |
| Waist circumference (cm) | 91.7 ± 13.9 | 92.9 ± 14.0 | 93.0 ± 15.0 | 0.25 |
| Systolic blood pressure (mmHg) | 124.9 ± 18.1 | 125.0 ± 17.1 | 129.8 ± 15.9 | 0.38 |
| Diastolic blood pressure (mmHg) | 74.1 ± 9.9 | 75.9 ± 9.5 | 75.9 ± 8.6 | 0.003 |
| Fasting glucose (mmol/l) | 5.26 ± 0.52 | 5.35 ± 0.57 | 5.56 ± 0.59 | 0.0004 |
| Fasting insulin (pmol/l) | 175.2 ± 65.4 | 186.0 ± 70.2 | 171.6 ± 97.2 | 0.02 |
| 2-h glucose (mmol/l) | 6.11 ± 1.55 | 6.38 ± 1.94 | 6.76 ± 1.84 | 0.002 |
| CIR | 4.1 ± 7.4 | 3.9 ± 5.5 | 2.1 ± 1.3 | 0.35 |
| HOMA-IR | 6.9 ± 3.0 | 7.5 ± 3.2 | 7.3 ± 5.1 | 0.007 |
| HDL-cholesterol (mmol/l) | 1.31 ± 0.39 | 1.27 ± 0.39 | 1.37 ± 0.33 | 0.11 |
| Triacylglycerol (mmol/l) | 1.60 ± 1.10 | 1.71 ± 1.52 | 1.50 ± 1.01 | 0.22 |
| MSS (range 0–4) | 1.5 ± 1.2 | 1.6 ± 1.2 | 1.9 ± 1.0 | 0.03 |
| Diet score (range 4–20)a | 12.0 ± 2.7 | 12.2 ± 2.7 | 11.2 ± 2.7 | 0.14 |
| Physical activity index (range 24–120)b | 34.8 ± 6.3 | 34.9 ± 5.9 | 34.9 ± 6.0 | 0.94 |
| GRS (0–124 score) | 66.5 ± 5.2 | 67.6 ± 5.5 | 69.3 ± 5.7 | <0.0001 |
Total n=2,159 for diet score
Total n=2,098 for physical activity index
Association between putative mediators and incident type 2 diabetes and parental diabetes
Figure 1 shows a schematic representation of mediation analysis and its application to this study of metabolic, genetic and lifestyle mediators of the effect of exposure to parental diabetes on the outcome of incident diabetes in offspring. In order for a variable to mediate the effect of a predictor on an outcome it must be associated with both the predictor and the outcome. To verify that each of our putative mediators could lie on a causal path between parental diabetes and incident diabetes in offspring, we first tested whether HOMA-IR, MSS, CIR, GRS, diet score and physical activity index were associated with incident offspring diabetes in our study population. As expected, HOMA-IR, MSS, GRS and diet score were significantly associated with incident diabetes (Table 2). CIR and physical activity index were not significantly associated with incident diabetes, making them less compelling candidate mediators of the association between parental diabetes and incident diabetes in our study population (Table 2).
Table 2.
Association between putative mediators and incident diabetes
| Mediator | OR (95% CI)a | p value |
|---|---|---|
| CIR | 0.95 (0.86, 1.06) | 0.37 |
| HOMA-IR | 1.29 (1.23, 1.36) | <0.0001 |
| MSS | 2.64 (2.30, 3.02) | <0.0001 |
| Diet score | 1.06 (1.01, 1.12) | 0.03 |
| Physical activity index | 1.01 (0.99, 1.03) | 0.34 |
| GRS | 1.07 (1.04, 1.10) | <0.0001 |
OR of incident type 2 diabetes per unit increase in each of the indicated indices, adjusting for age and sex
We examined whether each putative mediator was associated with parental diabetes status. All of the mediators representing metabolic function—CIR, HOMA-IR and MSS—and genetic risk, but not diabetogenic diet or physical activity, were associated with parental diabetes; that is, individuals with 0, one or two diabetic parents differed significantly in CIR, HOMA-IR, MSS and GRS (Table 3). Given that the diabetogenic diet score was not correlated with parental diabetes, and that physical activity index was not associated with either parental diabetes or incident offspring diabetes in our study population, these indices of lifestyle factors were not included in subsequent mediation analyses.
Table 3.
Association between putative mediators and parental diabetes
| Mediator | Number of parents with diabetes |
p value | ||
|---|---|---|---|---|
| None (n=1965) | One (n=370) | Two (n=26) | ||
| CIR | 2.31 [1.47, 4.00] | 2.30 [1.42, 4.25] | 1.79 [0.96, 2.86] | 0.01 |
| HOMA-IR | 6.22 [5.11, 7.86] | 6.59 [5.40, 8.33] | 6.41 [4.71, 7.99] | 0.01 |
| MSS | 1 [1, 2] | 1 [1, 3] | 2 [1, 3] | 0.03 |
| Diet score | 12 [10, 14] | 12 [10, 14] | 11 [9, 14] | 0.20 |
| Physical activity | 33.2 [30.4, 36.8] | 33.8 [30.7, 36.7] | 33.1 [31.4, 36.7] | 0.93 |
| GRS | 66.4 [63.1, 69.9] | 67.6 [63.5, 71.4] | 71.1 [66.1, 72.5] | 0.004 |
Data are presented as median (interquartile range)
Mediation analysis of familial diabetes risk
As a strong correlation between mediators can lead to biased estimates of mediation, we tested pairwise correlations between CIR, HOMA-IR, MSS and GRS. As expected, HOMA-IR and MSS were strongly positively correlated (r=0.535; p<0.0001), and CIR was negatively correlated with MSS (r=−0.113; p<0.0001) and uncorrelated with HOMA-IR (r=−0.035; p=0.089) (Table 4). GRS was weakly correlated positively with MSS (r=0.058; p=0.005) and negatively with CIR (r=−0.061; p=0.003), but was not correlated with HOMA-IR. As it can be challenging to dissect the contributions of strongly correlated mediators, we considered all three metabolic mediators together in our primary analyses, limiting inferences to the combined effects of metabolic dysfunction. We chose to examine the GRS as an independent mediator as well as jointly with metabolic risk factors.
Table 4.
Correlation between metabolic risk factors
| Risk factor | CIR | HOMA-IR | MSS | GRS | |
|---|---|---|---|---|---|
| CIR | r | 1 | −0.035 | −0.113 | −0.061 |
| p value | - | 0.089 | <0.0001 | 0.003 | |
| HOMA-IR | r | −0.035 | 1 | 0.535 | −0.009 |
| p value | 0.089 | - | <0.0001 | 0.68 | |
| MSS | r | −0.113 | 0.535 | 1 | 0.058 |
| p value | <0.0001 | <0.0001 | - | 0.005 | |
| GRS | r | −0.061 | −0.0086 | 0.058 | 1 |
| p value | 0.003 | 0.68 | 0.005 | - | |
To estimate the proportion of familial diabetes risk mediated by metabolic factors or genetic risk, we compared the ORs for incident offspring diabetes per parent with diabetes in models adjusting only for age and sex, and in models adjusting for age, sex and putative mediators. The odds of incident type 2 diabetes in offspring increased by 2.13-fold per parent with diabetes, and metabolic factors and genetic risk mediated 10.9% and 9% of this parental risk, respectively (Fig. 2). Examined together, metabolic factors and genetic risk mediated 18% of incident offspring diabetes risk associated with an increasing number of diabetic parents (Fig. 2). The indirect effect (i.e. the component of offspring diabetes risk associated with parental diabetes that is mediated by each putative mediator) was significant for MSS, HOMA-IR and GRS individually in models adjusting for age and sex (Table 5).
Fig. 2.

Analysis of mediators of parental diabetes association with incident diabetes. Association of parental diabetes with incident offspring diabetes. ORs for offspring diabetes per diabetic parent in models adjusted for covariates in the ‘base model’ and ‘mediators’, if any, are shown. Proportion of parental history effect mediated was calculated as 1 – [loge(ORBase model + Mediators) / loge(ORBase model)]
Table 5.
Indirect effect of parental diabetes mediated by individual mediators in models unadjusted for GRS (above) and adjusted for GRS (below)
| Base model | Mediator | Indirect effect, OR (95% CI) | Proportion mediated, % (95% CI) | p value |
|---|---|---|---|---|
| Age, sex | MSS | 1.20 (1.07, 1.33) | 21.0 (7.8, 32.9) | 0.001 |
| HOMA-IR | 1.13 (1.04, 1.22) | 14.4 (4.6, 23.5) | 0.002 | |
| CIR | 1.02 (0.98, 1.06) | 2.6 (−2.6, 7.6) | 0.26 | |
| GRS | 1.07 (1.03, 1.12) | 8.9 (3.9, 14.9) | 0.002 | |
| Age, sex, GRS | MSS | 1.18 (1.06, 1.32) | 20.5 (7.2, 34.4) | 0.003 |
| HOMA-IR | 1.14 (1.05, 1.23) | 16.8 (6.3, 26.6) | 0.002 | |
| CIR | 1.01 (0.98, 1.05) | 1.4 (−2.9, 7.0) | 0.35 |
The proportion mediated by GRS and metabolic factors together exceeded the proportion mediated by either alone, suggesting that these factors mediated non-overlapping proportions of risk associated with parental diabetes. To confirm that familial type 2 diabetes risk might decompose into separate genetic and metabolic proportions, we examined the impact of including metabolic factors on diabetes risk associated with parental diabetes in models adjusted for age, sex and GRS. Thus, after adjusting for GRS by including it in the base model, we were able to examine whether or not metabolic factors mediated familial risk of diabetes independently from the GRS. The OR for incident offspring diabetes was reduced from 1.99 to 1.84 per diabetic parent comparing a model adjusted for age, sex and GRS to one adjusted for age, sex, GRS and metabolic factors. This finding suggests that metabolic factors mediate approximately 11% of the parental diabetes risk independent of genetic risk (Fig. 2). Similarly, when examining metabolic factors individually, MSS and HOMA-IR, but not CIR, demonstrated statistically significant indirect effects as mediators of offspring diabetes risk associated with parental diabetes after adjusting for age, sex and GRS (Table 5).
Discussion
In this work we attempted to understand the pathways through which parental diabetes confers risk of incident diabetes on offspring. Using mediation analysis techniques, we found that metabolic factors associated with insulin resistance and beta cell dysfunction mediated approximately 11% of familial diabetes risk, a proportion comparable with that mediated by a count of 62 common genetic risk alleles. Furthermore, metabolic factors and genetic risk alleles appeared to mediate separate aspects of familial type 2 diabetes risk as the proportion of offspring diabetes risk associated with parental diabetes mediated by metabolic factors was not substantially affected by adjusting for a 62 common variant GRS.
This approach contributes to recent research efforts to better understand diabetes risk associated with family history of diabetes that have examined mediation by lifestyle factors, including diet, physical activity, education and smoking, as well as genetic risk [14]. We found that a more substantial proportion of familial diabetes risk was mediated by metabolic abnormalities than was previously observed for lifestyle factors [14]. In our study population, however, diabetogenic dietary habits and physical activity were not significantly associated with parental diabetes, and so could not be effectively analysed as putative mediators of familial diabetes risk. Overall, the results of this study agree qualitatively with recent work [14] in that lifestyle factors do not appear to mediate a substantial proportion of familial diabetes risk, genetic risk mediates a minor proportion of familial diabetes risk and a substantial proportion of familial diabetes risk is unexplained by lifestyle or genetic risk. The present study expands upon previous work by more comprehensively examining metabolic risk; although metabolic factors do mediate a portion of familial risk, the majority of offspring diabetes risk associated with parental diabetes remains unexplained. The finding that metabolic risk factors that mediate familial risk are separable from common genetic risk alleles suggests that metabolic dysfunction in non-diabetic offspring with at least one diabetic parent is not explained solely by known genetic loci associated with type 2 diabetes risk.
There are a number of possible explanations for the substantial unexplained proportion of diabetes risk associated with parental diabetes. First, our understanding of the genetic basis of diabetes is still at an early stage and much of the unexplained familial risk of diabetes may be due to as yet undiscovered genetic causes. While significant effort has been expended to discover genetic loci associated with type 2 diabetes and other glycaemic traits, including fasting glucose and insulin, the common genetic risk alleles discovered to date explain a relatively small proportion of estimated heritability of their respective glycaemic traits [9-11]. Age of parental diabetes onset may be a useful tool for discriminating genetic and metabolic familial risk, and prior work has demonstrated that onset of parental diabetes prior to 50 years of age is more strongly associated with increased risk of offspring diabetes than parental diabetes at any age; we lacked power, however, to examine this in the mediation framework [19]. Second, epigenetic factors that are associated with diabetes risk through mechanisms not captured by the metabolic measurements used here could mediate familial diabetes risk. It is notable that prior work has demonstrated a differential effect of maternal and paternal diabetes on offspring diabetes risk, suggesting that there may be parent-of-origin effects influencing genetic and epigenetic effects in diabetes [32]. Finally, there may be important aspects of the familial environment that are not captured in the lifestyle metrics employed here and that are not translated into metabolic disturbances captured in the measurements used in this study. We attempted to address shared behaviours resulting from the familial environment, but we did not detect an association between parental diabetes and offspring diet or physical activity, a limitation that suggests that health behaviours transmitted within families may be better assessed in a younger study population.
This study has a number of strengths. Most importantly, the family-based study structure allows direct assessment of parental diabetes, enabling accuracy in exposure assessment possible in few studies of family history. Furthermore, the measurements of all covariates/mediators are performed uniformly for all participants in the study. In particular, few studies of this size have direct parental history diabetes assessment, uniform genotyping at diabetes risk loci and uniform measurement of clinical, laboratory, physical activity and FFQ-derived diet data. Finally, the long duration of the FHS affords substantial follow-up time in our study participants to assess development of type 2 diabetes.
This study also has several limitations. Most importantly, the measures of lifestyle factors, in particular physical activity, are limited. The physical activity index estimated from questionnaire responses is not correlated with incident diabetes; prior analyses of physical activity in other studies clearly demonstrates association with type 2 diabetes incidence [33], and the absence of such an association in our data calls into question the accuracy of the physical activity index applied in this study. More broadly, the instruments to assess behavioural risk factors, namely structured questionnaires, are inherently subjective and thus have greater inter- and intra-individual variability, though they have previously been effectively used in this study population [3]. Similarly, CIR and HOMA of beta cell function (HOMA-β) are the only proxies for beta cell function that can be estimated in the FHS; previous work has indicated that CIR is only moderately correlated with better estimators of beta cell function but more strongly than HOMA-β [23]. Finally, the mediators are measured at exam five of the FOS, the baseline for our study, when participants are a mean of 54 years old; as such, many factors that may be associated with the familial environment at a younger age when the study participants live with or in close proximity to parents with diabetes are likely diluted by time.
Despite these limitations, this work demonstrates that metabolic disturbances in non-diabetic offspring mediate a substantive proportion of type 2 diabetes risk associated with parental diabetes. Furthermore, metabolic and genetic risk only partially mediate familial diabetes risk, suggesting that as yet undiscovered aspects of the familial environment or genetics underlie risk associated with parental diabetes. Complete decomposition of familial diabetes risk might allow both a better mechanistic understanding of pathways of diabetes risk transmission and levers upon which to intervene in individuals with parental diabetes. At present, however, familial diabetes risk cannot be completely substituted with assessment of genetic and metabolic risk, and, thus, family history remains an important factor in assessing an individual's risk of incident type 2 diabetes.
Acknowledgments
Funding
SR is supported by National Institutes of Health National Research Service Award T32HP10251 and the Ryoichi Sasakawa Fellowship Fund. JBM and JD are supported by NIH RO1DK78616, and JBM is also supported by K24 DK080140. We gratefully acknowledge the FHS study participants and staff for their contributions. This work was partially supported by the National Heart, Lung and Blood Institute's Framingham Heart Study (Contract No. N01-HC-25195) and its contract with Affymetrix for genotyping services (Contract No. N02-HL-6-4278). NM is supported by USDA agreement number 1950-51530-011-00D.
Abbreviations
- CIR
Corrected insulin response
- DIAGRAM
Diabetes Genetics Replication and Meta-Analysis
- FFQ
Food Frequency Questionnaire
- FHS
Framingham Heart Study
- FOS
Framingham Offspring Study
- GRS
Genetic risk score
- HOMA-β
HOMA of beta cell function
- MSS
Metabolic syndrome score
- SNP
Single nucleotide polymorphism
Footnotes
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author contributions
SR and JBM conceived and designed the study. SR, BP and NM contributed to data analysis and interpretation. JBM, NM, CSF and JD contributed to data acquisition and interpretation. All authors contributed to drafting and critical revisions of the manuscript, as well as final approval of the version submitted for review and publication. JBM is the guarantor of this work.
References
- 1.Danaei G, Finucane MM, Lu Y, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet. 2011;378:31–40. doi: 10.1016/S0140-6736(11)60679-X. [DOI] [PubMed] [Google Scholar]
- 2.Wilson PWF, Meigs JB, Sullivan L, Fox CS, Nathan DM, D'Agostino RB. Prediction of incident diabetes mellitus in middle-aged adults: the Framingham Offspring Study. Archives of internal medicine. 2007;167:1068–1074. doi: 10.1001/archinte.167.10.1068. [DOI] [PubMed] [Google Scholar]
- 3.Kannel WB, Sorlie P. Some health benefits of physical activity. The Framingham Study. Arch Intern Med. 1979;139:857–861. [PubMed] [Google Scholar]
- 4.Hu FB, Manson JE, Stampfer MJ, et al. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N Engl J Med. 2001;345:790–797. doi: 10.1056/NEJMoa010492. [DOI] [PubMed] [Google Scholar]
- 5.Barnett AH, Eff C, Leslie RD, Pyke DA. Diabetes in identical twins. A study of 200 pairs. Diabetologia. 1981;20:87–93. doi: 10.1007/BF00262007. [DOI] [PubMed] [Google Scholar]
- 6.Elbein SC, Hasstedt SJ, Wegner K, Kahn SE. Heritability of pancreatic beta-cell function among nondiabetic members of Caucasian familial type 2 diabetic kindreds. J Clin Endocrinol Metab. 1999;84:1398–1403. doi: 10.1210/jcem.84.4.5604. [DOI] [PubMed] [Google Scholar]
- 7.Poulsen P, Kyvik KO, Vaag A, Beck-Nielsen H. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance--a population-based twin study. Diabetologia. 1999;42:139–145. doi: 10.1007/s001250051131. [DOI] [PubMed] [Google Scholar]
- 8.Hanson RL, Imperatore G, Narayan KM, et al. Family and genetic studies of indices of insulin sensitivity and insulin secretion in Pima Indians. Diabetes/metabolism research and reviews. 2001;17:296–303. doi: 10.1002/dmrr.213. [DOI] [PubMed] [Google Scholar]
- 9.Manning AK, Hivert MF, Scott RA, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nature genetics. 2012;44:659–669. doi: 10.1038/ng.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scott RA, Lagou V, Welch RP, et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nature genetics. 2012;44:991–1005. doi: 10.1038/ng.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium. Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium. South Asian Type 2 Diabetes (SAT2D) Consortium et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nature Genetics. 2014;46:234–244. doi: 10.1038/ng.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vassy JL, Durant NH, Kabagambe EK, et al. A genotype risk score predicts type 2 diabetes from young adulthood: the CARDIA study. Diabetologia. 2012;55:2604–2612. doi: 10.1007/s00125-012-2637-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vassy JL, Hivert MF, Porneala B, et al. Polygenic type 2 diabetes prediction at the limit of common variant detection. Diabetes. 2014;63:2172–2182. doi: 10.2337/db13-1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.InterAct Consortium. Scott RA, Langenberg C, et al. The link between family history and risk of type 2 diabetes is not explained by anthropometric, lifestyle or genetic risk factors: the EPIC-InterAct study. Diabetologia. 2013;56:60–69. doi: 10.1007/s00125-012-2715-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eriksson J, Franssila-Kallunki A, Ekstrand A, et al. Early metabolic defects in persons at increased risk for non-insulin-dependent diabetes mellitus. The New England journal of medicine. 1989;321:337–343. doi: 10.1056/NEJM198908103210601. [DOI] [PubMed] [Google Scholar]
- 16.Gulli G, Ferrannini E, Stern M, Haffner S, DeFronzo RA. The metabolic profile of NIDDM is fully established in glucose-tolerant offspring of two Mexican-American NIDDM parents. Diabetes. 1992;41:1575–1586. doi: 10.2337/diab.41.12.1575. [DOI] [PubMed] [Google Scholar]
- 17.Haffner SM, Miettinen H, Gaskill SP, Stern MP. Decreased insulin action and insulin secretion predict the development of impaired glucose tolerance. Diabetologia. 1996;39:1201–1207. doi: 10.1007/BF02658507. [DOI] [PubMed] [Google Scholar]
- 18.Leslie RD, Volkmann HP, Poncher M, Hanning I, Orskov H, Alberti KG. Metabolic abnormalities in children of non-insulin dependent diabetics. British medical journal. 1986;293:840–842. doi: 10.1136/bmj.293.6551.840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meigs JB, Cupples LA, Wilson PW. Parental transmission of type 2 diabetes: the Framingham Offspring Study. Diabetes. 2000;49:2201–2207. doi: 10.2337/diabetes.49.12.2201. [DOI] [PubMed] [Google Scholar]
- 20.Srinivasan SR, Frontini MG, Berenson GS, Bogalusa Heart S. Longitudinal changes in risk variables of insulin resistance syndrome from childhood to young adulthood in offspring of parents with type 2 diabetes: the Bogalusa Heart Study. Metabolism: clinical and experimental. 2003;52:443–450. doi: 10.1053/meta.2003.50065. discussion 451-443. [DOI] [PubMed] [Google Scholar]
- 21.Warram JH, Martin BC, Krolewski AS, Soeldner JS, Kahn CR. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann Intern Med. 1990;113:909–915. doi: 10.7326/0003-4819-113-12-909. [DOI] [PubMed] [Google Scholar]
- 22.Warram JH, Martin BC, Soeldner JS, Krolewski AS. Study of glucose removal rate and first phase insulin secretion in the offspring of two parents with non-insulin-dependent diabetes. Advances in experimental medicine and biology. 1988;246:175–179. doi: 10.1007/978-1-4684-5616-5_21. [DOI] [PubMed] [Google Scholar]
- 23.Hanson RL, Pratley RE, Bogardus C, et al. Evaluation of simple indices of insulin sensitivity and insulin secretion for use in epidemiologic studies. American journal of epidemiology. 2000;151:190–198. doi: 10.1093/oxfordjournals.aje.a010187. [DOI] [PubMed] [Google Scholar]
- 24.Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–1645. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 25.Rimm EB, Giovannucci EL, Stampfer MJ, Colditz GA, Litin LB, Willett WC. Reproducibility and validity of an expanded self-administered semiquantitative food frequency questionnaire among male health professionals. Am J Epidemiol. 1992;135:1114–1126. doi: 10.1093/oxfordjournals.aje.a116211. discussion 1127-1136. [DOI] [PubMed] [Google Scholar]
- 26.McKeown NM, Meigs JB, Liu S, et al. Dietary carbohydrates and cardiovascular disease risk factors in the Framingham offspring cohort. Journal of the American College of Nutrition. 2009;28:150–158. doi: 10.1080/07315724.2009.10719766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molenaar EA, Massaro JM, Jacques PF, et al. Association of lifestyle factors with abdominal subcutaneous and visceral adiposity: the Framingham Heart Study. Diabetes Care. 2009;32:505–510. doi: 10.2337/dc08-1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nature genetics. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White IR, Royston P, Wood AM. Multiple imputation using chained equations: Issues and guidance for practice. Statistics in medicine. 2011;30:377–399. doi: 10.1002/sim.4067. [DOI] [PubMed] [Google Scholar]
- 30.Vanderweele TJ, Vansteelandt S. Odds ratios for mediation analysis for a dichotomous outcome. Am J Epidemiol. 2010;172:1339–1348. doi: 10.1093/aje/kwq332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valeri L, Vanderweele TJ. Mediation analysis allowing for exposure-mediator interactions and causal interpretation: theoretical assumptions and implementation with SAS and SPSS macros. Psychological methods. 2013;18:137–150. doi: 10.1037/a0031034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kong A, Steinthorsdottir V, Masson G, et al. Parental origin of sequence variants associated with complex diseases. Nature. 2009;462:868–874. doi: 10.1038/nature08625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manson JE, Rimm EB, Stampfer MJ, et al. Physical activity and incidence of non-insulin-dependent diabetes mellitus in women. Lancet. 1991;338:774–778. doi: 10.1016/0140-6736(91)90664-b. [DOI] [PubMed] [Google Scholar]