

Abstract
Structural and functional details of the N-terminal activation function 1 (AF1) of most nuclear receptors are poorly understood due to the highly dynamic intrinsically disordered nature of this domain. A hydrogen/deuterium exchange (HDX) mass spectrometry based investigation of TATA box binding protein (TBP) interaction with various domains of progesterone receptor (PR) demonstrate that agonist bound PR interaction with TBP via AF1 impacts the mobility of the C-terminal AF2. Results from HDX and other biophysical studies involving agonist and antagonist bound full length PR and isolated PR domains reveals the molecular mechanism underlying synergistic transcriptional activation mediated by AF1 and AF2, dominance of PR-B isoform over PR-A, and the necessity of AF2 for full AF1-mediated transcriptional activity. These results provide a comprehensive picture elaborating the underlying mechanism of PR-TBP interactions as a model for studying NR-transcription factor functional interactions.
Keywords: Hydrogen deuterium exchange mass spectrometry, nuclear receptors, intrinsically disordered proteins, conformational flexibility, protein dynamics, progesterone receptor, activation function, allosteric communication
The progesterone receptor (PR) is the cognate steroid receptor (SR) for the hormone progesterone belonging to the nuclear receptor (NR) superfamily. This receptor plays a pivotal role in a range of biological functions including development and maintenance of female reproductive tissue (Anderson and Clarke, 2004; Graham and Clarke, 1997; Li and O’Malley, 2003; Obr and Edwards, 2012). There are two major isoforms of PR, PR-A and PR-B, with the latter having extra 164 amino acids at the N-terminus. For a majority of PR target genes, PR-B is a more potent transcriptional activator as compared to PR-A (Tung et al., 1993; Vegeto et al., 1993). The functional differences and tissue specificity of both isoforms are evident from knockout studies in mice (Fernandez-Valdivia et al., 2005). It has been reported that in breast cancer cells the majority of PR target genes are regulated by the B isoform (Mote et al., 1999; Mote et al., 2002; Richer et al., 2002).
Steroid NRs are multi-domain proteins containing an; N-terminal domain (NTD), DNA binding domain (DBD), hinge region, and a C-terminal ligand-binding domain (LBD). Both the LBD and DBD of PR and other SRs are globular in nature and they have been well studied by a wide range of structural techniques. Moreover, several co-crystal structures of ligand-bound LBD in complex with coregulatory peptides have been solved (Bledsoe et al., 2002; Brzozowski et al., 1997; Madauss et al., 2007; Raaijmakers et al., 2009; Shiau et al., 1998; Williams and Sigler, 1998). Typical of SRs, PR consists of an unusually large intrinsically disordered (ID) NTD that comprises nearly half of the receptor. PR contains two activation function domains (AF1 and AF2) that provide interaction surfaces for transcriptional coregulatory proteins to bind. The N-terminal AF1 is ligand-independent whereas the C-terminal AF2 is ligand-dependent. Unfortunately, no high resolution structures of full length SRs have been reported, partly due to the highly disordered nature of their NTDs (Hill et al., 2012; Khan et al., 2012; Kumar and McEwan, 2012; Kumar and Thompson, 2012; McEwan et al., 2007). Thus, efforts to develop steroid receptor modulators (SRMs) for endocrine-based therapies have been mostly based on their ability to modulate AF2/LBD surfaces for coregulatory protein interactions, thus neglecting the AF1/NTD despite the fact that this region of SRs contribute significantly to the transcriptional activity of these receptors.
Despite the lack of defined structure, ID regions of proteins are known to play important roles in molecular recognition and assembly formations. They carry out functions through a process called coupled binding and folding where upon interaction with its target binding partner, the ID protein or region undergoes a disorder-ordered transition (Dyson and Wright, 2002; Wright and Dyson, 2009). Consistent with this, a core C-terminal domain of the transcription factor TBP (TBPc, aa 159-339) has been reported to bind and fold the AF1/NTD regions of several SRs by promoting the ordered structure formation that facilitates AF1-mediated activity(Khan et al., 2011; Kumar et al., 2004; Kumar et al., 2013; Warnmark et al., 2001). In a recent report, we demonstrated a subregion of PR AF1/NTD (aa 350-428) is required for TBPc binding and TBPc dependent PR transcriptional activation (Kumar et al., 2013). However, these previous studies have examined the influence TBP on structure and folding of isolated NTDs and thus do not account for mechanisms and potential conformational flexibility associated with interdomain interactions of full-length receptors. It has been suggested that SRs are dynamic ensembles of structures responding to a variety of target molecules via synergistic actions of AF1 and AF2. Both the AFs are regulated through allosteric coupling to produce differential selection and/or activation of gene expression (Billas and Moras, 2013; Hilser and Thompson, 2011). However, direct detection of coordinated actions of AF1 and AF2 to bring about synergistic effects on gene activation remains elusive (Tetel et al., 1999; Tung et al., 2006).
Since AF1/NTDs have eluded crystallization and no high resolution structures have been obtained of intact SRs, several groups have applied solution phase techniques to study SR structures including the use of hydrogen/deuterium exchange mass spectrometry (HDX-MS). HDX-MS has emerged as a powerful technique to characterize conformational flexibility associated with protein-protein or protein-ligand interaction in solution state (Chalmers et al., 2006; Englander, 2006; Konermann et al., 2011; Landgraf et al., 2013; Zhang et al., 2013). HDX-MS has advantages for large molecular proteins such as SRs since it is not limited by protein size and can localize changes in conformational flexibility to specific sequences. This technique has been used previously to characterize NRs’ interaction with other binding partners and small molecule dependent activation mechanisms (Chalmers et al., 2011; Dai et al., 2009; Devarakonda et al., 2011; Goswami et al., 2013; Harms et al., 2013; Zhang et al., 2011). Here we report the use of HDX-MS to provide an analysis of the solution state flexibility of full length PR-A and PR-B and the conformational ensemble of PR upon binding the co-regulatory protein TBP and hormonal agonist and antagonist ligands. These studies demonstrate that although TBP directly binds PR through AF1/NTD, conformational rearrangements in the intact PR involve both AF1/NTD and AF2/LBD indicating intramolecular interdomain interactions. Changes in structural flexibility in regions of TBP were also observed upon interaction with PR- NTD/AF1 indicating the potential of the NTD/AF1 to modulate conformation and activity of co-regulatory proteins.
RESULTS
Sequence coverage of full length progesterone receptor
Intact PR-A and PR-B complexed with either hormone agonist ligand (R5020) or antagonist (RU486) were expressed in the baculovirus insect cell system and purified to near homogeneity as previously described (Kumar et al., 2013; Wardell et al., 2005). Purified full length PR-A and PR-B have previously been shown by biochemical and biophysical analyses to form dimers in solution in the absence of DNA (Connaghan-Jones et al., 2006; Heneghan et al., 2005; Tetel et al., 1997). By size exclusion chromatography purified PR-A and PR-B in the 1–2 uM range used here for HDX analysis behaved as a stable dimeric protein (data not shown). Pepsin digestion of PR-A (~750 aa) and PR-B (~930 aa) liganded with R5020 under HDX compatible conditions resulted in 75% and 78% sequence coverage, respectively (Supplementary Fig. S1a). While sequence coverage of the LBD was nearly complete, sequence coverage of the heavily post-transcriptionally modified (including phosphorylation, acetylation, methylation, and others) AF1/NTD was significantly less (59% PR-A, 69% PR-B). Sequence coverage for PR-A was similar to that previously reported while this is the first report of HDX-MS of the larger full length PR-B. As anticipated, sequence coverage decreased slightly as the perturbation map (Supplementary Fig. S1b) contains only peptides that are detected in all the injections (all 45 injections for a 3 replicate experiment). Nevertheless, the overall sequence coverage is sufficient to provide meaningful insight into the flexibility of such large proteins in the 85–100kDa range.
Conformational flexibility of full length PR and TBPc
We investigated the conformational flexibility of full length PR-A and PR-B bound to the hormone agonist R5020 and the results are displayed in Figure 1 (also Supplementary Fig. S2 and Supplementary Fig. S3). The average percentage deuterium exchange corrected for back exchange (see Methods) over a 1hr time period is overlaid onto the crystal structure of PR LBD (PDB ID: 1A28) and PR DBD (PDB ID: 2C7A). Since no crystal structure is available containing the NTD and hinge for either PR-A or PR-B, these regions are illustrated schematically. The exchange dynamics of agonist bound PR-B and PR-A are displayed in a sequence overlay format in Supplementary Figure S2 and Supplementary S3. As expected, the ID AF1/NTD and hinge domains of the receptors afforded little protection to H/D exchange whereas the folded globular DBD and LBD contain regions with significant protection to solvent exchange. Representative deuterium build up plots for peptides from AF1/NTD show that at the earliest time point measured (10s), their backbone amide hydrogens are near fully exchanged with solvent (Fig 1). The conformational flexibility of TBPc, expressed in bacterial cells and purified as previously described (Kumar et al., 2013), was also examined by HDX-MS. TBPc is a folded globular structure and thus most sequence regions are significantly protected from solvent exchange. However, TBPc contains regions that are highly dynamic and not protected to exchange (Supplementary Fig. S4).
Figure 1. Solution state conformational flexibility of full length PR-B.

Agonist (R5020) bound full length PR-B was analyzed by hydrogen deuterium exchange studies and the average % of deuterium incorporation across six different time points (0,10,30,60,300,900 and 3600 sec) is overlaid onto the crystal structure of PR LBD (PDB ID: 1A28) and PR DBD (PDB ID: 2C7A). The color is according to the color code at the bottom of the figure. The hinge region and N terminal domain are represented by schematics due to lack of atomic structure. Representative peptide deuterium build up curves from each domain are indicated at top.
Influence of TBPc interaction on conformational flexibility of intact PR-liganded with hormone agonist
Next we utilized differential HDX-MS to investigate the effect of TBPc interaction on conformational flexibility of the hormone agonist (R5020) occupied intact receptor by comparing exchange kinetics in peptide regions of either PR-A or PR-B in the absence (apo) and presence of TBPc. Interaction of agonist-liganded PR-B with TBPc did not result in significant changes in HDX in any region of AF1/NTD detected. However, perturbations in exchange kinetics were observed in the carboxyl terminal LBD of the receptor. Specifically, regions in the LBD including (helix 1) H1 (aa 680-690), H9-H10 (aa 850-870) and H12 showed reduced solvent exchange upon interaction with TBPc. The deuterium build up curves of selected peptides along with the differential exchange data overlaid onto the LBD crystal structure are shown in Figure 2a. AF2 is a well-structured protein fold composed of helices 3–5 and 12 of the LBD that forms a pocket for complementary binding of LXXLL motifs of coactivators. Conformational positioning of H12 is critical for ligand-agonist dependent activation of AF2. The strongest effect of TBPc on stabilizing deuterium exchange kinetics in the LBD was with peptides in H12 suggesting that TBPc influences this critical region of PR either directly or indirectly through allosteric interactions or through physical interactions between N and C-terminus of intact PR. Similar effects of TBPc were observed on stabilizing exchange kinetics of H12 in PR-A (Fig. 2b). Fig 2c shows a comparative view of H12 stabilization in agonist bound PR-A and PR-B upon TBPc binding.
Figure 2. Agonist (R5020) bound full length PR when interacted with TBPc shows stabilization at the C terminal ligand binding domain (LBD).

(a) Agonist (R5020) bound PR-B isoform when interacted with TBPc, shows protection from exchange at several regions in LBD as depicted by schematic representations. The average % of deuterium uptake values across six different time points is overlaid (color coded) onto LBD atomic structure (PDB ID: 1A28). The color is according to the color bar at the bottom of the figure. The representative deuterium build-up curves of protected regions of the LBD, including the helix 12 is shown also. (b) Schematic representation along with atomic structure (LBD) overlay and deuterium build-up curve of agonist bound full length PR-A and TBPc interaction. (c) Comparison of deuterium uptake of AF2 regions in agonist bound PR-B and PR-A interaction with TBPc after 900 sec exposure to heavy water. Asterisks indicate significant differences as calculated by the processing software (Pascal et al., 2009). (d) HDX footprint of truncated agonist bound PR-B (233-933) - TBP interaction and corresponding deuterium build-up curve.
In previous reports TBPc interaction with NTD of PR and other SRs was demonstrated to induce an increase in secondary structure as detected by circular dichroism spectroscopy and tertiary folding by fluoresence emission and partial proteolysis (Kumar et al., 2004; Kumar et al., 2013; Kumar and Thompson, 2012). Therefore the failure to observe significant changes in HDX kinetics within AF1/NTD upon interaction with TBPc was somewhat unexpected but may be due in part to the highly dynamic nature of this domain. The deuterium build-up curves from NTD (Fig. 1) show most of the amide hydrogens are fully exchanged at the earliest time point (10s) measured. Thus, it is possible that helical structures or tertiary folding detected by other methods within the NTD upon interaction with TBPc (Kumar et al., 2013) interconverts to unstructured conformers at a time scale faster than 10s. This phenomenon of transient “coupled folding and binding” process, where an IDP undergoes disorder-order transition upon interaction with a folded binding partner has been reported (Wright and Dyson, 2009). Previously we reported a strategy to characterize the transient protein-protein interaction involving an IDP and its folded binding partner by lowering the exchange pH and in turn expanding the time window to the millisecond level (Goswami et al., 2013). Appling a similar approach here (interaction tested at pH 6.0) did not result in a perturbation in HDX kinetics upon TBPc interaction for any peptide detected from the AF1/NTD of hormone agonist bound full length PR. It is possible that the affinity between PR and TBPc of 0.17uM at pH 7.5 (Kumar et al., 2013), is reduced further at pH 6.0. Another possibility is that the sequence region that undergoes change in conformation may be in a region that is not covered by MS/MS sequencing in HDX experimental conditions (Supp Fig. S1). To further address this we used a fragment of PR with truncation of NTD to aa 233 that is a shortened version of PR-A lacking an additional 68 aa from the N-terminus. Differential HDX-MS was performed with this 233-PR in the presence and absence of TBPc. TBPc interaction failed to affect exchange in any regions of NTD and resulted in protection to solvent exchange in AF2, with the magnitude of protection being similar to that observed for TBPc interaction with PR-A (Fig. 2d). Similarity of results with PR-A and 233-PR further emphasizes the importance of the unique N-terminal sequence of PR-B as responsible for stronger effect of TBPc on structural flexibility in the AF2 LBD and that failure to detect perturbation in NTD is likely due to transient nature of stabilized structure.
TBP interaction with antagonist liganded PR
Next we investigated the interaction of TBPc with antagonist bound PR. Previous crystallography and other protein biophysical methods have shown a significant change in conformation of H12 of PR LBD upon binding the antagonist RU486 versus hormone agonist (Raaijmakers et al., 2009; Vegeto et al., 1993; Weigel et al., 1992). Interestingly TBPc interaction with RU486 liganded PR did not result in changes in the HDX kinetics in H12 or other helices of the LBD (Fig. 3a and 3b). However, unlike that observed for agonist bound receptor, TBPc interaction with RU486 liganded PR resulted into a slight but statistically significant increase in solvent exchange in the region containing aa 319-328 within the AF1/NTD of both PR-A and PR-B. This increase in solvent exchange likely results from a rearrangement of hydrogen bonding in this region upon interaction with TBPc. Together these results indicate that antagonist (RU486) binding with PR blocks the allosteric communication between the NTD bound TBPc with AF2/LBD.
Figure 3. Antagonist (RU486) bound full length PR interaction with TBPc.
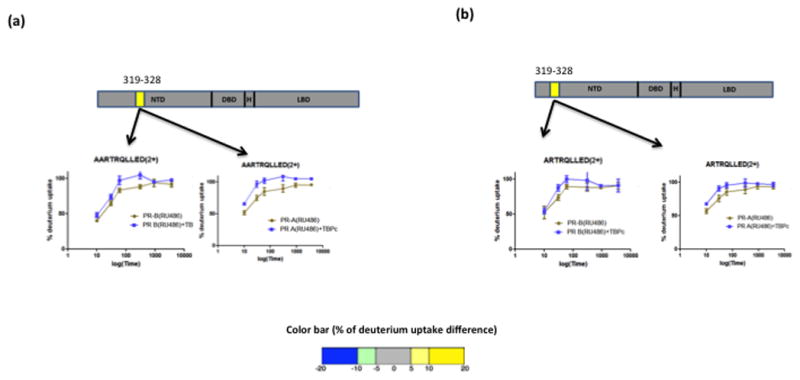
Antagonist bound full length PR-B (a) and PR-A (b) when interacted with TBPc showed slight destabilization at the N terminal domain. Corresponding build-up curves are shown with the schematic representation.
HDX of TBPc in the presence and absence of PR
Results from HDX analysis of the isolated AF1/NTD of PR in the presence and absence of TBPc failed to detect any footprint on NTD and hence was consistent with the data obtained using full length receptor (Supplementary Fig. S5). Therefore, we investigated the impact of AF1/NTD interaction on the conformational flexibility of TBPc by analyzing the differential HDX kinetics of TBPc in the presence and absence of these domains. These experiments were performed with both isolated NTDs and full length receptors. Results demonstrate that the isolated AF1/NTDs of PR-A and PR-B interact directly with TBPc with the AF1/NTD of PR-B inducing more stabilization of TBPc when compared to AF1/NTD of PR-A (Fig. 4a, 4b). The region of TBPc containing aa 243-253 was stabilized by both NTDs; however, interaction with PR-B AF1/NTD resulted in additional regions being protected from solvent exchange (aa 275-290) suggesting that AF1/NTD of PR-B makes a stronger interaction with TBPc as compared with the NTD of PR-A. Full length PR-B induced greater perturbation of exchange on TBPc than PR-A (Fig. 4c, 4d), an observation that is consistent with HDX data obtained on the isolated AF1/NTDs. However, closer inspection of the data shows additional regions within TBPc are influenced in the presence of full length PR-B as compared to its isolated AF1/NTD suggesting that there are contributions from other regions of PR-B in binding to TBPc.
Figure 4. Stabilization of TBPc structure upon interaction with isolated N terminal domain (NTD) and agonist bound full length PR.
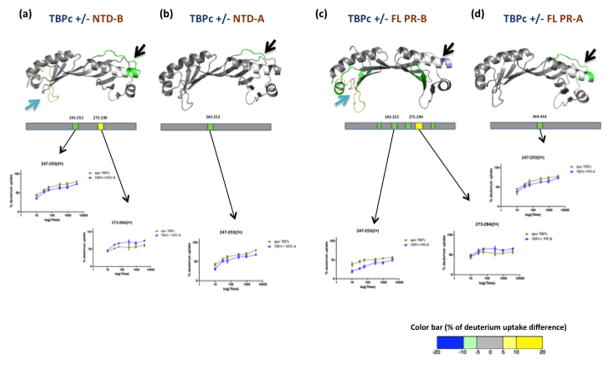
TBP shows protection from exchange when interacted with isolated NTD of PR-B (a), NTD of PR-A (b), agonist bound full length PR-B (c) and agonist bound full length PR-A (d). Regions of protection are represented by schematics and the average % of deuterium uptake values across six different time points is overlaid (color coded) onto the TBPc crystal structure (PDB ID: 1TGH) according to the color bar at the bottom of the figure. The deuterium build-up curves are shown below the atomic models.
Isolated PR LBD does not interact with TBPc
The observation that TBPc alters solvent exchange in both AF1/NTD and AF2 (LBD) in the intact receptor encouraged us to investigate if the LBD alone can interact with the TBPc and if so whether this interaction can alter conformation in AF2/H12. To this end, isolated PR LBD liganded with R5020 was analyzed by HDX in the presence and absence of TBPc. As shown in Figure 5a and 5b, no statistically significant changes in HDX kinetics were observed in either the PR-LBD or TBPc. These data indicate either a lack of direct physical interaction of TBPc with the LBD or that this interaction is not sufficient for altering conformational flexibility of either LBD/AF2 or TBPc. In previous studies with isolated PR LBD we were unable to detect direct binding with TBPc (Kumar et al., 2013). These data collectively suggest that TBPc only influences the flexibility of AF2 in the context of the full length PR and that there is no direct TBPc-AF2 interaction.
Figure 5. Ligand binding domain (LBD) of PR shows no significant interaction with TBPc by itself.
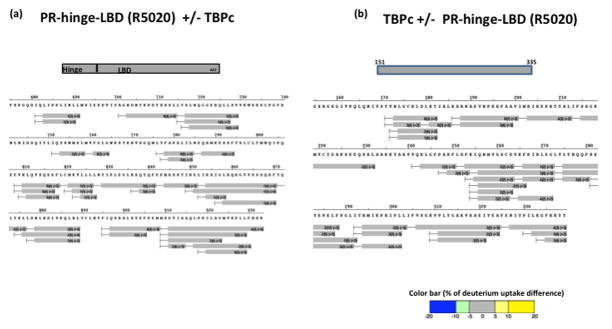
Differential HDX data of agonist bound PR hinge-LBD region with TBPc (a) and vice versa (b) showed no significant interactions. The color coding is according to the color bar at the bottom of the figure.
Limited proteolysis analysis of intact PR vs. isolated domains confirms that conformational change is dependent upon TBPc interaction with the NTD
Previously we have shown that when bound to TBPC, the isolated AF1/NTD of PR is protected against limited proteolysis, suggesting that TBPC binding induces a more compact tertiary structure in AF1/NTD (Kumar et al., 2013). To determine the influence of TBPc interaction on folding of the NTD and other domains in the context of full length PR, experiments were extended to include limited proteolysis by trypsin of full length PR-A, and isolated PR LBD and NTD in the absence and presence of TBPc. As shown by Coomassie stained SDS gels, the PR-NTD was highly susceptible to proteolysis resulting in nearly complete degradation with little intact NTD remaining (Fig. 6a, compare lanes 2 and 6). PR-A also showed a high susceptibility to proteolysis except for a protected band corresponding to the size of LBD (Fig 6a, compare lanes 1 and 5). The isolated PR LBD and TBPC are highly resistant to limited proteolysis (Fig. 6a compare lanes 3 and 7; and lanes 4 and 8, respectively) consistent with stable globular structure of these polypeptides. Limited proteolysis of mixtures of PR-A or PR-NTD and TBPC resulted in some residual intact PR-A and NTD plus smaller fragments not detected by digestion with either the PR-A or PR-NTD alone (Fig. 6a lanes 9 and 10). To determine whether protected fragments generated from the PR-A:TBPC mixture were coming from the PR-NTD or LBD, tryptic digests were also analyzed by immunoblotting with monoclonal antibodies (clone 1294 and 636) to different epitopes within the PR-NTD (Fig. 6b; upper and middle Panels) or the C-terminus of PR-LBD (clone 10A9)(Fig. 6b, lower panel). Digests of PR-A in the absence of TBPC showed a few fragments larger than LBD that are reactive with NTD specific MAbs (1294 and 636) plus a predominant protected protein band the size of the LBD (~ 27kDa) that was reactive only with the C-terminal specific MAb (10A9) (Fig 6b, 4). As expected digests of NTD alone generated only trace protected bands reactive with NTD specific MAbs and none that are reactive with the LBD specific MAb (Fig 6b, lane 5). When PR-A or PR-NTD and TBPC were mixed together, several unique protected PR fragments were detected with the PR NTD specific MAbs and the patterns were distinct with each antibody indicating that multiple sites within the NTD change their accessibility to trypsin in the presence of TBPC (Fig 6b compare lanes 4 and 7; and 5 and 8, respectively). Protection against partial proteolysis indicates that the NTD folds into a more compact conformation in the context of intact PR-A when complexed with TBPC. Analysis of PR-A digests in the presence TBPc with the PR LBD specific MAb (10A9) showed an increase in relative amount of intact PR-A, a stronger protected band corresponding to LBD plus several predominant unique fragments between the size of intact PR and the LBD not detected in the absence of TBPc (Fig. 6b compare lanes 4 and 7). As expected no bands were detected with the LBD specific MAb (10A9) with digests of the NTD either in the absence or presence of TBPc (Fig. 6b lanes 5 & 8). These results are consistent with regions of both the NTD and LBD undergoing protection from limited proteolysis when intact PR-A is complexed with TBPC.
Figure 6. Folding NTD in the context of full length PR-A in the presence of TBPC as detected by protection against partial proteolysis.
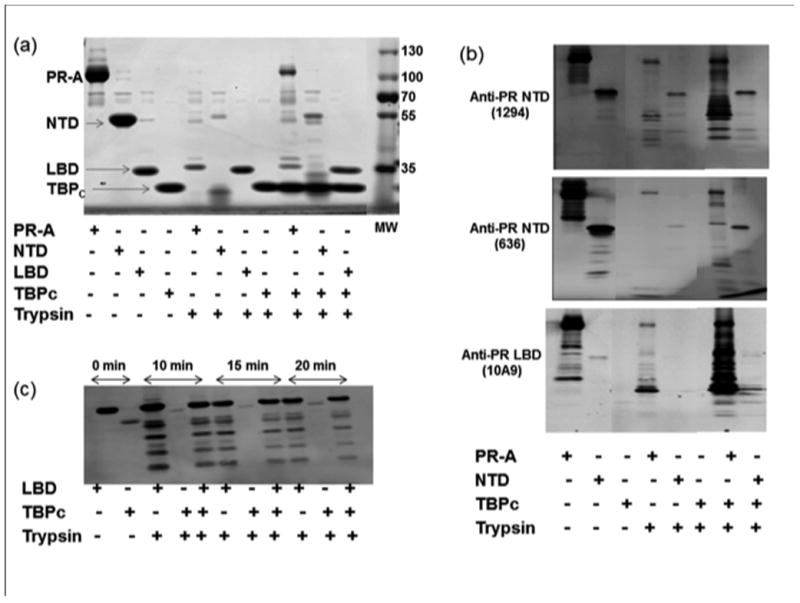
Purified PR-A, PR-NTD, PR-LBD, TBPC or an equal molar mixture of PR-A:TBPC, PR-A NTD:TBPC, and PR-LBD:TBPC proteins were subjected to limited proteolysis with trypsin and samples were analyzed by Coomassie blue-stained SDS-PAGE or by immunoblotting. a) Coomassie-stained SDS-PAGE. b) Immunoblotting of limited proteolytic products derived from either full length PR-A or PR-A NTD with antibodies (1294 and 636) specific to distinct epitopes in the NTD or the C-terminal LBD (10A9), c) Immunoblotting with the C-terminal MAb (10A9) of limited proteolytic products generated from the isolated PR-LBD at different time points indicated (0 to 20 min).
We previously showed that TBPC directly binds to NTD and not LBD in vitro (Kumar et al., 2013), therefore we carried out another set of experiments to confirm that the conformational changes observed in the PR-LBD are as a result of direct NTD-TBPc binding in the full length receptor. Since no detectable cleavage of PR LBD alone was detected under conditions above (Fig. 6a and 6b) we carried out limited tryptic digestion of PR-LBD with and without TBPc at room temperature for 10, 15, and 20 min to allow a higher degree of digestion, and resolved the products of digestion by immunoblot with the PR LBD specific antibody. As expected a strong reaction for intact PR-LBD was seen with the antibody (Fig. 6c; lane 1) with no reaction with TBPc. Under these conditions several smaller protected fragments of PR LBD were generated by limited digestion at each time point and there were no evident differences in the patterns of LBD cleavage in the absence or presence of TBPC (Fig 6c). These results demonstrate that protection from proteolysis in the LBD in the presence of TBP occurs only with intact PR-A and not with the isolated LBD (Fig. 6b; lower Panel) indicating this effect on structure in the LBD is dependent on NTD-TBPC interaction and an interdomain communication.
DISCUSSION
The strategy for the development of structure-based SR modulators has focused largely on ligand control of AF2 despite the fact that the AF1/NTD region of SRs contribute significantly to the receptor’s transcriptional activity, and functional synergy between AF1 and AF2 is essential to SR-mediated target gene regulation. This is not surprising due to the limited knowledge about the structure and conformational flexibility of AF1-AF2 interaction within full length SRs. Similarly there is lack of structural insight into coregulatory protein interactions with AF1/NTD. It is generally thought that the ID nature of AF1/NTD enables this region of the receptor to adapt different structures depending on the context of the interacting partner. This structural plasticity likely contributes to the functional diversity of the receptor and may provide a strategy to modulate target gene promoter or tissue specific effects.
Previous reports demonstrate that TBPc binding with the NTD of steroid receptors induces secondary structure within AF1/NTD by coupled binding and folding mechanism (Fischer et al., 2010; Khan et al., 2011; Warnmark et al., 2001). Based on deletion mutation experiments a subregion of PR AF1/NTD (aa 350-428) has been identified to undergo disorder-order conformational transition upon its interaction with TBP and in cell transfection assays this subregion was also required for TBPc enhancement of AF1/NTD dependent transcriptional activity (Kumar et al., 2013). In addition to independent effects of TBP on structure and function of the isolated PR NTD, effects of TBP on hormone-dependent AF2 activity in the context of full length PR were previously observed. With a constitutively active two domain PR DBD/AF1-NTD construct that lacks LBD/AF2, deletion of aa 323-427 of the NTD completely abrogated TBP stimulation of AF1/NTD mediated transcriptional activity whereas deletion of this same region in full length PR-B only partially reduced TBP dependent hormone-induced transcriptional activation (Kumar et al., 2013). These functional data suggested to us that TBP induced folding of the NTD has the potential to mediate an allosteric inter-domain interaction, thus prompting HDX-MS analysis to further explore this question.
HDX-MS has been used to probe the conformational flexibility of intact multidomain proteins in solution even in the absence of a high resolution atomic structure and has the potential to detect long range allosteric effects (Zhang et al., 2011). In this study we applied HDX to probe the conformational flexibility of intact PR-A and PR-B. As expected, the HDX results show that AF1/NTD of both PR isoforms is highly dynamic as compared to the DBD and LBD for both agonist and antagonist bound receptor. In both receptor isoforms the hinge region appears to be unstructured as well as determined by rapid deuterium incorporation in peptides representative of this domain. These observations are consistent with another report on full length nuclear receptors (Chandra et al., 2008). Our present study shows that full length PR-B (bound to hormone agonist) has higher degree of stabilization at helix 12 and more perturbation sites compared to PR-A upon interaction with TBPc (Fig 2a–c). Isolated NTDs of PR-A and PR-B follow the same norm (Fig. 4a–b). Moreover, when the first 232 aa of PR-B NTD is deleted, it shows a more PR-A like interaction with TBP (Fig 2d). Considering all these observation of higher global protection in PR-B-TBPc interactions, our HDX study supports previous findings that PR-B is the dominant transcription activator and acts through a distinct structural conformation of the NTD of B isoform than that of the PR-A isoform (Bain et al., 2001; Hill et al., 2012; Tung et al., 2006).
Despite the fact that TBPc interaction with isolated AF1/NTDs of PR (and other SRs) has been demonstrated to induce a stabilization of secondary helical structure and folding by other biophysical methods, we were not able to detect TPBc dependent protection to solvent exchange in AF1/NTD in the context of either the intact PR (Fig. 2 and 3) or the isolated NTD (Supplementary Fig. S5). This apparent discrepancy is likely due to the extreme flexibility and disorder of the NTD and to the transient nature of TBPc induced folded structures that may not be detectable in the time frame of the HD exchange kinetics. Interaction of the TIF2 co-activator with AF1 of glucocorticoid receptor (GR) gave similar results wherein HDX-MS was unable to detect changes in conformational flexibility whereas other biophysical methods detected structural changes (Khan et al., 2012). Interestingly we were able to detect perturbation in HDX kinetics of TBPc upon interaction with either the isolated AF1/NTD or full length PR (Fig. 4a–b). Additionally, both full length PR-B and the isolated NTD of PR-B gave stronger interactions and effects on stabilizing structural conformations of TBPc than either intact PR-A or isolated NTD respectively of PR-A (Fig. 4). These novel results indicate that the AF1/NTD can affect structural conformation of an interacting protein and maybe a mechanism by which the intrinsically disordered AF1/NTD can affect activity of co-regulatory proteins associated with receptors. Another novel finding of this study was that TBPc interaction mediated stabilization of AF2/LBD but only through interaction with NTD of the intact PR occupied with hormone agonist. Additionally, the magnitude of perturbation of AF2 conformation and number of protected sites was greater with PR-B than PR-A (Fig. 2). Consistent with previous reports that TBPc binds only to NTD and not with LBD (Kumar et al., 2013), no change in HD exchange kinetics was observed with either an isolated PR LBD (+ hinge) or TBPc when incubated together (Fig. 5). With intact PR (either isoform) bound to the hormone antagonist (RU486), TBPc induced perturbation of AF2/LBD was prevented and unique perturbation of a short region between aa 319-328 within AF1/NTD was observed (Fig. 3). These data collectively support the conclusion that TBP interaction with AF1/NTD impacts the AF2 surface via long-range allostery, or that binding TBP alters the proximity of AF1/NTD to AF2/LBD within the intact receptor. Physical interaction between AF1 and AF2, both in vivo and in vitro has been reported before (Tetel et al., 1999), resulting in a functional synergy between AF1 and AF2 that is an essential component of SR-mediated target gene regulation (Chen et al., 2006; Choudhry et al., 2006). This synergy is believed to be mediated by inter-domain allosteric pathways that may involve the conformation flexibility of the NTD but through mechanisms that remain undefined experimentally (Hilser and Thompson, 2011). Our limited proteolysis experiments further support HDX results and this hypothesis (Fig 6).
Hormone agonists as well as the antagonist RU486 induce dimerization and binding of PR to consensus progesterone response element (PRE) DNA. However, PR bound to target DNA in the presence of RU486 has impaired transcription activity due to an altered conformation in PR that does not permit optimal recruitment of co-regulatory proteins (Hill et al., 2012). Ligand dependent dimerization of PR in solution is thought to occur as a requisite step prior to DNA binding. Although controversial because the dimerization constant for purified PR in solution is in the uM range above the concentration of PR in most cells (Connaghan-Jones et al., 2006; Heneghan et al., 2005), ligand induced dimerization independent of DNA binding has been shown in cell based assays (Carbajo et al., 1996; Hill et al., 2012; Tetel et al., 1997). The PR dimer interface in the absence of DNA involves surfaces in both the NTD (Bain et al., 2001; Tetel et al., 1997) and the LBD (Williams and Sigler, 1998). Crystal structure of the PR LBD showed dimerization to be mediated by helix 10 and 11 (Williams and Sigler, 1998) but sequences in the NTD have not been defined. An additional DNA dependent dimerization interface is present in the DNA binding domain that functions to stabilize receptor dimers upon binding DNA (Hill et al., 2012). HDX analysis of full length PRs in the present study was performed in the absence of DNA with purified PR in a dimeric form. Therefore our results reflect conformational flexibility of pre-formed PR dimers and do not provide insight into dimerization process. HDX-MS experiments for analysis of solution dimerization will require isolation of PR monomers and in vitro conditions to promote monomer-dimer assembly. DNA interaction has been demonstrated to induce changes in structural conformation in either the NTD or LBD/AF2s of steroid receptors. Moreover, different target DNA sequences have been observed to induce distinct conformations indicating DNA can act as a regulatory ligand for steroid receptors (Hill et al., 2012; Kumar and Thompson, 2012; Meijsing et al., 2009). In the present study we did not analyze the effect of DNA on conformational flexibility of PR. Studies focused on the effects of hormonal ligands and a protein binding partner. It will be important to perform differential HDX-MS analysis of PR-DNA complexes to gain a more complete assessment of the structural state of transcriptionally active receptors.
Based on the results from the studies presented here we developed a model for how TBP interaction with the flexible NTD can allosterically mediate structural rearrangement in the NTD and the LBD to potentially affect synergistic transcriptional activation between AF1 and AF2 modulated by ligand (Fig. 7). Although HDX-MS analysis was done only with PR dimers in solution, DNA is included in the model as the convention for PR as a sequence specific DNA binding transcription factor. Both hormone agonists and the antagonist RU486, are known to induce dimerization and binding of PR to progesterone response element DNA. Upon binding DNA PR recruits assembly of co-regulatory proteins required for transcriptional activation and due to distinct conformations in PR, co-regulators are different in the presence of hormone agonist and antagonist. HDX results provide new information on potential allosteric regulation of LBD/AF2 through TBP-PR NTD interaction. In the presence of hormone agonist, TBP-PR interaction results in structural re-organization of both the NTD and LBD/AF2. In the presence of the antagonist RU486, TBP interaction resulted in a small destabilization of NTD and failed to mediate the inter-domain communication that results in structural reorganization of LBD/AF2.
Figure 7. A model of TBP-PR NTD interaction to allosterically mediate structural changes in LBD/AF2.
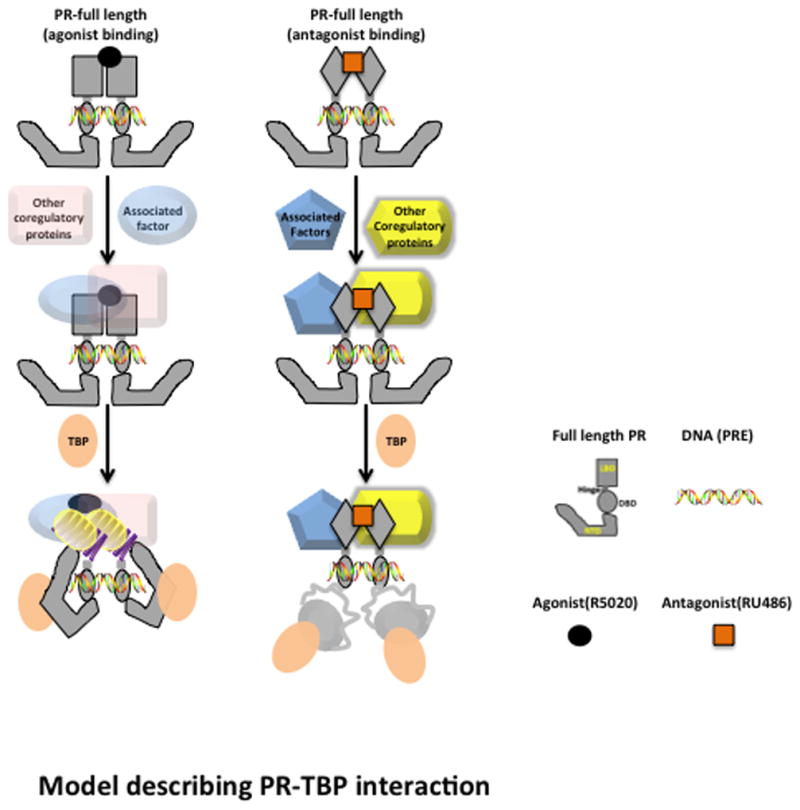
Both hormonal agonist and antagonist promote dimerization and binding of PR to progesterone response element (PRE) target DNA. Once DNA-bound, receptor dimers assemble a multi-protein complex of coregulators which differ for an agonist vs. antagonist due at least in part to distinct conformations in the LBD/AF2. In the presence of agonist, TBP binding to the NTD results in conformational rearrangements in the NTD and in the LBD/AF2 through allosteric regulation. These structural rearrangements facilitate the binding and assembly of other co-activators at either LBD/AF2 or AF1 in the NTD. In the presence of antagonist, TBP binding with NTD resulted in a slight destabilization of NTD structure and the interdomain communication with LBD/AF2 was lost.
An emerging picture is that the entire SR signaling spectrum involves allosteric interactions between AF1/NTD and AF2/LBD surfaces for coregulatory protein interactions. It has also been suggested that the tissue-specific residual activity of selective steroid receptor modulators (SRMs) used to therapeutically target SRs is mediated primarily via AF1 and that the relative functional importance of AF1 may be decided by specific SRM-induced conformational changes in either LBD or transmitted allosterically to the NTD (Berry et al., 1990; Halachmi et al., 1994; Lonard and O’Malley, 2012; Simons, 2010; Wu et al., 2005). Our present studies support this notion as HDX analysis of RU486-bound full length PR:TBP complex shows that antagonist both negates the TBP impact on AF2 flexibility and destabilizes a region within AF1/NTD that was not detected with intact PR occupied by hormone agonist (Fig. 3). There is precedent for RU486 promoting a structural conformation in the AF1/NTD of PR distinct from that of hormone agonist. Partial agonist activity of RU486 mediated by a co-activator that binds the PR DBD and allostrically affects NTD structure, was shown to require sequence regions within NTD (aa 327 to 427) that are not required for functional coactivator response in the presence of hormone agonist (Wardell et al., 2010).
The significance of these findings lies in the possibility of therapeutically targeting AF1/NTD surfaces directly or indirectly, by allosteric modulations, to achieve tissue-restricted effects. Since TBP is a common binding partner for AF1/NTD of all SRs and does not bind to the AF2/LBD, yet leads to conformational changes in the AF2/LBD, we hypothesize that TBP-induced disorder-order transition opens AF1/NTD protein surfaces for its interactions with specific coactivators. Consistent with this concept, we previously reported that structural reorganization of AF1/NTD of both GR and PR induced by TBP enhanced binding of SRC-1 to AF1/NTD and TBP and SRC-1 acted synergistically to functionally stimulate AF1/NTD-dependent transcriptional activity (Khan et al., 2011; Kumar et al., 2013). Further understanding the structural and functional consequences of the AF1/NTD-TBP interaction sites may provide potential avenues to modify both AF1 and AF2 activities simultaneously. This level of control is needed for additional selectivity to target cell-tissue specific gene regulations in current endocrine-based therapies that could complement or replace existing SRMs actions. Targeting ID proteins by small molecules/peptides to block protein-protein interactions is a rapidly evolving field, and the above findings suggest that compounds that bind to NTD/AF1 could be promising molecules for SR-based therapeutics (Dunker and Uversky, 2010; Ferreon et al., 2013; Simons et al., 2013). Drugs have been defined that interfere with intrinsic disorder-to-order transition induced by a binding protein by directly interacting with ID regions of the transcription factor (Dunker and Uversky, 2010). Meaningful screens for small molecules that could modify AF1/NTD-TBP binding may provide the additional selectivity needed to target SR-selective genes and thereby reduce the number of undesirable side effects in current endocrine-based cancers.
Materials and Methods
Reagents, chemicals and purified proteins
HPLC grade H2O, D2O ((9.9%), acetonitrile, formic acid, iso-propanol, Tris, NaCl was purchased from Sigma-Aldrich (St. Louis, MO), Trifluroacetic acid (TFA, sequanal grade) was obtained from Pierce (Rockford, IL). The procine pepsin-immobilized POROS 20 AL beads (Applied Biosystems, Foster City, CA) of particle size 20 μm was used to pack immobilized pepsin columns. Full length PR-A and PR-B as well as isolated PR NTD domains were expressed from baculovirus vectors as recombinant proteins in Sf9 insect cells and purified as previously described with >95% purity at a concentration range of 10–20uM (Kumar et al., 2013; Wardell et al., 2005). By size exclusion chromatography (S-200 column) purified full length PR-A and PR-B fractionated as a single symmetrical peak with a retention volume consistent with that of a PR dimer. For isolation and purification of intact PR bound to either hormone agonist (R5020) or antagonist (RU486) ligands were added to Sf9 insect cell cultures for 24hr prior to cell harvest as previously described (Wardell et al., 2005). The C-terminal core DNA binding domain (aa 159-339) of human TATA-binding protein (TBPc) was expressed from a pET-21d bacterial expression vector and purified as previously described (Kumar et al., 2013). GST tagged PR LBD (675-693) was purchased from Invitrogen (Catalogue no. P2899, Invitrogen, USA).
Hydrogen/Deuterium Exchange and Mass Spectrometry
Solution-phase amide HDX was carried out with a fully automated system as described previously (Chalmers et al., 2006; Goswami et al., 2013). Briefly, 4 μl of 10 μM full length PR or PR constructs or TBPC was diluted to 20 μl with D2O-containing HDX buffer and incubated at 4 °C for 10 s, 30 s, 60 s, 900 s or 3,600 s. Following on exchange, unwanted forward or back exchange was minimized, and the protein was denatured by dilution to 50 μl with 0.1% (v/v) TFA in 5 M urea (held at 1 °C). Samples were then passed across an immobilized pepsin column at 50 μl min−1 (0.1% v/v TFA, 15 °C); the resulting peptides were trapped on a C8 trap cartridge (Hypersil Gold, Thermo Fisher). Peptides were then gradient-eluted (4% (w/v) CH3CN to 40% (w/v) CH3CN, 0.3% (w/v) formic acid over 5 min, at 2 °C) across a 1 mm × 50 mm C18 HPLC column (Hypersil Gold, Thermo Fisher) and electro sprayed directly into an Orbitrap mass spectrometer (LTQ Orbitrap with ETD, Thermo Fisher). Peptide ion signals were confirmed if they had a MASCOT score of 20 or greater and had no ambiguous hits using a decoy (reverse) sequence in a separate experiment using a 60 minute gradient. The intensity weighted average m/z value (centroid) of each peptide’s isotopic envelope was calculated with the in-house developed software (Pascal et al., 2012) and corrected for back-exchange on an estimated 70% recovery and accounting for the known deuterium content of the on-exchange buffer. To measure the difference in exchange rates, we calculated the average percentage deuterium uptake for say full length PR following 10, 30, 60, 900 and 3,600 s of on exchange. From this value, we subtracted the average percent deuterium uptake measured for the TBPc bound PR (at 5:1 molar ratio).
For back exchange correction, experiments were repeated with full deuterium controls (run in triplicate). Proteins were diluted to 10 μM, pre-digested with pepsin offline and incubated at 37 °C for 16 hr with D2O containing HDX buffer with the same ratio as the above mentioned HDX experiments. Then 20 μL of aliquots were added to 30 μL of cold quench buffer and subsequent sample analysis was carried out in automated fashion as described for HDX samples (above), except no correction for estimated deuterium recovery was applied. Instead, the average percent deuterium values from each triplicate sample was divided by the average percent deuterium values from the full deuterium controls to correct for individual peptide back exchange
Limited proteolysis
Three sets of partial proteolytic experiments were carried out. In first set, purified proteins (PR-A, PR-NTD, PR-LBD, TBPC, PR-A:TBPC, PR-NTD:TBPC, and PR-LBD:TBPC mixture) were digested by using trypsin (Promega). Digestions were carried out at 4°C for 15 min by using a protein:enzyme mass ratio of 100:1. Reactions were terminated by adding SDS loading buffer and placing the sample tubes in boiling water for 5 min. The proteolytic digestion products were resolved on SDS-PAGE followed by Coomassie Blue R-250 staining. In second set, following digestion of PR-A, PR-NTD, TBPC, and PR-A:TBPC and PR-NTD:TBPC mixtures, proteolytic digestion products were resolved on SDS-PAGE and proceeded with immunoblotting with PR-NTD- or LBD- specific- antibodies. Mouse monoclonal antibodies (MAbs) to human PR (clone #1294 and 636), that detect distinct epitopes in the NTD (Press et al., 2002), and clone 10A9 (Immunotech, Hamburg, Germany) elicited against amino acids 922–933, which form the extreme C-terminus of the human PR were used to detect the peptide products of PR. In the third set, PR-LBD, TBPC, and PR-LBD:TBPC mixture were digested with trypsin at room temperature for 10, 15, and 20 min. and proteolytic digestion products were resolved on SDS-PAGE followed by immunoblotting with PR-LBD- specific- antibody (10A9).
Supplementary Material
Supplementary Figure S1, Related to Figure 1. Sequence coverage of full length PR-A and PR-B at different stages of HDX experiments. (a) MSMS sequence coverage of PR-B and PR-A. Letters in red are amino acid sequence detected by analyzing pepsin digested MS/MS spectra using MASCOT. (b) Sequence coverage (%) obtained during MS/MS, 0 second time point injection (t0) and on-exchange experiments of full length and N terminal domain of PR-B and PR-A.
Supplementary Figure S2, Related to Figure 1. Solution state conformational flexibility of agonist bound full length PR-B with different domains indicated by arrow head. The back exchange corrected average % of deuterium incorporation across six different time points (0,10,30,60,300,900 and 3600 sec) are represented is sequence lay out format where each horizontal bar represents each peptide. Colors are according to the color bar at the bottom of the figure.
Supplementary Figure S3, Related to Figure 1. Solution state conformational flexibility of agonist bound full length PR-A with different domains indicated by arrow head. The back exchange corrected average % of deuterium incorporation across six different time points (0,10,30,60,300,900 and 3600 sec) are represented is sequence lay out format where each horizontal bar represents each peptide. Colors are according to the color bar at the bottom of the figure.
Supplementary Figure S4, Related to Figure 4. Solution state conformational flexibility of TBPc. The back exchange corrected average % of deuterium incorporation across six different time points (0,10,30,60,300,900 and 3600 sec) are represented is sequence lay out format where each horizontal bar represents each peptide. Colors are according to the color bar at the bottom of the figure.
Supplementary Figure S5, Related to Figure 4. Differential HDX of isolated N terminal domain (PR-A) showed no perturbation upon interaction with TBPC.
Highlights.
Interdomain communication in with PR upon coactivator binding detected by HDX
Molecular basis of AF1-AF2 synergism for TBP dependent PR activation
Conformational mobility of full length PR isoforms (~100 kDa) characterized by HDX
isoform specific interactions detected by HDX
Acknowledgments
This work was supported in part by NIH grant CA046938 (DPE) and GM84041 (PRG). We acknowledge the Monoclonal Antibody and Recombinant Protein Core services of the Proteomics Shared Resource of the Dan L. Duncan Cancer Center at Baylor College of Medicine for expression of PR in the baculovirus insect cell system and for providing monoclonal antibodies to PR. The Core is supported by NCI Cancer Center CCSG (P30CA123125).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson E, Clarke RB. Steroid receptors and cell cycle in normal mammary epithelium. J Mammary Gland Biol Neoplasia. 2004;9:3–13. doi: 10.1023/B:JOMG.0000023584.01750.16. [DOI] [PubMed] [Google Scholar]
- Bain DL, Franden MA, McManaman JL, Takimoto GS, Horwitz KB. The N-terminal region of human progesterone B-receptors: biophysical and biochemical comparison to A-receptors. J Biol Chem. 2001;276:23825–23831. doi: 10.1074/jbc.M102611200. [DOI] [PubMed] [Google Scholar]
- Berry M, Metzger D, Chambon P. Role of the two activating domains of the oestrogen receptor in the cell-type and promoter-context dependent agonistic activity of the anti-oestrogen 4-hydroxytamoxifen. EMBO J. 1990;9:2811–2818. doi: 10.1002/j.1460-2075.1990.tb07469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billas I, Moras D. Allosteric controls of nuclear receptor function in the regulation of transcription. J Mol Biol. 2013;425:2317–2329. doi: 10.1016/j.jmb.2013.03.017. [DOI] [PubMed] [Google Scholar]
- Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, Consler TG, Parks DJ, Stewart EL, Willson TM, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 2002;110:93–105. doi: 10.1016/s0092-8674(02)00817-6. [DOI] [PubMed] [Google Scholar]
- Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- Carbajo P, Christensen K, Edwards DP, Skafar DF. Binding of [3H]progesterone to the human progesterone receptor: differences between individual and mixed isoforms. Endocrinology. 1996;137:2339–2346. doi: 10.1210/endo.137.6.8641184. [DOI] [PubMed] [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, He Y, Hendrickson CL, Marshall AG, Griffin PR. Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2006;78:1005–1014. doi: 10.1021/ac051294f. [DOI] [PubMed] [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, West GM, Griffin PR. Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert Rev Proteomics. 2011;8:43–59. doi: 10.1586/epr.10.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, Rastinejad F. Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature. 2008;456:350–356. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Rogatsky I, Garabedian MJ. MED14 and MED1 differentially regulate target-specific gene activation by the glucocorticoid receptor. Mol Endocrinol. 2006;20:560–572. doi: 10.1210/me.2005-0318. [DOI] [PubMed] [Google Scholar]
- Choudhry MA, Ball A, McEwan IJ. The role of the general transcription factor IIF in androgen receptor-dependent transcription. Mol Endocrinol. 2006;20:2052–2061. doi: 10.1210/me.2005-0486. [DOI] [PubMed] [Google Scholar]
- Connaghan-Jones KD, Heneghan AF, Miura MT, Bain DL. Hydrodynamic analysis of the human progesterone receptor A-isoform reveals that self-association occurs in the micromolar range. Biochemistry. 2006;45:12090–12099. doi: 10.1021/bi0612317. [DOI] [PubMed] [Google Scholar]
- Dai SY, Burris TP, Dodge JA, Montrose-Rafizadeh C, Wang Y, Pascal BD, Chalmers MJ, Griffin PR. Unique ligand binding patterns between estrogen receptor alpha and beta revealed by hydrogen-deuterium exchange. Biochemistry. 2009;48:9668–9676. doi: 10.1021/bi901149t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarakonda S, Gupta K, Chalmers MJ, Hunt JF, Griffin PR, Van Duyne GD, Spiegelman BM. Disorder-to-order transition underlies the structural basis for the assembly of a transcriptionally active PGC-1alpha/ERRgamma complex. Proc Natl Acad Sci U S A. 2011;108:18678–18683. doi: 10.1073/pnas.1113813108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunker AK, Uversky VN. Drugs for ‘protein clouds’: targeting intrinsically disordered transcription factors. Curr Opin Pharmacol. 2010;10:782–788. doi: 10.1016/j.coph.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- Englander SW. Hydrogen exchange and mass spectrometry: A historical perspective. J Am Soc Mass Spectrom. 2006;17:1481–1489. doi: 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Valdivia R, Mukherjee A, Mulac-Jericevic B, Conneely OM, DeMayo FJ, Amato P, Lydon JP. Revealing progesterone’s role in uterine and mammary gland biology: insights from the mouse. Semin Reprod Med. 2005;23:22–37. doi: 10.1055/s-2005-864031. [DOI] [PubMed] [Google Scholar]
- Ferreon AC, Ferreon JC, Wright PE, Deniz AA. Modulation of allostery by protein intrinsic disorder. Nature. 2013;498:390–394. doi: 10.1038/nature12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer K, Kelly SM, Watt K, Price NC, McEwan IJ. Conformation of the mineralocorticoid receptor N-terminal domain: evidence for induced and stable structure. Mol Endocrinol. 2010;24:1935–1948. doi: 10.1210/me.2010-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami D, Devarakonda S, Chalmers MJ, Pascal BD, Spiegelman BM, Griffin PR. Time window expansion for HDX analysis of an intrinsically disordered protein. J Am Soc Mass Spectrom. 2013;24:1584–1592. doi: 10.1007/s13361-013-0669-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JD, Clarke CL. Physiological action of progesterone in target tissues. Endocr Rev. 1997;18:502–519. doi: 10.1210/edrv.18.4.0308. [DOI] [PubMed] [Google Scholar]
- Halachmi S, Marden E, Martin G, MacKay H, Abbondanza C, Brown M. Estrogen receptor-associated proteins: possible mediators of hormone-induced transcription. Science. 1994;264:1455–1458. doi: 10.1126/science.8197458. [DOI] [PubMed] [Google Scholar]
- Harms MJ, Eick GN, Goswami D, Colucci JK, Griffin PR, Ortlund EA, Thornton JW. Biophysical mechanisms for large-effect mutations in the evolution of steroid hormone receptors. Proc Natl Acad Sci U S A. 2013;110:11475–11480. doi: 10.1073/pnas.1303930110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneghan AF, Berton N, Miura MT, Bain DL. Self-association energetics of an intact, full-length nuclear receptor: the B-isoform of human progesterone receptor dimerizes in the micromolar range. Biochemistry. 2005;44:9528–9537. doi: 10.1021/bi050609i. [DOI] [PubMed] [Google Scholar]
- Hill KK, Roemer SC, Churchill ME, Edwards DP. Structural and functional analysis of domains of the progesterone receptor. Mol Cell Endocrinol. 2012;348:418–429. doi: 10.1016/j.mce.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilser VJ, Thompson EB. Structural dynamics, intrinsic disorder, and allostery in nuclear receptors as transcription factors. J Biol Chem. 2011;286:39675–39682. doi: 10.1074/jbc.R111.278929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SH, Awasthi S, Guo C, Goswami D, Ling J, Griffin PR, Simons SS, Jr, Kumar R. Binding of the N-terminal region of coactivator TIF2 to the intrinsically disordered AF1 domain of the glucocorticoid receptor is accompanied by conformational reorganizations. J Biol Chem. 2012;287:44546–44560. doi: 10.1074/jbc.M112.411330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SH, Ling J, Kumar R. TBP binding-induced folding of the glucocorticoid receptor AF1 domain facilitates its interaction with steroid receptor coactivator-1. PLoS One. 2011;6:e21939. doi: 10.1371/journal.pone.0021939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L, Pan J, Liu YH. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev. 2011;40:1224–1234. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- Kumar R, Betney R, Li J, Thompson EB, McEwan IJ. Induced alpha-helix structure in AF1 of the androgen receptor upon binding transcription factor TFIIF. Biochemistry. 2004;43:3008–3013. doi: 10.1021/bi035934p. [DOI] [PubMed] [Google Scholar]
- Kumar R, McEwan IJ. Allosteric modulators of steroid hormone receptors: structural dynamics and gene regulation. Endocr Rev. 2012;33:271–299. doi: 10.1210/er.2011-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Moure CM, Khan SH, Callaway C, Grimm SL, Goswami D, Griffin PR, Edwards DP. Regulation of the Structurally Dynamic N-terminal Domain of Progesterone Receptor by Protein-induced Folding. J Biol Chem. 2013;288:30285–30299. doi: 10.1074/jbc.M113.491787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Thompson EB. Folding of the glucocorticoid receptor N-terminal transactivation function: dynamics and regulation. Mol Cell Endocrinol. 2012;348:450–456. doi: 10.1016/j.mce.2011.03.024. [DOI] [PubMed] [Google Scholar]
- Landgraf RR, Goswami D, Rajamohan F, Harris MS, Calabrese MF, Hoth LR, Magyar R, Pascal BD, Chalmers MJ, Busby SA, et al. Activation of AMP-Activated Protein Kinase Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry. Structure. 2013 doi: 10.1016/j.str.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, O’Malley BW. Unfolding the action of progesterone receptors. J Biol Chem. 2003;278:39261–39264. doi: 10.1074/jbc.R300024200. [DOI] [PubMed] [Google Scholar]
- Lonard DM, O’Malley BW. Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat Rev Endocrinol. 2012;8:598–604. doi: 10.1038/nrendo.2012.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madauss KP, Grygielko ET, Deng SJ, Sulpizio AC, Stanley TB, Wu C, Short SA, Thompson SK, Stewart EL, Laping NJ, et al. A structural and in vitro characterization of asoprisnil: a selective progesterone receptor modulator. Mol Endocrinol. 2007;21:1066–1081. doi: 10.1210/me.2006-0524. [DOI] [PubMed] [Google Scholar]
- McEwan IJ, Lavery D, Fischer K, Watt K. Natural disordered sequences in the amino terminal domain of nuclear receptors: lessons from the androgen and glucocorticoid receptors. Nucl Recept Signal. 2007;5:e001. doi: 10.1621/nrs.05001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mote PA, Balleine RL, McGowan EM, Clarke CL. Colocalization of progesterone receptors A and B by dual immunofluorescent histochemistry in human endometrium during the menstrual cycle. J Clin Endocrinol Metab. 1999;84:2963–2971. doi: 10.1210/jcem.84.8.5928. [DOI] [PubMed] [Google Scholar]
- Mote PA, Bartow S, Tran N, Clarke CL. Loss of co-ordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast Cancer Res Treat. 2002;72:163–172. doi: 10.1023/a:1014820500738. [DOI] [PubMed] [Google Scholar]
- Obr AE, Edwards DP. The biology of progesterone receptor in the normal mammary gland and in breast cancer. Mol Cell Endocrinol. 2012;357:4–17. doi: 10.1016/j.mce.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal BD, Chalmers MJ, Busby SA, Griffin PR. HD desktop: an integrated platform for the analysis and visualization of H/D exchange data. J Am Soc Mass Spectrom. 2009;20:601–610. doi: 10.1016/j.jasms.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, Novick S, Goswami D, Chalmers MJ, Griffin PR. HDX workbench: software for the analysis of H/D exchange MS data. J Am Soc Mass Spectrom. 2012;23:1512–1521. doi: 10.1007/s13361-012-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press M, Spaulding B, Groshen S, Kaminsky D, Hagerty M, Sherman L, Christensen K, Edwards DP. Comparison of different antibodies for detection of progesterone receptor in breast cancer. Steroids. 2002;67:799–813. doi: 10.1016/s0039-128x(02)00039-9. [DOI] [PubMed] [Google Scholar]
- Raaijmakers HC, Versteegh JE, Uitdehaag JC. The X-ray structure of RU486 bound to the progesterone receptor in a destabilized agonistic conformation. J Biol Chem. 2009;284:19572–19579. doi: 10.1074/jbc.M109.007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem. 2002;277:5209–5218. doi: 10.1074/jbc.M110090200. [DOI] [PubMed] [Google Scholar]
- Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- Simons SS., Jr Glucocorticoid receptor cofactors as therapeutic targets. Curr Opin Pharmacol. 2010;10:613–619. doi: 10.1016/j.coph.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons SS, Jr, Edwards DP, Kumar R. Dynamic Structures of Nuclear Hormone Receptors: New Promises and Challenges. Mol Endocrinol. 2013 doi: 10.1210/me.2013-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetel MJ, Giangrande PH, Leonhardt SA, McDonnell DP, Edwards DP. Hormone-dependent interaction between the amino- and carboxyl-terminal domains of progesterone receptor in vitro and in vivo. Mol Endocrinol. 1999;13:910–924. doi: 10.1210/mend.13.6.0300. [DOI] [PubMed] [Google Scholar]
- Tetel MJ, Jung S, Carbajo P, Ladtkow T, Skafar DF, Edwards DP. Hinge and amino-terminal sequences contribute to solution dimerization of human progesterone receptor. Mol Endocrinol. 1997;11:1114–1128. doi: 10.1210/mend.11.8.9963. [DOI] [PubMed] [Google Scholar]
- Tung L, Abdel-Hafiz H, Shen T, Harvell DM, Nitao LK, Richer JK, Sartorius CA, Takimoto GS, Horwitz KB. Progesterone receptors (PR)-B and -A regulate transcription by different mechanisms: AF-3 exerts regulatory control over coactivator binding to PR-B. Mol Endocrinol. 2006;20:2656–2670. doi: 10.1210/me.2006-0105. [DOI] [PubMed] [Google Scholar]
- Tung L, Mohamed MK, Hoeffler JP, Takimoto GS, Horwitz KB. Antagonist-occupied human progesterone B-receptors activate transcription without binding to progesterone response elements and are dominantly inhibited by A-receptors. Mol Endocrinol. 1993;7:1256–1265. doi: 10.1210/mend.7.10.8123133. [DOI] [PubMed] [Google Scholar]
- Vegeto E, Shahbaz MM, Wen DX, Goldman ME, O’Malley BW, McDonnell DP. Human progesterone receptor A form is a cell- and promoter-specific repressor of human progesterone receptor B function. Mol Endocrinol. 1993;7:1244–1255. doi: 10.1210/mend.7.10.8264658. [DOI] [PubMed] [Google Scholar]
- Wardell SE, Kwok SC, Sherman L, Hodges RS, Edwards DP. Regulation of the amino-terminal transcription activation domain of progesterone receptor by a cofactor-induced protein folding mechanism. Mol Cell Biol. 2005;25:8792–8808. doi: 10.1128/MCB.25.20.8792-8808.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardell SE, Narayanan R, Weigel NL, Edwards DP. Partial agonist activity of the progesterone receptor antagonist RU486 mediated by an amino-terminal domain coactivator and phosphorylation of serine400. Mol Endocrinol. 2010;24:335–345. doi: 10.1210/me.2008-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnmark A, Wikstrom A, Wright AP, Gustafsson JA, Hard T. The N-terminal regions of estrogen receptor alpha and beta are unstructured in vitro and show different TBP binding properties. J Biol Chem. 2001;276:45939–45944. doi: 10.1074/jbc.M107875200. [DOI] [PubMed] [Google Scholar]
- Weigel NL, Beck CA, Estes PA, Prendergast P, Altmann M, Christensen K, Edwards DP. Ligands induce conformational changes in the carboxyl-terminus of progesterone receptors which are detected by a site-directed antipeptide monoclonal antibody. Mol Endocrinol. 1992;6:1585–1597. doi: 10.1210/mend.6.10.1448113. [DOI] [PubMed] [Google Scholar]
- Williams SP, Sigler PB. Atomic structure of progesterone complexed with its receptor. Nature. 1998;393:392–396. doi: 10.1038/30775. [DOI] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YL, Yang X, Ren Z, McDonnell DP, Norris JD, Willson TM, Greene GL. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol Cell. 2005;18:413–424. doi: 10.1016/j.molcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Zhang J, Chalmers MJ, Stayrook KR, Burris LL, Wang Y, Busby SA, Pascal BD, Garcia-Ordonez RD, Bruning JB, Istrate MA, et al. DNA binding alters coactivator interaction surfaces of the intact VDR-RXR complex. Nat Struct Mol Biol. 2011;18:556–563. doi: 10.1038/nsmb.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Goswami D, Wang D, Wang TS, Sen S, Magliery TJ, Griffin PR, Wang F, Schultz PG. An Antibody with a Variable-Region Coiled-Coil “Knob” Domain. Angew Chem Int Ed Engl. 2013 doi: 10.1002/anie.201307939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1, Related to Figure 1. Sequence coverage of full length PR-A and PR-B at different stages of HDX experiments. (a) MSMS sequence coverage of PR-B and PR-A. Letters in red are amino acid sequence detected by analyzing pepsin digested MS/MS spectra using MASCOT. (b) Sequence coverage (%) obtained during MS/MS, 0 second time point injection (t0) and on-exchange experiments of full length and N terminal domain of PR-B and PR-A.
Supplementary Figure S2, Related to Figure 1. Solution state conformational flexibility of agonist bound full length PR-B with different domains indicated by arrow head. The back exchange corrected average % of deuterium incorporation across six different time points (0,10,30,60,300,900 and 3600 sec) are represented is sequence lay out format where each horizontal bar represents each peptide. Colors are according to the color bar at the bottom of the figure.
Supplementary Figure S3, Related to Figure 1. Solution state conformational flexibility of agonist bound full length PR-A with different domains indicated by arrow head. The back exchange corrected average % of deuterium incorporation across six different time points (0,10,30,60,300,900 and 3600 sec) are represented is sequence lay out format where each horizontal bar represents each peptide. Colors are according to the color bar at the bottom of the figure.
Supplementary Figure S4, Related to Figure 4. Solution state conformational flexibility of TBPc. The back exchange corrected average % of deuterium incorporation across six different time points (0,10,30,60,300,900 and 3600 sec) are represented is sequence lay out format where each horizontal bar represents each peptide. Colors are according to the color bar at the bottom of the figure.
Supplementary Figure S5, Related to Figure 4. Differential HDX of isolated N terminal domain (PR-A) showed no perturbation upon interaction with TBPC.