

SUMMARY
T cell metabolism plays a central role to support and shape immune responses and may play a key role in anti-tumor immunity. T cell metabolism is normally held under tight regulation in an immune response of glycolysis to promote effector T cell expansion and function. However, tumors may deplete nutrients, generate toxic products, or stimulate conserved negative feedback mechanisms, such as through PD-1, to impair effector T cell nutrient uptake and metabolic fitness. In addition, regulatory T cells are favored in low glucose conditions and may inhibit anti-tumor immune responses. Here we review how the tumor microenvironment modifies metabolic and functional pathways in T cells and how these changes may uncover new targets and challenges for cancer immunotherapy and treatment.
Keywords: T cell metabolism, glycolysis, IDO, PD-1, CTLA4, tumor microenvironment, anti-tumor immunity, checkpoint blockade
Introduction
The ability of the adaptive immune system to eliminate invading pathogens has long suggested that T cells may have the capacity to respond to the neoantigens or inflammatory and damage signals to eliminate tumors[1]. However, tumor microenvironments can pose particular challenges for anti-tumor T cell responses. It was first recognized in the early 1920s that tumors utilize glucose at a high rate and produce lactate in a process termed the Warburg effect[2]. This metabolic program differs from that used by most normal tissues in that nutrients such as glucose are not readily oxidized in mitochondria for maximal ATP generation, but are instead conserved for biosynthesis of nucleic acids, lipids, and amino acids to support cell growth[3]. While a key benefit of this metabolic program is to protect tumor cells in hypoxic regions, oncogenes that drive this mode of metabolism do so even in the presence of oxygen in a metabolic program termed aerobic glycolysis. Cancer cells utilize aerobic glycolysis to differing extents, and high levels of glycolytic activity coupled with poor angiogenesis can lead to near glucose depletion and accumulation of waste products, such as up to 20 or more millimolar levels of lactate in both vital and peri-necrotic tumor zones[4]. T cells infiltrating the tumor microenvironment thus face significant metabolic challenges to mount and sustain an anti-tumor response.
Despite these barriers, approaches to interfere with inhibitory immunomodulatory signals in immune checkpoint therapies have shown that tumor infiltrating CD4 and CD8 T cells can play key roles to control or mediate anti-tumor immunity[5]. Specifically, inhibition of Cytotoxic T Lymphocyte Associated Protein 4 (CTLA4) and Programmed Cell Death 1 (PD-1) interactions with ligands have enhanced anti-tumor responses that can lead to curative therapy in some cancers, most successfully in melanoma[5]. These findings suggest that, at least in some cases, T cell mediated anti-tumor responses have initiated even in potentially nutrient limited tumor microenvironment conditions, but are held in check by immunosuppressive mechanisms.
It is now widely appreciated that T cell differentiation and effector function are coupled to metabolic reprogramming processes, and that interfering with these reprogramming pathways can impair T cell responses[6]. This has suggested that tumor-mediated immune suppression may be associated with alterations to the metabolic pathways that would normally support T cell effector function. Here we review evidence in support of this notion, and propose that T cell “metabolic fitness” is central to effective anti-tumor immunity, and is modulated by both the tumor nutrient microenvironment and by immune checkpoints.
Metabolic reprogramming in T cell differentiation and effector function
To exert function, activated T cells must undergo metabolic reprogramming to shift from an energy-oriented oxidative metabolism that supports quiescence and immune surveillance, to primarily anabolic and biosynthetic to support rapid growth. After pathogen clearance, T cells are eliminated in clonal contraction or return to a primarily catabolic metabolism as long-lived memory cells[6]. Naïve T cells are small and quiescent cells that require relatively small amounts of glucose, amino acids and fatty acids to maintain basic energetic and minimal replacement biosynthesis demands. Encounter with cognate antigen in the context of appropriate costimulation triggers T cell activation and differentiation into effector T (Teff), which reduce lipid oxidation and instead rely on high intake of glucose and amino acids[7] to support proliferation and effector functions such as cytotoxicity and cytokine production. The pathways that control these metabolic transitions are now beginning to be unraveled. The T cell receptor (TCR) with CD28 costimulation activate the Phosphatidylinositide 3-kinase (PI3K)/Akt/mTOR, cMyc, and Hypoxia Inducible Factor (HIF1α) signaling pathways which promote glycolytic gene expression and post-translational regulation essential to induce aerobic glycolysis and amino acid metabolism of Teff[8-11] (Figure 1). In particular, activated effector T cells sharply increase glycolysis and glutamine metabolism to support anabolic pathways of nucleotide and lipid synthesis essential for cell growth[6, 7, 11, 12]. Inhibition of glucose or glutamine metabolic enzymes can reduce effector T activation and function, as glucose deprivation or treatment with inhibitors of glycolysis can impair Teff proliferation and cytokine secretion [11, 13-15]. Indeed, inhibitors of nucleotide synthesis are potent immunosuppressive agents that are standard of care for a variety of rheumatologic diseases[16]
Figure 1. Metabolic regulation and feedback in T cell immunity.
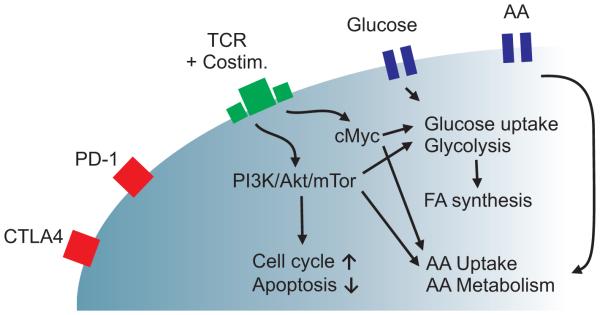
T cell activation and signaling through T cell receptor (TCR) activates PI3K/Akt/mTOR and cMyc pathways, leading to increased glycolysis and metabolism in effector T cells (Teff). AA, amino acids; FA, fatty acids; Costim, costimulation.
The inability to gain access to appropriate nutrients, thus, poses a significant barrier to effector T cell function. To avoid this potential limitation T cell activation leads to upregulation of both glucose and amino acid transporters[17-19], and regulation of nutrient uptake is now appreciated to be a critical component of T cell activation and function. Indeed, genetic disruption of amino acid uptake through the transporters SLC7a5 or SLC1a5 impairs growth and proliferation of effector T cells[18, 19]. Likewise, T cells express a panel of glucose transporters and genetic deletion of the glucose transporter Glut1 (SLC2a1) sharply reduced T cell glycolysis and proliferation in vivo, preventing CD4 T cells from inducing multiple inflammatory diseases, including mouse models of graft versus host disease after allogeneic bone marrow transplant and colitis [15]. This was also true in peripheral human CD4 T cells, where reduction in Glut1 expression by siRNA impaired T cell growth and proliferation after activation[15]. Further supporting the notion that glucose uptake modulates T cell physiology after activation, transgenic overexpression of Glut1 to increase glucose uptake in T cells was sufficient to augment T cell stimulation by enhancing proliferation and inflammatory cytokine production upon submitogenic stimulation[17]. Ultimately, this increased T cell activation lead to lymphadenopathy and a Systemic Lupus Erythematosus-like autoimmunity as mice aged[17, 20].
After activation T cells can differentiate into functional Teff and regulatory T cell (Treg) subsets that may play critical and differential roles to control tumors. These subsets have now been shown to utilize and require distinct metabolic programs[6, 21]. While Teff are key drivers of anti-tumor immunity, Treg can inhibit Teff to suppress immunity[22] and are generally associated with poor prognosis in many cancers[23-26]. An exception is colorectal carcinoma, where Treg have been reported in some cases to be associated with better patient outcomes[27]. This discrepancy may be due to a role for inflammation to promote the development and progression of colorectal carcinoma that Treg could suppress[28]. Metabolically, while activated CD8 T cells and CD4 Teff cells require high levels of Glut1 and glucose metabolism, Treg cells express low levels of Glut1 and can be Glut1-independent and not rely on high rates of glucose[10, 15, 20]. Treg are instead primarily oxidative and can efficiently metabolize pyruvate through the tricarboxylic acid cycle (TCA) or utilize lipid beta-oxidation as a primary metabolic mechanism[10, 15, 20]. Also unlike Teff, Treg do not require the mTOR kinase and are preferentially generated when T cells are deficient in mTOR kinase or if mTOR complex 1 (mTORC1) is inhibited by rapamycin[29]. Opposing mTORC1 is the AMP-activated Protein Kinase (AMPK), which is activated in conditions where nutrients are limiting and promotes oxidative metabolism[30]. AMPK can be highly phosphorylated and activated in Treg[20] and activation of AMPK by treatment with metformin can decrease Teff and increase Treg frequency in vivo[20, 31]. Conditions in the tumor microenvironment that restrict Teff nutrient uptake and metabolism may, therefore, alter mTORC1 and AMPK signaling to impair Teff or induce Treg that may suppress anti-tumor immunity[23-25]. Conversely, interventions that improve the ability of Teff to compete with tumor cells for uptake essential nutrients may increase the metabolic fitness of T cells to improve the functional capacity of Teff to mediate an anti-tumor immune response.
Glucose metabolism and nutrient microenvironment limit anti-tumor immunity
The abundance of glucose is critical for activated T cells and the potential depletion of glucose in tumors[4] may promote competition for nutrients and suppress Teff cell function. Metabolic stress with insufficient glucose can greatly alter T cell signaling and gene expression. Culture of T cells in glucose-limiting conditions has been shown to inhibit signaling through the mTORC1 pathway to reduce phospho-S6 and lower expression of up to a tenth of antigen-induced genes to impair cell adhesion, cell cycle progression and proliferation of CD8 T cells, including IFNγ, GM-CSF and granzyme B[6, 13]. IFNγ translation was recently shown to be regulated by sustained glycolysis, through glyceraldehyde 3-phosphate levels and the association of the glycolytic enzyme Glyceraldehyde 3-Phosphate Dehydrogenase (GAPDH) with the 3’UTR of the ifnγ mRNA[32]. In addition, inhibition of glycolysis or use of primarily oxidative fuels, such as galactose, can also lead to increased expression of immune regulatory receptors, such as PD-1[32]. PD-1 is associated with T cell exhaustion and non-responsiveness through inhibition of T cell receptor signaling and CD28-mediated costimulation (see Pauken and Wherry, this issue). Glucose limitation has also been demonstrated to lead activated T cells to enter a state of T cell functional inactivation or anergy[33].
Decreased glucose availability leads to changes in T cell effector function in part through modulation of metabolically-sensitive signaling pathways (Figure 2). An increase in AMP/ATP ratio activates AMPK to decrease anabolic pathways and favor oxidative catabolic pathways[30, 34, 35]. AMPK can directly impact the balance of Teff and Treg as it inhibits mTORC1 that is essential for Teff function[29] and instead promotes Treg[20, 31]. This regulation may occur in part through decreased Treg lineage stability, which was recently shown to be impaired in Treg with genetic loss of PTEN and constitutively active PI3K signaling in vivo[36, 37]. The Sirtuin histone deacetylases (Sirt) are regulated by NAD+ levels and have been shown to control T cell function through deacetylation and protein stability of the Treg transcription factor, FoxP3[38-40]. Sirt1, in particular, also plays a key role in Treg lineage stability and function. Sirt1 deletion or inhibition was reported to enhance FoxP3 acetylation and protein stabilization to increase Treg suppressive function in vitro and in vivo[39]. Thus, decreased nutrient availability within tumors may lead to decreased Teff function and to both improved or impaired Treg function depending on the balance of signals and context (Figure 2). In addition, metabolites themselves can act as signaling molecules. Decreased flux through in the Tricarboxylic Acid (TCA) cycle in the mitochondria may lower succinate levels. Succinate was recently show to act as a danger signal and may play a key role to induce inflammation through stabilization of HIF1α and inflammatory cytokine transcription[41] or via the succinate receptor GPR91 to synergize with Toll-like Receptor ligands[42]. Mitochondrial reactive oxygen species (ROS) are also essential for T cell signaling and activation, and impaired mitochondrial ROS production by disruption of mitochondrial electron transport blocked T cell activation[43]. Ultimately, insufficient glucose can also lead to Teff apoptosis induction via induction and activation of pro-apoptotic Bcl-2 family proteins Noxa and Bax and instability of the anti-apoptotic Bcl-2 family protein Mcl1[44, 45] (Figure 2).
Figure 2. Regulation of tumor associated T cells by changes in tumor microenvironment.

Proliferating tumor cells deplete critical nutrients such as tryptophan (Trp) or glucose. The increased metabolic activity of tumor cells results in high concentration of metabolites such as lactate or kynurenines (Kyn), which can activate the Aryl Hydrocarbon Receptor (AHR). Both nutrient depletion and metabolite production negatively impact T cell effector function, metabolism, and survival by modulation of the PI3K/Akt/mTOR pathway, AMPK, and SirT1. In addition, immunosuppressive regulatory T cells (Treg) are favored in tumor microenvironment. Kyn, kynurenines; Trp, tryptophan; AHR; Aryl Hydrocarbon Receptor.
Oxygen is critical for mitochondrial oxidative phosphorylation and, like glucose, can be significantly depleted in tumors. Hypoxia due to poor vascularization is a characteristic of many solid tumors, and gene signatures induced by HIF1α that indicate adaptation to hypoxia are correlated with poor prognosis[46]. Hypoxia has also been described in non-malignant diseases, such as in the gut in colitis[47] and in diseased joints in rheumatic arthritis[48]. Moreover, the thymus and secondary lymphoid organs can be hypoxic under physiological conditions[49]. The effect of hypoxia on T cells, however, appears context specific. Th17 CD4 T cells have been reported to require HIF1α as a co-factor for the Th17 transcription factor RORγT, whereas other CD4 subtypes were reported to be largely HIF1α independent or inhibited by HIF1α[9, 10]. Conversely, in vivo induction of T cell activation markers after injection of concanavolin A or anti-CD3 was shown to be greatest in the most highly oxygenated tissues[49], suggesting that depletion of oxygen in tumors may broadly impair Teff cell function. Hypoxia has also been reported to impact Treg by enhancing FoxP3 expression and the generation of induced Treg cells, and HIF1α-deficient Treg cells display impaired in vivo suppressive function in T cell-mediated colitis[50]. Importantly, hypoxia may protect tumor cells from anti-tumor immunity and was shown to promote HIF1α-dependent transcriptional upregulation of PD-1 ligand 1 (PD-L1) on cancer cells that may inhibit PD-1 expressing T cells[51]. Indeed, tumor cells showed greater resistance to T cell-mediated killing under hypoxic conditions[51].
In addition to decreased levels of essential nutrients, tumor cells utilizing aerobic glycolysis produce high levels of lactate through lactate dehydrogenase-mediated reduction of pyruvate. Lactic acid can suppress the proliferation and cytokine production of human cytotoxic T lymphocytes (CTLs) and reduce cytotoxic activity[52, 53]. Moreover, blockade of the lactate transporter MCT-1 leads to accumulation of intracellular lactate that can lower glycolytic flux [52, 54]. In part, these effects are due to inhibition of cell signaling and transcription. Lactic acidosis selectively inhibited JNK and p38-mediated stimulation of IFNγ production without affecting MEK1 and ERK that promote cytokine release and granule exocytosis[53]. Lactate also suppressed the PI3K/Akt/mTOR pathway in a feedback that inhibited glycolysis[8, 55, 56] (Figure 2). An additional consequence of lactate secretion is microenvironmental acidification. Although few studies have addressed the role of low pH on immunity, acidification has been reported to regulate macrophage polarization and induce Arginase 1, which can then deplete extracellular arginine levels to inhibit T cell amino acid uptake necessary for efficient proliferation and activation[57, 58]. Moreover, proton-pump inhibitors may reverse the dysfunction of tumor infiltrating lymphocytes and were shown to increase the therapeutic efficacy of both active and adoptive immunotherapy[59]. Despite these negative impacts, lactate itself can be consumed as a metabolic fuel through conversion back to pyruvate and oxidation to provide a fuel in times of nutrient depletion. While cancer cells can utilize lactate[60], the extent to which T cells oxidize lactate and this may impact Teff or Treg is not clear. Given the preference for oxidative metabolism for Treg[7, 10, 20], it may be anticipated that the availability of excess lactate as a fuel would preferentially support Treg.
Tryptophan metabolism in T cell mediated inflammation and cancer immune escape
Similar to glucose, amino acids are critical nutrients for activated Teff [18, 19]. Likewise, other immune cells, such as macrophages, also require essential amino acids to mediate inflammation[61]. As tumor cells consume or release enzymes that degrade amino acids, the accessibility of amino acids to support T cell and macrophage effector functions can become limiting and waste products can accumulate. In addition to arginine depletion[62], tryptophan has gained much attention as a potentially limiting amino acid in T cell activation and effector function[63] and is regulated by Indoleamine 2,3 dioxygenase (IDO) and tryptophan 2,3 dioxygenase. IDO metabolizes tryptophan to kynurenine in the kynurenine pathway and can significantly decrease tryptophan concentration to render this amino acid potentially limiting for T cells. In addition, kynurenine itself is a potent and active suppressor of T cell activation (Figure 2). Mechanistically, kynurenine has been shown to be an endogenous ligand to activate the Aryl hydrocarbon Receptor (AhR)[64], which can induce Treg in the gut and in neuroinflammation [64-67] and may also do so in tumors[68]. In addition to roles for IDO to modify allogeneic fetal rejection[63], autoimmune disorders such as diabetes[69], inflammatory bowel disease[70, 71], asthma[72], arthritis[73] and autoimmune encephalitis[74], it is now clear that many cancers can express high amounts of IDO[65]. Tumor IDO levels have been correlated with poor prognosis[75] and clinical trials are currently testing IDO inhibitors to promote anti-tumor immunity[65]. Decreased levels of essential amino acids and accumulation of byproducts of amino acid metabolism thus act in concert with microenvironmental changes induced by tumor aerobic glycolysis and hypoxia to form a barrier to anti-tumor immunity.
Immunomodulatory receptors and checkpoints on T cell metabolic fitness
Just as it is critical for T cells to have access to appropriate nutrients including glucose and essential amino acids, it is also crucial that T cells have the capacity to internalize and utilize those available nutrients. Indeed, nutrient uptake and flux through metabolic pathways are highly regulated and can be limiting in T cell activation regardless of nutrient availability in the surrounding microenvironment[15, 18, 19]. T cell activation leads to upregulation of cMyc and induction of HIF1α, which promote expression of glycolytic and anabolic metabolic genes including those that regulate nutrient transport, such as Glut1, Slc7a5, and Slc1a5[9-11, 18, 19, 76]. cMyc plays a broad role early to support glycolytic and glutamine metabolism in T cell activation while HIF1α appears to play a more selective role in inflammatory Th17 CD4 T cell subsets[9, 10] and cytolytic CD8 T cells[77]. In addition, costimulation through CD28 plays a key role to stimulate the PI3K/Akt/mTORC1 pathway, which plays key transcriptional and post-transcriptional roles to promote anabolic gene expression and intracellular trafficking of nutrient transporters[15, 55, 76, 78]. Inhibition of the signaling and regulatory pathways that control nutrient transporters can sharply reduce T cell metabolic fitness and ability to utilize or compete with tumor cells for available nutrients, thus impairing effector T cell function while favoring Treg[10, 20].
Inhibitory signals play key roles to control and moderate T cell responses in both normal and anti-tumor immunity[5] and may do so in part by limiting the nutrient uptake and metabolic fitness of effector T cells. CTLA4 is upregulated in T cell activation and both opposes CD28-mediated costimulation of effector T cells and promotes the function of Treg[79]. While the context of CTLA4 signaling appears important, it was proposed that CTLA4 can inhibit CD28-mediated activation of Akt through sequestration of CD28 ligands and recruitment of the phosphatase SHP[79, 80] to reduce the ability of Akt to stimulate Glut1 expression, glucose uptake, and aerobic glycolysis in activated T cells[17, 81, 82]. Similar to CTLA4, PD-1 has been shown to restrain effector T cell activation and function (see Pauken and Wherry, this issue).
PD-1 is induced in T cell activation and is a marker and mediator of T cell exhaustion, such occurs as in chronic viral infection[83, 84]. Upon ligation with PD-1 ligand (PD-L1) or PD-1 ligand 2 (PD-L2), PD-1 induces changes leading to T cell inhibition that modify the sensitivity of T cells to antigenic stimulation[85]. PD-1 signaling is associated with reduced cMyc expression[84, 86] and inhibits activity of the PI3K/Akt/mTOR pathway[80, 87], possibly by regulation of PTEN phosphorylation and increased phosphatase activity[87]. Through reduced cMyc expression and PI3K/Akt/mTORC1 signaling, PD-1 can lower the capacity of T cells to express Glut1, uptake glucose, and perform glycolysis[80, 88] necessary for effector function[11, 15] (Figure 3). In particular, treatment of T cells with agonist anti-PD-1 antibodies prevented T cell receptor and CD28-mediated costimulation from activating Akt and increasing T cell glucose uptake or glycolysis[80]. Further, T cells in allogeneic PD-L1−/− bone marrow transplant recipients had elevated levels of Glut1 and lactate production, suggesting a normal in vivo role for PD-1 signaling to restrain T cell glucose metabolism[88]. CTLA4 and PD-1 can also both promote Treg[79, 89, 90], which are Glut1-independent and rely primarily on oxidative metabolism[7, 10, 15, 20]. The extent to which impaired glucose metabolism contributes to PD-1 regulation of Treg induction, however, is unclear.
Figure 3. Immune checkpoint blockade may regulate metabolic reprogramming of T cells.
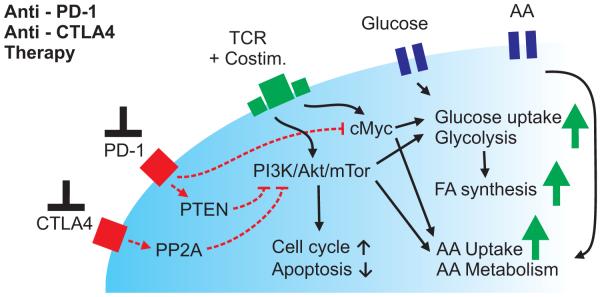
Blockade of PD-1 and CTLA4 may increase the metabolic fitness of Teff by enhancing the activity of PI3K/Akt/mTor and increasing cMyc expression that then stimulate aerobic glycolysis and anabolic metabolism. AA, amino acids; FA, fatty acids; Costim, costimulation.
PD-L1 is expressed on a wide array of non-hematopoietic tissues[91] and can be induced by IFNγ[92] or hypoxia[51]. Importantly, many cancers have been now found to express PD-L1 or PD-L2 and are associated with intratumoral PD-1+ T cells that appear exhausted[5]. Indeed, blockade of PD-1 interaction with ligand has emerged as an exciting new approach to reactivate T cells and promote anti-tumor immunity[5, 93-96]. While this approach has been remarkably successful in some cases, a significant portion of patients do not respond and some cancer types are less susceptible than others[5]. In addition to differential expression of PD-1 ligands in tumors, mechanisms by which PD-1 inhibits T cells may provide new approaches to identify patients that may respond or to enhance the efficacy of PD-1 checkpoint blockade therapy. One mechanism by which CTLA4 or PD-1 blockade may act to promote anti-tumor immunity is to relieve inhibition of T cell metabolism and thus improve T cell nutrient uptake and metabolic fitness. By blocking CTLA4 or PD-1 and allowing increased cMyc expression and PI3K/Akt/mTORC1 activity, T cells would be expected to elevate expression of Glut1 and glycolytic proteins. This improved metabolic fitness would augment the ability of T cells to compete with tumor cells for limiting nutrients that are essential for effector function and may counteract the negative effects of tumor cell waste products (Figure 4). It would be essential, however, that some nutrients are available for even T cells with improved metabolic fitness to access. Indeed, some tumors can deplete glucose to very low levels[4] that may impair an anti-tumor T cell response regardless of immunomodulatory signals. It remains to be determined, however, to what extent improved metabolic fitness and nutrient availability contribute to the patient responses and the sensitivity of specific tumor types to CTLA4 and PD-1 blockade.
Figure 4. T cell metabolic fitness and anti-tumor immunity.

Effector T cells require high levels of glucose and amino acid uptake and are inhibited by lactate and other metabolic waste products. Solid tumor that consume nutrients and accumulate waste products may limit the capacity of T cells to mount an anti-tumor immune response. Blockade of immune checkpoint modulatory signals, such as inhibition of PD-1 or CTLA4, can enhance effector T cell signaling and metabolism to improve T cell metabolic fitness and capacity to compete for nutrients in increasingly challenging metabolic environments.
Concluding remarks
The recent excitement of advances in promoting anti-tumor immunity by checkpoint blockade raises significant questions about how tumors and the tumor microenvironment inhibit T cells and how this can be overcome. Understanding the signaling and physiological limitations to T cell and macrophage functions in a tumor may reveal improved approaches and better identify patients that may respond to these treatments. This is particularly important given the high rate of patients that fail to respond to current cancer immune checkpoint blockade therapy[5]. Regulation of cell metabolism is now known to play a critical role in T cell function and fate[6]. Given the metabolic changes of decreased nutrients and increased waste products within tumors and the ability of tumors to express immunomodulatory ligands that impair T cell metabolic fitness and capacity to uptake and utilize nutrients, it is highly likely that restrictions on T cell metabolism play critical roles in the anti-tumor response. An important goal for future studies is to better define the metabolic programs of T cells within tumors and establish how these pathways reflect the potential success in patients of checkpoint blockade therapies. Moreover, data on metabolism of other regulatory cells such as myeloid-derived suppressor cells or mesenchymal stem cells is scarce. Metabolic limitations to T cell physiology may also play roles in chimeric antigen receptor (CAR) T cell therapies in which T cells are genetically engineered to express tumor-directed antigen receptors[97]. The increased number of tumor-reactive T cells in CAR therapy could be offset by limited nutrients and conditions that restrict T cell metabolic fitness. In addition to T cells, it is also important in assessing the tumor microenvironment to consider macrophages. Indeed, activated macrophage subsets are metabolically similar to T cells[98]. Levels of glucose uptake can influence macrophage inflammatory function, as genetically increased Glut1 expression stimulated greater cytokine production and stimulated classically activated inflammatory M1 macrophages[99]. Thus, approaches to modify metabolism may have dual effects on T cells and macrophage populations. These conditions are also likely not limited to solid tumors, as leukemic cells can often express immunomodulatory proteins and are associated with an exhausted T cell phenotype not dissimilar to that which occurs in chronic viral infection or in solid tumors[100]. In this case, decreased T cell metabolic fitness may impair Graft-vs-Leukemia responses[88]. Together, evidence now shows that physiologic requirements of T cells and T cell metabolic fitness are crucial to understand Teff and Treg function and that immune metabolism and cancer metabolism are inexorably linked in efforts to optimize and maximize anti-tumor immunity.
HIGHLIGHTS.
Effector T cells and tumors utilize similar metabolic programs
Nutrient depletion and accumulation of waste products in tumors can limit T cells
Immunomodulatory signals can reduce T cell metabolic fitness
Blockade of PD-1 and CTLA4 can enhance T cell metabolism and function
ACKNOWLEDGEMENTS
This work was supported by a Clinical and Laboratory Integration Program grant from the Cancer Research Institute (J.C.R.) and by the German Research Foundation (Deutsche Forschungsgemeinschaft; P.J.S.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Melero I, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nature reviews. Clinical oncology. 2014;11:509–524. doi: 10.1038/nrclinonc.2014.111. [DOI] [PubMed] [Google Scholar]
- 2.Koppenol WH, et al. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 3.Vander Heiden MG, et al. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schroeder T, et al. Spatial heterogeneity and oxygen dependence of glucose consumption in R3230Ac and fibrosarcomas of the Fischer 344 rat. Cancer Res. 2005;65:5163–5171. doi: 10.1158/0008-5472.CAN-04-3900. [DOI] [PubMed] [Google Scholar]
- 5.Page DB, et al. Immune modulation in cancer with antibodies. Annu Rev Med. 2014;65:185–202. doi: 10.1146/annurev-med-092012-112807. [DOI] [PubMed] [Google Scholar]
- 6.MacIver NJ, et al. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerriets VA, et al. Metabolic Programming and PDHK1 Control CD4 T-Cell Subsets and Inflammation. J Clin Invest Accepted. 2014 doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev. 2012;249:43–58. doi: 10.1111/j.1600-065X.2012.01152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dang EV, et al. Control of T(H)17/T(reg) Balance by Hypoxia-Inducible Factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi LZ, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang RN, et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang R, Green DR. Metabolic reprogramming and metabolic dependency in T cells. Immunol Rev. 2012;249:14–26. doi: 10.1111/j.1600-065X.2012.01155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cham CM, et al. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008;38:2438–2450. doi: 10.1002/eji.200838289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cham CM, Gajewski TF. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. J Immunol. 2005;174:4670–4677. doi: 10.4049/jimmunol.174.8.4670. [DOI] [PubMed] [Google Scholar]
- 15.Macintyre AN, et al. The Glucose Transporter Glut1 is Selectively Essential for CD4 T Cell Activation and Effector Function. Cell Metab. 2014;20:61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Floreth T, et al. Conventional and novel approaches to immunosuppression. Clin Chest Med. 2011;32:265–277. doi: 10.1016/j.ccm.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 17.Jacobs SR, et al. Glucose Uptake Is Limiting in T Cell Activation and Requires CD28-Mediated Akt-Dependent and Independent Pathways. J Immunol. 2008;180:4476–4486. doi: 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakaya M, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40:692–705. doi: 10.1016/j.immuni.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinclair LV, et al. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. 2013;14:500–508. doi: 10.1038/ni.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Michalek RD, et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pearce EL, et al. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohkura N, et al. Development and maintenance of regulatory T cells. Immunity. 2013;38:414–423. doi: 10.1016/j.immuni.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Bauer CA, et al. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J Clin Invest. 2014;124:2425–2440. doi: 10.1172/JCI66375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jie HB, et al. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer. 2013;109:2629–2635. doi: 10.1038/bjc.2013.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waight JD, et al. Cutting Edge: Epigenetic Regulation of Foxp3 Defines a Stable Population of CD4+ Regulatory T Cells in Tumors from Mice and Humans. J Immunol. 2015;194:878–882. doi: 10.4049/jimmunol.1402725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. 2014;27:1–7. doi: 10.1016/j.coi.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 27.Zhuo C, et al. FOXP3+ Tregs: heterogeneous phenotypes and conflicting impacts on survival outcomes in patients with colorectal cancer. Immunol Res. 2015 doi: 10.1007/s12026-014-8616-y. [DOI] [PubMed] [Google Scholar]
- 28.Grivennikov SI, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254–258. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delgoffe GM, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Son HJ, et al. Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediators of inflammation. 2014;2014:973986. doi: 10.1155/2014/973986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang CH, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng Y, et al. Anergic T cells are metabolically anergic. J Immunol. 2009;183:6095–6101. doi: 10.4049/jimmunol.0803510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 35.Blagih J, et al. The Energy Sensor AMPK Regulates T Cell Metabolic Adaptation and Effector Responses In Vivo. Immunity. 2015;42:41–54. doi: 10.1016/j.immuni.2014.12.030. [DOI] [PubMed] [Google Scholar]
- 36.Huynh A, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol. 2015;16:188–196. doi: 10.1038/ni.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shrestha S, et al. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol. 2015;16:178–187. doi: 10.1038/ni.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akimova T, et al. Targeting sirtuin-1 alleviates experimental autoimmune colitis by induction of Foxp3+ T-regulatory cells. Mucosal immunology. 2014;7:1209–1220. doi: 10.1038/mi.2014.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beier UH, et al. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Science signaling. 2012;5:ra45. doi: 10.1126/scisignal.2002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Loosdregt J, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood. 2010;115:965–974. doi: 10.1182/blood-2009-02-207118. [DOI] [PubMed] [Google Scholar]
- 41.Tannahill GM, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rubic T, et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol. 2008;9:1261–1269. doi: 10.1038/ni.1657. [DOI] [PubMed] [Google Scholar]
- 43.Sena LA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38:225–236. doi: 10.1016/j.immuni.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alves NL, et al. The Noxa/Mcl-1 Axis Regulates Susceptibility to Apoptosis under Glucose Limitation in Dividing T Cells. Immunity. 2006;24:703–716. doi: 10.1016/j.immuni.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 45.Coloff JL, et al. Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res. 2011;71:5204–5213. doi: 10.1158/0008-5472.CAN-10-4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glover LE, et al. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proc Natl Acad Sci U S A. 2013;110:19820–19825. doi: 10.1073/pnas.1302840110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X, et al. Hypoxia-induced transcription factor 1alpha: a potent driving force behind rheumatoid arthritis. Clin Exp Rheumatol. 2014;32:760. [PubMed] [Google Scholar]
- 49.Ohta A, et al. In vivo T cell activation in lymphoid tissues is inhibited in the oxygen-poor microenvironment. Frontiers in immunology. 2011;2:27. doi: 10.3389/fimmu.2011.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clambey ET, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A. 2012;109:E2784–2793. doi: 10.1073/pnas.1202366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barsoum IB, et al. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74:665–674. doi: 10.1158/0008-5472.CAN-13-0992. [DOI] [PubMed] [Google Scholar]
- 52.Fischer K, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 53.Mendler AN, et al. Tumor lactic acidosis suppresses CTL function by inhibition of p38 and JNK/c-Jun activation. Int J Cancer. 2012;131:633–640. doi: 10.1002/ijc.26410. [DOI] [PubMed] [Google Scholar]
- 54.Pahlman C, et al. Immunosuppressive properties of a series of novel inhibitors of the monocarboxylate transporter MCT-1. Transplant international : official journal of the European Society for Organ Transplantation. 2013;26:22–29. doi: 10.1111/j.1432-2277.2012.01579.x. [DOI] [PubMed] [Google Scholar]
- 55.Duvel K, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen JL, et al. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet. 2008;4:e1000293. doi: 10.1371/journal.pgen.1000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Colegio OR, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ohashi T, et al. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int J Cancer. 2013;133:1107–1118. doi: 10.1002/ijc.28114. [DOI] [PubMed] [Google Scholar]
- 59.Calcinotto A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012;72:2746–2756. doi: 10.1158/0008-5472.CAN-11-1272. [DOI] [PubMed] [Google Scholar]
- 60.Kennedy KM, et al. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PLoS One. 2013;8:e75154. doi: 10.1371/journal.pone.0075154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsumura H, et al. The role of CD98hc in mouse macrophage functions. Cellular immunology. 2012;276:128–134. doi: 10.1016/j.cellimm.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 62.Raber P, et al. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunol Invest. 2012;41:614–634. doi: 10.3109/08820139.2012.680634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Munn DH, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 64.Opitz CA, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 65.Platten M, et al. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Frontiers in immunology. 2014;5:673. doi: 10.3389/fimmu.2014.00673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mezrich JD, et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–3198. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Quintana FJ, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107:20768–20773. doi: 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murray IA, et al. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer. 2014;14:801–814. doi: 10.1038/nrc3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fallarino F, et al. IDO mediates TLR9-driven protection from experimental autoimmune diabetes. J Immunol. 2009;183:6303–6312. doi: 10.4049/jimmunol.0901577. [DOI] [PubMed] [Google Scholar]
- 70.Furuzawa-Carballeda J, et al. Indoleamine 2,3-dioxygenase: expressing cells in inflammatory bowel disease-a cross-sectional study. Clinical & developmental immunology. 2013;2013:278035. doi: 10.1155/2013/278035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gurtner GJ, et al. Inhibition of indoleamine 2,3-dioxygenase augments trinitrobenzene sulfonic acid colitis in mice. Gastroenterology. 2003;125:1762–1773. doi: 10.1053/j.gastro.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 72.Hayashi T, et al. Inhibition of experimental asthma by indoleamine 2,3-dioxygenase. J Clin Invest. 2004;114:270–279. doi: 10.1172/JCI21275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bernard NJ. Rheumatoid arthritis: Who knows why regulatory T cells are defective in RA … IDO. Nature reviews. Rheumatology. 2014;101:381. doi: 10.1038/nrrheum.2014.96. [DOI] [PubMed] [Google Scholar]
- 74.Kwidzinski E, et al. Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. Faseb J. 2005;19:1347–1349. doi: 10.1096/fj.04-3228fje. [DOI] [PubMed] [Google Scholar]
- 75.Prendergast GC, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer immunology, immunotherapy : CII. 2014;63:721–735. doi: 10.1007/s00262-014-1549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wieman HL, et al. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18:1437–1446. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Finlay DK, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. 2012;209:2441–2453. doi: 10.1084/jem.20112607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McCracken AN, Edinger AL. Nutrient transporters: the Achilles' heel of anabolism. Trends Endocrinol Metab. 2013;24:200–208. doi: 10.1016/j.tem.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Walker LS, Sansom DM. Confusing signals: Recent progress in CTLA-4 biology. Trends Immunol. 2015 doi: 10.1016/j.it.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Parry RV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frauwirth KA, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- 82.Gubser PM, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. 2013;14:1064–1072. doi: 10.1038/ni.2687. [DOI] [PubMed] [Google Scholar]
- 83.Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 84.Doering TA, et al. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37:1130–1144. doi: 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wei F, et al. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci U S A. 2013;110:E2480–2489. doi: 10.1073/pnas.1305394110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shimatani K, et al. PD-1+ memory phenotype CD4+ T cells expressing C/EBPalpha underlie T cell immunodepression in senescence and leukemia. Proc Natl Acad Sci U S A. 2009;106:15807–15812. doi: 10.1073/pnas.0908805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Patsoukis N, et al. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol. 2013;33:3091–3098. doi: 10.1128/MCB.00319-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Saha A, et al. Host programmed death ligand 1 is dominant over programmed death ligand 2 expression in regulating graft-versus-host disease lethality. Blood. 2013;122:3062–3073. doi: 10.1182/blood-2013-05-500801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen X, et al. PD-1 regulates extrathymic regulatory T-cell differentiation. Eur J Immunol. 2014;44:2603–2616. doi: 10.1002/eji.201344423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Francisco LM, et al. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dai S, et al. The PD-1/PD-Ls pathway and autoimmune diseases. Cellular immunology. 2014;290:72–79. doi: 10.1016/j.cellimm.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 92.Kronig H, et al. Interferon-induced programmed death-ligand 1 (PD-L1/B7-H1) expression increases on human acute myeloid leukemia blast cells during treatment. Eur J Haematol. 2014;92:195–203. doi: 10.1111/ejh.12228. [DOI] [PubMed] [Google Scholar]
- 93.Ansell SM, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372:311–319. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lipson EJ, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. 2013;19:462–468. doi: 10.1158/1078-0432.CCR-12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Topalian SL, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–1030. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gill S, June CH. Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev. 2015;263:68–89. doi: 10.1111/imr.12243. [DOI] [PubMed] [Google Scholar]
- 98.Johnson A, et al. The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev. 2012 doi: 10.1111/j.1600-065X.2012.01151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Freemerman AJ, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem. 2014;289:7884–7896. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yamamoto R, et al. PD-1-PD-1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood. 2008;111:3220–3224. doi: 10.1182/blood-2007-05-085159. [DOI] [PubMed] [Google Scholar]