

Abstract
Epilepsy and autism spectrum disorder (ASD) are common comorbidities of one another. Despite the prevalent correlation between the two disorders, few studies have been able to elucidate a mechanistic link. We demonstrate that forebrain specific Tsc1 deletion in mice causes epilepsy and autism-like behaviors, concomitant with disruption of 5-HT neurotransmission. We find that epileptiform activity propagates to the raphe nuclei, resulting in seizure-dependent hyperactivation of mTOR in 5-HT neurons. To dissect whether mTOR hyperactivity in 5-HT neurons alone was sufficient to recapitulate an autism-like phenotype we utilized Tsc1flox/flox;Slc6a4-cre mice, in which mTOR is restrictively hyperactivated in 5-HT neurons. Tsc1flox/flox;Slc6a4-cre mice displayed alterations of the 5-HT system and autism-like behaviors, without causing epilepsy. Rapamycin treatment in these mice was sufficient to rescue the phenotype. We conclude that the spread of seizure activity to the brainstem is capable of promoting hyperactivation of mTOR in the raphe nuclei, which in turn promotes autism-like behaviors. Thus our study provides a novel mechanism describing how epilepsy can contribute to the development of autism-like behaviors, suggesting new therapeutic strategies for autism.
Keywords: mTOR, Autism, Epilepsy, 5-HT, TSC
Introduction
Autism spectrum disorders (ASD) are neurodevelopmental disorders which present with core behavioral deficits including impaired social interactions, repetitive behaviors and restricted interests (1). Epilepsy and autism are common comorbidities of one another, with a striking 30% of patients with autism also having epilepsy (2–4). These data indicate a prominent link between autism and epilepsy, which has led to the hypothesis that the two disorders share common molecular pathologies.
One candidate pathway is the mechanistic target of rapamycin (mTOR), which functions to control neurite outgrowth, neuronal plasticity, myelination, autophagy and cell survival in the brain (5–10). Epilepsy and autism are common clinical manifestations of numerous TORopathies including tuberous sclerosis complex (TSC), focal cortical dysplasia, and hemimegalencephaly, with TSC being among the most well studied. TSC is a developmental disorder arising from loss of function of either hamartin or tuberin (encoded by TSC1 and TSC2 respectively), which complex together to inhibit mTOR signaling (11, 12). Epilepsy is the most common neurological disorder associated with TSC, with a prevalence of 80–90%. The incidence of autism in patients with TSC is approximately 50%, which is strikingly elevated above the general population (13). Furthermore, deletion of another mTOR suppressor, PTEN, has been linked to autism and epilepsy, clinically and in animal models (14–17). Together these data provide compelling evidence for the role of mTOR in causing both epilepsy and ASD, making it an ideal model for studying the relationship between the two disorders. Deletion of Tsc1 in purkinje cells has been shown to result in autism-like behaviors independent of seizure, suggesting a direct role of mTOR in autism (18). However, evidence of cerebellar pathology is found in approximately 25% of patients, presenting a discrepancy between the high incidence of autism (19, 20). Therefore, exploring other possible contributing factors is warranted. One possibility is that uncontrolled seizures also contribute to the development of autism. In support of this are recent findings which demonstrate early onset seizures are associated with development of autism in humans (21). Additionally, antiepileptic drugs have successfully reduced the incidence of autism-like behaviors in a mouse model of Dravet syndrome (22). Moreover, recent studies have shown using chemoconvulsants, including pilocarpine or flurothyl, to induce seizures is sufficient to promote behavioral alterations in rodent models (23, 24). Despite evidence in support of seizures promoting autism-like behaviors, a mechanistic link remains to be elucidated.
One hypothesis is that autism can be caused by dysfunction of the serotonergic system. Corroborating this claim is the observation that altered levels of serotonin in the blood plasma remain one of the few biomarkers for autism (25–27). Moreover, recent findings demonstrate autism-like behaviors are observed in mice following impairment of 5-HT biosynthesis (28, 29), or altered 5-HT reuptake (30).
We report herein that forebrain specific Tsc1 deletions causes autism and epilepsy. Furthermore, the serotonergic system is indirectly modified following recurrent seizure, including a hyperactivation of mTOR in 5-HT neurons. We demonstrate that altered mTOR activity in serotonergic neurons alone is sufficient to recapitulate autism-like behaviors. Thus, we propose a model in which uncontrolled seizure activity promotes changes in serotonergic signaling, via hyperactivation of mTOR in 5-HT neurons, which in turn contributes to the development of autism.
Materials and Methods
Animal Care
Tsc1 mice (31)[from The Jackson Laboratory (Bar Harbor, ME)] were crossed with CaMKIIα-Cre mice [strain T29-1 (Tsien et al., 1996)] or Slc6a4-Cre mice[from Mutant Mouse Region Resource Center (MMRRC) (UC Davis, CA)]. The CaMKIIα-Cre line was received and maintained on a C57BL/6J background. The Tsc1 line was received on a mixed background (C57BL/6J, BALB/cJ, and 129/SvJae) and was backcrossed onto C57BL/J6 for 8 generations in our lab. The Slc6a4-Cre mice were on a C57BL/6J background. Animals were housed in a pathogen-free, temperature- and humidity-controlled facility with a 12 h light cycle (lights on at 7:00 A.M.) and ad libitum access to food and water. All experiments were performed according to the guidelines set by the Animal Care and Use Committee as well as the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Efforts were made to minimize suffering and unnecessary use of animals.
Test Animal Breeding and Housing Conditions
The breeding scheme utilized to generate knockout mice included harems of 2–3 females per male. Cre transmission was accomplished by the male (Tsc1flox/+; CaMKIIα-Cre or Tsc1flox/+;Slc6a4-Cre), and female mice carried the floxed allele (Tsc1flox/flox or Tsc1flox/+). Cages were regularly checked for pregnancy and pregnant females were moved to single housing with bedding and a mouse hut for enrichment. Litters were maintained with the dam until postnatal day 21 when the litter was weaned and separated by gender. Separated litters were housed in groups of 3–5 gender and age matched mice, containing both control and knockout mice. To minimize the effect of environmental variation littermates were compared whenever possible. Two nights, approximately 48 hrs, were allowed to pass between performing any behavioral tests to allow mice to rest.
Behavioral Testing
The 3 chamber tests and marble burying tests were repeated three times per mouse and the average was calculated per individual mouse. Averages per mouse within an experimental group were then averaged. Notably, a novel C57BL/J6 stimulus mouse was presented to each individual test mouse for each repetition of the test. Experiments were performed by the same investigator (J.M.) in the morning between 8:00AM and 12:00PM. Male and female adult mice were tested and were evenly split between groups, such that the following experimental groups were utilized: Figure 1: Control (6M, 5F), Tsc1flox/flox;CaMKIIα-cre (6M, 4F), Figure 6: Control (5M, 5F), Tsc1flox/flox;Slc6a4-cre (6M, 5F), Figure 8: Control (6M, 4F), Tsc1flox/flox;Slc6a4-cre (5M, 5F), Tsc1flox/flox;Slc6a4-cre + Rapamycin (5M,6F), Tsc1flox/flox;Slc6a4-cre + Fluoxetine (5M,5F). Exact details on the litters used can be found in supplemental information (Supplemental Table 1). Video recording was utilized for the three chamber test with subsequent blinding prior to analysis. Marble burying tests were scored immediately. Following testing mice were returned to their home cages.
Figure 1.

Tsc1;CaMKIIα-cre Mice Develop Core Autism-like Behaviors. (a–b) Fraction of time control and Tsc1;CaMKII-cre mice spent in the animal (black bars), middle (white bars), and object (gray bars) chambers in the three chamber sociability test. (c) Amount of time the test mouse spent actively approaching the caged mouse. (d) Number of times the test mouse crossed chambers (e) Fraction of marbles buried by control (black bars) and Tsc1;CaMKII-cre (white bars) mice in a marble burying test. a.u. indicates arbitrary units, n=10–11 * indicates p<0.05, ** indicates p<0.01 by ANOVA or Students t-test.
Figure 6.

Tsc1;Slc6a4-cre Mice Recapitulate Autism-like behaviors. (a)Representative image of the DRN of Slc6a4-cre;eGFP mice stained with 5-HT. (b) Representative P-s6 and total s6 western blot. (c) Quantification of P-s6/s6. (d–e) Fraction of time control and Tsc1;Slc6a4-cre mice spent in the animal (black bars), middle (white bars), and object (gray bars) chambers in the three chamber sociability test. (f) Amount of time the test mouse spent actively approaching the caged mouse. (g) Number of times the test mouse crossed chambers (h) Fraction of marbles buried by control (black bars) and Tsc1;Slc6a4-cre (white bar) mice in a marble burying test. a.u. indicates arbitrary units, n=3–5 (immunostaining and western) n=10–11 (behavior) * indicates p<0.05, ** indicates p<0.01 by ANOVA or Students t-test.
Figure 8.

Pharmacological mTOR Inhibition is Sufficient to Rescue Altered Behaviors. (a) Representative P-s6 and total s6 western blot in control or Tsc1;Slc6a4-cre with vehicle or rapamycin treatment. (b) Quantification of P-s6/s6. (c–f) Fraction of time spent in each chamber in the three chamber test by control, Tsc1;Slc6a4-cre, Tsc1;Slc6a4-cre + Rapamycin, and Tsc1;Slc6a4-cre + fluoxetine. (g) Amount of time the test mouse spent actively approaching the caged mouse. (h) Fraction of marbles buried by control, Tsc1;Slc6a4-cre, Tsc1;Slc6a4-cre + Rapamycin, and Tsc1;Slc6a4-cre + fluoxetine. a.u. indicates arbitrary units, n=10–11 * indicates p<0.05, ** indicates p<0.01 by ANOVA.
The Crawley 3 chamber test for social preference was performed as previously described (32). Briefly, a test mouse was placed into the center chamber of a three chamber system and allowed to explore for 5 min. At this time an empty cage and a cage containing a gender and age matched mouse of equal or lesser body weight were placed into opposite chambers (the sides were systematically switched to avoid a side preference bias). The caged mouse was on a C57BL/6J background. The test mouse was allowed to freely move between chambers during a 10 min exposure, and the number of times the animal moved from one chamber to another was recorded. Mice were counted to be in one chamber when all four limbs entered that chamber. Social approach was determined by the time a test mouse spent actively interacting (sniffing and nose pokes) with the caged mouse.
The marble burying test was used to assay repetitive behaviors (33) and was performed by placing mice into 30cmx 30cm chambers containing 5cm of corn cob bedding, with 20 marbles spaced evenly in a 5×4 grid. Mice were then placed into the center of the chamber and allowed to freely move about the chamber for a 20 minute period. After 20 minutes mice were immediately removed and the buried marbles were counted. Only marbles that were entirely buried were counted.
8-OH-DPAT Induced Hypothermia
Basal temperature was taken rectally 5 and 10 minutes prior to drug injection with a rectal probe (RET3; ThermoWorks) connected to a thermometer (Microtherma 2T; ThermoWorks). DPAT was administered by subcutaneous injection at 0.1 mg/kg in saline. Immediately following injection temperature was taken at time 0. Subsequently temperature was taken every 10 minutes over a 60 minute period.
DOI Injection and Head Twitch Assessment
Mice were habituated to a clear test chamber for 10 minutes. DOI was diluted in saline and injected intraperitoneal (i.p.) at 1 mg/kg. Mice were then placed into a chamber and recorded with a digital camera for 30 minutes. Videos were scored blindly by trained researchers. Head twitches were defined as a quick, distinct and clear back and forth movement of the head. The number of head twitch events were counted between 10 and 20 minutes after injection of DOI.
Genotyping
DNA extraction was accomplished by digestion of 2mm mouse tail snips in 500 μl of digestion buffer (50 mM KCl, 10 mM Tris-HCl, 2.5 mM MgCl2, 0.45% v/v NP-40, and 0.45% v/v Tween 20) with 100 μg/ml proteinase K at 56°C overnight. On the next day, the samples were heated to 95°C for 5 min. PCR was performed using the following primers: Tsc1 [detects wild-type (WT) and mutant] sense 5′-gtcacgaccgtaggagaagc, antisense 5′-gaatcaaccccacagagcat; and Cre sense 5′-gcatttctggggattgtta, antisense 5′-cccggcaaaacaggtagtta.
Fixation and Immunohistochemistry
Mice were anesthetized by i.p. injection of pentobarbital (30 mg/kg) supplemented with inhalation of isoflurane. Mice were first perfused with 30 ml of PBS and subsequently perfused with 4% paraformaldehyde. Brains were removed and held in 4% paraformaldehyde for 24 h at 4°C and transferred into 30% sucrose solution at 4°C until they were completely submerged. Brain tissues were then frozen and sectioned into 40 μm slices using a cryostat and stored at 4°C in PBS with sodium azide. For staining, slides were blocked with 5% BSA with 0.25% Triton X-100 for 1 hour at room temperature (RT). Slices were incubated in a primary antibody overnight at 4°C [anti-phospho-S6 (Cell Signaling); anti-SerT and anti-5-HT(Immunostar)]. Slices were incubated with secondary antibody for 45 min at RT and mounted in fluoromount G. C-fos staining was performed using the ABC elite kit (Vector Labs), and was performed as follows: Endogenous peroxidase activity was quenched in 1% H2O2 in water. Slices were blocked in BSA with triton and incubated in primary antibody (1:10,000, Millipore). Subsequently, slices were incubated in biotinylated anti-rabbit secondary (1:200, Vector) followed by ABC reagent. DAB was used to visualize staining and slides were dehydrated in an ethanol gradient, followed by xylene. Mounting was performed using permount. ImageJ was used for image analysis.
Western blot
Mice were sacrificed, and the brains were removed and sliced into 2mm thick sections in a stainless steel matrix. Similar anatomic regions were identified, and a 2 mm-diameter hole punch was used to isolate two punches from the desired regions for each animal. Isolated tissues were homogenized in lysis buffer consisting of 50 mM Tris, pH 7.4, 2 mM EDTA, and a proteinase inhibitor set (Roche). The lysates were then mixed with equal volumes of 2× lithium dodecyl sulfate sample buffer composed of 20% β-mercaptoethanol, 20 mM NaF, 20 mM of Na3VO4, and proteinase inhibitor and heated at 95°C for 5 min. Insoluble cell debris was removed by centrifugation at 10,000 × g for 10 min. Protein samples were resolved in an 8% Bis-Tris gel in MES buffer (50 mM MES, 50 mM Tris-HCl, 1 mM EDTA, and 0.1% SDS) and transferred to a 0.45 μm nitrocellulose membrane in transfer buffer (25 mM Tris, 200 mM glycine, and 20% methanol). Membranes were subsequently blocked in 5% nonfat dry milk in TBST (25 mM Tris-HCl, pH 7.4, 1.5 M NaCl, and 0.05% Tween 20) for 1 hour at RT and incubated with rabbit anti-S6, anti-phospho S6(Ser235/236), or anti-GAPDH antibodies (Cell Signaling Inc.) at 1:1000 dilution at 4°C overnight. Membranes were washed in TBST and incubated with an anti-rabbit HRP-conjugated secondary antibody (1:10,000) in 5% milk in TBST for 1 h at RT. Membranes were washed in TBST, followed by a final wash in TBS. Signals were visualized with ECL reagent (Pierce Chemical) and an LAS-4000 luminescent image analyzer (Fujifilm). ImageJ software was used to subtract background and to perform densitometry.
Video/EEG recording of spontaneous seizures
Spontaneous seizure activities were detected by continuous video monitoring and simultaneous EEG monitoring of Tsc1flox/flox;CaMKIIα-cre mice from 8:00 P.M. until 8:00 A.M., starting at 3–4 weeks of age. 12 hour long video monitoring sessions were administered 3–4 times per week. For EEG surgery mice were sedated using isoflurane inhalation and placed into a stereotaxic chamber. Two electrodes were linked to stainless steel surgical screws (J.I. Morris) which were implanted at AP −1.8, ML 1.8 (left and right) and served as differential recording electrodes. For deep recordings from the dorsal raphe nuclei an additional electrode was lowered into position using stereotaxic coordinates from lambda (AP −0.5, ML 0.0 DV −3.5). A third screw was placed into the skull serving as a ground. Electrodes were held in place using dental cement (Harvard Apparatus). EEG recordings were performed in free-moving mice hooked to a Pinnacle 8200 EEG system (Pinnacle Technology). EEG data were analyzed with the Sirenia Seizure program (Pinnacle Technology), and seizures were manually identified by characteristic high-frequency and high-amplitude firing. Additionally, most seizures were followed by brief postictal suppression. All electrographic seizures were verified behaviorally by video recording. Videos were captured using DVR video surveillance software. Of note, mice used for EEG/video monitoring consisted of a distinct cohort from those used for behavioral testing. Behavioral testing on Tsc1flox/flox;CaMKIIα-cre mice was performed following the identification of spontaneous seizures. Briefly, seizures were detected during routine handling, or during a brief video monitoring sessions lasting less than 4 hours. Mice which were not observed to display seizures were excluded from future studies.
Electrophysiology
The excitability of serotonergic neurons in DRN (dosal raphe nuclei) in the brainstem of Tsc1flox/flox;CaMKIIα-cre and control (Tsc1flox/flox)mice was examined via electrophysiological recording. Briefly, following rapid decapitation, brains were removed and blocked in cold sucrose-substituted artificial cerebrospinal fluid (aCSF). Brainstem at 0 to 1.5 mm caudal to the lambda, which contains the DRN, was cut in coronal sections at 200-μm thickness using a vibratome at ice-cold temperature. The slices were maintained in aCSF at room temperature for 1 hr before recording. Neurons were visualized with a microscope equipped for near-IR differential interference contrast. We performed cell-attached extracellular recordings using voltage-clamp. The spontaneous, basal firing rate was recorded and measured in the presence of 3 μM phenylephrine hydrochloride and 40 μM L-tryptophan in all solutions to maintain the spontaneous ring rate. 5-HT1A-mediated suppression of excitability (basal firing) is a feature of serotonergic neurons. Accordingly, following recording of basal firing, slices were further perfused with 5-HT at various concentrations.
Drug Treatment
For rescue experiments vehicle or rapamycin was given to adult Tsc1;Slc6a4-cre mice via i.p. injection at 5 mg/kg diluted to 1 mg/ml in 4%EtOH and 5% Tween-20 from a stock dilution in DMSO. Rapamycin was given daily for 3 days and then every other day for the duration of the testing. Behavioral tests were conducted starting on the fourth day of rapamycin treatment and were performed on days when animals did not receive injections, or prior to injections to avoid effects of stress related to the injection. Fluoxetine was added to the drinking water of mice at 77mg/L as previously described(29) and behavioral testing began 7 days after fluoxetine treatment began.
Statistical analysis
Statistics were performed using GraphPad Prism and included t tests for comparisons between two groups and one-way ANOVA with a Tukey’s post hoc test for three or more groups.
Results
Forebrain Tsc1 Deletion Causes Epilepsy and Autism-like Behaviors in Mice
To identify whether autism-like behaviors were present in a mouse model with spontaneous seizures we utilized a conditional Tsc1flox/flox;CaMKIIα-cre line. This line has been previously characterized to manifest severe recurrent spontaneous seizures, but has not been reported to show autism-like behaviors (34). Seizures in Tsc1flox/flox;CaMKIIα-cre were severe and progressive starting as early as week 3–4 (Supplemental Figure 1). All Tsc1flox/flox;CaMKIIα-cre mice were confirmed to develop behavioral seizures by 6 weeks of age via video recording and routine monitoring. Mice identified to display spontaneous seizures were subject to behavioral testing starting at 4–6 weeks of age. We found Tsc1flox/flox;CaMKIIα-cre mice showed a loss of social preference in the three chamber sociability test, without displaying any significant change in locomotor activity as assayed by chamber crosses (Figures 1a–d). Tsc1flox/flox;CaMKIIα-cre mice also displayed significantly increased repetitive behaviors as assayed by the marble burying test (Figure 1e). These results demonstrate that Tsc1flox/flox;CaMKIIα-cre mice recapitulate the core behavioral changes associated with ASD.
Serotonergic Signaling is Disrupted in Tsc1flox/flox;CaMKIIα-cre Mice
As alterations of 5-HT signaling have been demonstrated to induce autism-like behaviors in mice (28–30), we next determined whether 5-HT signaling was disrupted in the Tsc1flox/flox;CaMKIIα-cre model. First, we performed immunohistostaining for the serotonin transporter (SerT) to assay SerT projections in the prefrontal cortex region. Tsc1flox/flox;CaMKIIα-cre mice displayed a profound loss of SerT projections (Figure 2a, b), indicating either a reduction of cortical projections or depletion of SerT expression. We also detected a significant reduction in the levels of 5-HT in the cortex (Figure 2c). Next we monitored for changes in the serotonergic system by evaluating the response of mice to 5-HT agonists. DOI is an agonist which has been shown to elicit a dose dependent head twitch response via activation of the 5-HT2C receptors (35, 36). We found Tsc1flox/flox;CaMKIIα-cre mice displayed a significant increase in head twitches (Figure 2d). Tsc1flox/flox;CaMKIIα-cre mice also displayed an exaggerated hypothermic response following administration of the 5-HT1A agonist 8-OH-DPAT (Figure 2e). Together these data show a pronounced alteration of the serotonergic system. To distinguish whether reduced 5-HT levels reflected altered serotonergic excitability we performed whole cell electrophysiology in brain slices to assay intrinsic changes in firing properties of serotonergic neurons. 5-HT neurons of Tsc1flox/flox;CaMKIIα-cre mice displayed a reduced threshold for excitation and an increased rate of firing compared to controls (Figures 2f–h).
Figure 2.

Disruption of the 5-HT System in Tsc1;CaMKIIα-cre Mice (a) Serotonin transporter (SerT) staining in the cortex of control and Tsc1;CaMKII-cre mice. (b) Relative expression levels of SerT in the cortex of control and Tsc1;CaMKII-cre mice. (c) Levels of 5-HT in cortical tissue by HPLC in control and Tsc1;CaMKII-cre mice. (d) DOI (1 mg/kg, i.p.) induced head twitch response in control and Tsc1;CaMKII-cre mice. (e) Hypothermic response to 8-OH-DPAT (0.1 mg/kg, s.c.) in control and Tsc1;CaMKII-cre mice. (f) Firing rate of control and Tsc1;CaMKII-cre mice when held at a constant resting membrane potential (RMP) of 55 mV. (g–h) Representative firing traces and quantification of control and Tsc1;CaMKII-cre mice during constant current injection. n=3–7 * indicates p<0.05, ** indicates p<0.01 by Students t-test.
Seizure-dependent Hyperactivation of mTOR Occurs in the Raphe Nuclei
We next sought to determine what molecular changes were occurring in 5-HT neurons. One possibility was that forebrain Tsc1 deletion was driving an increase in mTOR activity. To test this we looked at levels of phosphorylated s6 (P-s6), a marker of mTOR activity. We find P-s6 accumulation occurs in serotonergic neurons of Tsc1flox/flox;CaMKIIα-cre mice, as identified by costaining for 5-HT and P-s6 (Figure 3a). Western blot of lysates from the raphe nuclei confirmed a significant increase in the levels of P-s6 compared to controls (Figures 3b and c). To delineate whether the disinhibition of mTOR was direct (a cell-autonomous response to Tsc1 deletion in 5-HT neurons) or indirect, we utilized CaMKIIα-cre;GFPstoplox reporter mice. We observed GFP expression in the forebrain (Data Not Shown), but GFP was absent from neurons in the raphe (Figure 4a), suggesting that mTOR activation in serotonergic neurons is not directly from cell-autonomous Tsc1 deletion. To confirm the increase in P-s6 was due to the development of seizures we dissected the temporal expression of mTOR hyperactivation in the raphe as it related to the onset of seizures in Tsc1flox/flox;CaMKIIα-cre mice. We observed that in mice 3–4 weeks of age, in which Tsc1 deletion has occurred but no overt seizure phenotype had yet developed, we found no significant hyperactivation of mTOR in the raphe (Figure 4b and c). When P-s6 was monitored in Tsc1flox/flox;CaMKIIα-cre mice who had previously developed seizures we observed a significant increase in P-s6 in the raphe (Figure 4b and c). These findings support the notion that seizures are a major contributor to hyperactivation of mTOR in 5-HT neurons.
Figure 3.

Hyperactivation of mTOR Occurs in Serotonergic Neurons Following Repeated Seizure (a) Co-immunofluorescent staining for phospo-s6 (P-s6) and 5-HT in the dorsal raphe nuclei (DRN) of control and Tsc1;CaMKII-cre. (b) Representative western blot of P-s6 and total s6. (c) Quantification of P-s6 in the raphe nuclei. a.u. indicates arbitrary units, n=3–4 * indicates p<0.05, ** indicates p<0.01 by Students t-test.
Figure 4.

Tsc1 Deletion Does Not Occur in the DRN and is Not the Primary Cause of Hyperactivation of mTOR in the DRN. (a) Representative image showing a lack of Cre driven GFP expression in the DRN of CaMKII-cre;eGFP mice. (b) Representative western blot for P-s6 in Tsc1;CaMKII-cre prior to the onset of seizure (upper panel) and after repeated seizure episodes (bottom panel). (c) Quantification of P-s6 expression in (b). a.u. indicates arbitrary units, n=3–4 ** indicates p<0.01 by Students t-test.
Recurrent Seizures Propagate to the Brainstem
In Tsc1flox/flox;CaMKIIα-cre mice seizures presumably originate in the forebrain, the site of Tsc1 deficient neurons. As seizure events have been demonstrated to induce mTOR activation, one possibility is that epileptiform activity spreads to the brainstem, leading to mTOR hyperactivity (37–39). We performed c-fos immunostaining, as a readout of neuronal activity, and found a striking increase in c-fos accumulation in the raphe (Figure 5a). In order to confirm epileptiform activity occurred in the brainstem we implanted recording electrodes in the cortex and dorsal raphe nuclei of Tsc1flox/flox;CaMKIIα-cre mice. EEG showed initial ictal events were detected in the cortex (Figure 5b) and not in the brainstem. Following repeated severe seizures, epileptiform activity was also detected in the brainstem (Figure 5b). Thus brainstem epileptiform events correlate well with aberrant mTOR activation in the raphe.
Figure 5.
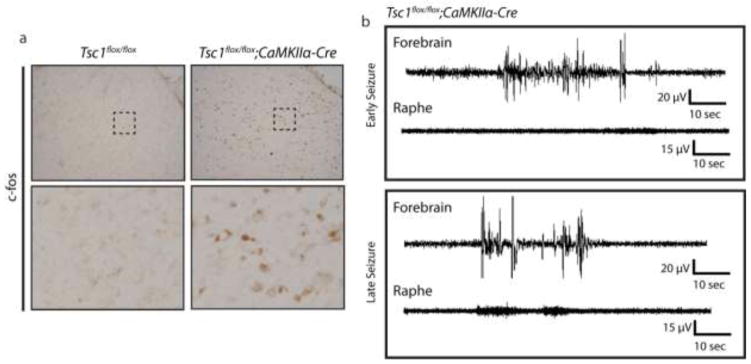
Tsc1;CaMKII-cre Mice Display Ictal Events in the Raphe Nucleus Following Repeated Seizure. (a) c-fos staining in the DRN from control and Tsc1;CaMKII-cre mice. (b) Representative forebrain (cortical) and raphe EEG traces during an early and late seizure event. n=4–5
Hyperactivation of mTOR in 5-HT Neurons is Sufficient to Promote Autism-like Behaviors
To elucidate whether hyperactivation of mTOR in serotonergic neurons alone was sufficient to promote autism-like behaviors we generated Tsc1flox/flox;Slc6a4-cre mice, in which disinhibition of mTOR occurs selectively in serotonergic neurons. Slc6a4-cre;GFPstoplox reporter mice displayed GFP expression restricted to 5-HT positive neurons in the raphe nucleus, indicating that Slc6a4-cre-mediated deletion of Tsc1 occurs selectively in serotonergic neurons (Figure 6a). Increased P-s6 levels were observed in the raphe nuclei of Tsc1flox/flox;Slc6a4-cre mice, confirming mTOR hyperactivation (Figure 6b–c). Importantly Tsc1flox/flox;Slc6a4-cre mice were not observed to develop spontaneous seizures. In the 3 chamber test Tsc1flox/flox;Slc6a4-cre mice show a loss of social preference (Figure 6d–g). Tsc1flox/flox;Slc6a4-cre mice also showed increased repetitive behaviors in the marble burying test (Figure 6h). To ascertain whether perturbation of the serotonergic system occurred in Tsc1flox/flox;Slc6a4-cre mice, we performed SerT immunostaining, electrophysiological recording and agonist challenge using 8-OH-DPAT and DOI. We found that Tsc1flox/flox;Slc6a4-cre mice did not display a marked decrease in serotonergic SerT projections in the cortex (Figure 7a–b), suggesting that cortical seizures may contribute to the loss of SerT projections seen in Tsc1flox/flox;CaMKIIα-cre mice. Additionally, we did not see a significant change in firing rates of 5-HT neurons in Tsc1flox/flox;Slc6a4-cre (data not shown). However, Tsc1flox/flox;Slc6a4-cre mice did display exaggerated responses to both 8-OH-DPAT induced hypothermia and DOI head twitch response (Figure 7c and d). Furthermore, 5-HT levels were significantly reduced within the cortex of Tsc1flox/flox;Slc6a4-cre mice, when compared to controls (Figure 7e). Therefore, hyperactivation of mTOR in 5-HT neurons is sufficient to disrupt the serotonergic system and cause autism-like behaviors.
Figure 7.

Disruption of the Serotonergic System in Tsc1;Slc6a4-cre Mice. (a) Serotonin transporter (SerT) staining in the cortex of control and Tsc1;Slc6a4-cre mice. (b) Relative expression levels of SerT in the cortex of control and Tsc1;Slc6a4-cre mice. (c) Hypothermic response to 8-OH-DPAT (0.1 mg/kg, s.c.) in control and Tsc1;Slc6a4-cre mice. (d) DOI (1 mg/kg, i.p.) induced head twitch response in control and Tsc1;Slc6a4-cre mice. (e) Levels of 5-HT in cortical tissue by HPLC in control and Tsc1;Slc6a4-cre mice. n=5–7 * indicates p<0.05, ** indicates p<0.01 by Students t-test.
We next determined whether either pharmacological inhibition of mTOR or SSRI treatment were sufficient to attenuate autism-like behaviors. To test this we first treated Tsc1flox/flox;Slc6a4-cre mice with vehicle, rapamycin (mTOR inhibitor) or fluoxetine (serotonin reuptake inhibitor). Rapamycin was successful in reducing mTOR hyperactivtion (Figure 8a and b). Behavioral tests revealed that rapamycin treatment was sufficient to attenuate changes in social preference and in marble burying. Fluoxetine on the other hand, displayed only a marginal, and statistically insignificant improvement in the social preference test (Figure 8c–g). However, fluoxetine was able to fully rescue from repetitive behaviors (Figure 8h). This data suggests that, in certain conditions, pharmacologic intervention with rapamycin or analogs may be beneficial in reversing the core behavioral symptoms associated with autism.
Discussion
Herein we report the Tsc1flox/flox;CaMKIIα-cre mouse model of TSC previously characterized to have spontaneous seizures also displays autism-like behaviors. We observe pronounced alterations of the 5-HT system, including loss of cortical 5-HT, exaggerated response to 5-HT agonists, and hyperactivation of mTOR in 5-HT neurons. Furthermore, we show evidence that hyperactivation of mTOR in 5-HT neurons is not caused directly by deletion of Tsc1 in our model. Rather, based on the detection of abnormal EEG signal and induction of c-fos in the brain stem, we demonstrate that the spread of epileptiform events into the raphe nuclei is the primary cause of elevated mTOR signaling. Moreover, hyperactivation of mTOR targeted to serotonergic neurons was sufficient to recapitulate autism-like behaviors in the absence of seizures, indicating that hyperactivation of mTOR in 5-HT neurons is sufficient to cause changes in 5-HT signaling and autism-like behaviors. Thus, we demonstrate that epilepsy-induced mTOR hyperactivation in the raphe promotes autism-like behavior.
The association between autism and epilepsy has been well documented, with recent findings showing 30% of patients with autism also have epilepsy. Furthermore, as many as 85% of patients with autism have been reported to display abnormal EEG patterns, including those who do not display overt epilepsy (40). Yet the mechanisms underlying these correlations remain largely enigmatic. Recent studies have begun to elucidate evidence supporting a causal role of epilepsy in the development of autism. For example, a clinical study demonstrates that early onset of epilepsy is correlated with a substantial increase in the likelihood of developing autism later in life (21). Additionally, in a mouse model of dravet syndrome the use of antiepileptic drugs has been demonstrated to reverse autistic-like behaviors in epileptic mice. Furthermore, a rodent status epilepticus model of acquired epilepsy also demonstrated reduced social behaviors (23). Recent reports also show pharmacological inhibition of either AMPAR or mTOR can prevent autism-like behaviors following neonatal seizures (41, 42). These finding are of particular interest as seizures precede changes in social behavior, allowing for dissection of cause and effect.
Due to the high incidence of autism and epilepsy in TORopathies, the mTOR pathway is often implicated as a likely common mechanism. Clinical conditions and mouse models causing loss of function of upstream inhibitors of mTOR, including TSC and PTEN, have been shown to develop epilepsy and/or autism-like behaviors (14–16, 18, 31, 34, 43–45). Despite this, in patients with TSC it remains unclear whether epilepsy is a major contributor to the development of autism. Our unique approach utilizes a Tsc1flox/flox;CaMKIIα-cre model, in which the direct effects of Tsc1 are primarily confined to the forebrain, allowing for dissection of indirect, seizure-dependent modifications to other brain regions. However, we are unable to discriminate in the Tsc1flox/flox;CaMKIIα-cre model whether seizure-dependent modifications are necessary to promote autism-like behaviors, or conversely, whether cell-autonomous deletion of Tsc1 in forebrain neurons contributes to the behavioral alterations. In order to address whether the seizure-dependent hyperactivation of mTOR in the raphe is sufficient to drive autism-like behaviors, we utilize 5-HT neuron selective Tsc1flox/flox ;Slc6a4-Cre mice. Tsc1flox/flox ;Slc6a4-Cre mice recapitulate hyperactivation of mTOR in the raphe in the absence of seizures.
Our data support that recurrent seizures promote pathogenic modifications to the brainstem. This is of particular importance as other generalized seizure disorders, with etiologies lying outside of mTOR disinhibition, may similarly cause aberrant changes to the raphe. Supporting this is a recent paper that discovered that pilocarpine induced status epilepticus resulted in a long term dramatic reduction of 5-HT and loss of serotonergic neurons (46). Moreover, the pilocarpine model of temporal lobe epilepsy has also been shown to induce altered social behaviors and aggressive behaviors (23, 47). These findings suggest the possibility of a more general role of seizure-dependent raphe modulation in autism. Clinically it has been observed that drug resistant and severe cases of epilepsy are more commonly associated with autism spectrum disorders (48–50). This is consistent with what we observed in our Tsc1flox/flox;CaMKIIα-cre model, in which mice tend to develop frequent and generalized seizures. Clinical evidence also supports the notion that serotonergic signaling is altered in cases of both autism and epilepsy (51, 52). Furthermore, dietary restriction of tryptophan, a 5-HT precursor, can worsen autism (53). Additionally, serotonin has recently been shown to be a major contributor to social reward (54). Taken together, previous studies provide precedent for implicating seizures in modifying 5-HT signaling and in causing autism.
Identifying molecular mechanisms through which seizures are capable of causing autism is vital to the identification of novel therapeutics which may benefit patients who suffer from epilepsy, autism or both. Importantly, in our model pharmacological intervention via rapamycin post-onset of the autism-like phenotype was sufficient to reverse the behavioral alterations, indicating it may hold therapeutic value in reversing autism behaviors. Fluoxetine proved efficacious in reducing repetitive behaviors, but was not significantly effective at improving social behavior. This is congruent with previous reports finding beneficial effects of SSRI treatment in patients and mice in alleviating specific autism-like behaviors (55–57), although the efficacy of SSRIs clinically is still under debate. Future preclinical and clinical studies addressing the efficacy of mTOR inhibitors in the treatment, prevention and reversal of epilepsy and/or autism will be essential in validating mTOR as a therapeutic target.
Supplementary Material
Highlights.
Tsc1 deletion in neurons causes epilepsy and autism-like behaviors in mice
Epileptiform activity spreads to the brainstem
mTOR becomes hyperactivated in 5-HT neurons following seizure onset
mTOR hyperactivity in 5-HT neurons causes autism behaviors
Autism-like behaviors can be reversed following treatment with rapamycin
Acknowledgments
This work was supported by the National Institutes of Health by the grant R01NS062068-04 (to Y.H.)
Abbreviations
- ASD
autism spectrum disorders
- TSC
tuberous sclerosis complex
- mTOR
mechanistic target of rapamycin
- PTEN
phospatase and tensin homolog
- 5-HT
5-hydroxytryptophan
- SerT/Slc6a4
serotonin transporter
- CaMKIIα
Ca2+/calmodulin-dependent protein kinase II alpha
- DRN
dorsal raphe nuclei
- 8-OH-DPAT
8-Hydroxy-DPAT hydrobromide
- DOI
2,5-Dimethoxy-4-iodoamphetamine
- EEG
electroencephalography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Happe F, Ronald A. The ‘fractionable autism triad’: a review of evidence from behavioural, genetic, cognitive and neural research. Neuropsychology review. 2008;18(4):287–304. doi: 10.1007/s11065-008-9076-8. [DOI] [PubMed] [Google Scholar]
- 2.Parmeggiani A, et al. Epilepsy and EEG paroxysmal abnormalities in autism spectrum disorders. Brain & development. 2010;32(9):783–789. doi: 10.1016/j.braindev.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Viscidi EW, et al. Clinical characteristics of children with autism spectrum disorder and co-occurring epilepsy. PloS one. 2013;8(7):e67797. doi: 10.1371/journal.pone.0067797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ekinci O, Arman AR, Isik U, Bez Y, Berkem M. EEG abnormalities and epilepsy in autistic spectrum disorders: clinical and familial correlates. Epilepsy & behavior : E&B. 2010;17(2):178–182. doi: 10.1016/j.yebeh.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 5.Grider MH, Park D, Spencer DM, Shine HD. Lipid raft-targeted Akt promotes axonal branching and growth cone expansion via mTOR and Rac1, respectively. Journal of neuroscience research. 2009;87(14):3033–3042. doi: 10.1002/jnr.22140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24(28):6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang SJ, et al. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(1):467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29(21):6860–6870. doi: 10.1523/JNEUROSCI.0232-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meikle L, et al. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28(21):5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nature genetics. 2004;36(6):585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 11.van Slegtenhorst M, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277(5327):805–808. doi: 10.1126/science.277.5327.805. [DOI] [PubMed] [Google Scholar]
- 12.European Chromosome 16 Tuberous Sclerosis C. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75(7):1305–1315. doi: 10.1016/0092-8674(93)90618-z. [DOI] [PubMed] [Google Scholar]
- 13.Jeste SS, Sahin M, Bolton P, Ploubidis GB, Humphrey A. Characterization of autism in young children with tuberous sclerosis complex. Journal of child neurology. 2008;23(5):520–525. doi: 10.1177/0883073807309788. [DOI] [PubMed] [Google Scholar]
- 14.Takeuchi K, et al. Dysregulation of synaptic plasticity precedes appearance of morphological defects in a Pten conditional knockout mouse model of autism. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(12):4738–4743. doi: 10.1073/pnas.1222803110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwon CH, et al. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50(3):377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Backman SA, et al. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nature genetics. 2001;29(4):396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- 17.Goffin A, Hoefsloot LH, Bosgoed E, Swillen A, Fryns JP. PTEN mutation in a family with Cowden syndrome and autism. American journal of medical genetics. 2001;105(6):521–524. doi: 10.1002/ajmg.1477. [DOI] [PubMed] [Google Scholar]
- 18.Tsai PT, et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488(7413):647–651. doi: 10.1038/nature11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eluvathingal TJ, et al. Cerebellar lesions in tuberous sclerosis complex: neurobehavioral and neuroimaging correlates. Journal of child neurology. 2006;21(10):846–851. doi: 10.1177/08830738060210100301. [DOI] [PubMed] [Google Scholar]
- 20.Vaughn J, et al. MRI characterization and longitudinal study of focal cerebellar lesions in a young tuberous sclerosis cohort. AJNR American journal of neuroradiology. 2013;34(3):655–659. doi: 10.3174/ajnr.A3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berg AT, Plioplys S, Tuchman R. Risk and correlates of autism spectrum disorder in children with epilepsy: a community-based study. Journal of child neurology. 2011;26(5):540–547. doi: 10.1177/0883073810384869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han S, et al. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012;489(7416):385–390. doi: 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marin JC, et al. Temporal lobe epilepsy and social behavior: an animal model for autism? Epilepsy & behavior : E&B. 2008;13(1):43–46. doi: 10.1016/j.yebeh.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Lugo JN, Swann JW, Anderson AE. Early-life seizures result in deficits in social behavior and learning. Experimental neurology. 2014;256:74–80. doi: 10.1016/j.expneurol.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mulder EJ, et al. Platelet serotonin levels in pervasive developmental disorders and mental retardation: diagnostic group differences, within-group distribution, and behavioral correlates. Journal of the American Academy of Child and Adolescent Psychiatry. 2004;43(4):491–499. doi: 10.1097/00004583-200404000-00016. [DOI] [PubMed] [Google Scholar]
- 26.Anderson GM, et al. Whole blood serotonin in autistic and normal subjects. Journal of child psychology and psychiatry, and allied disciplines. 1987;28(6):885–900. doi: 10.1111/j.1469-7610.1987.tb00677.x. [DOI] [PubMed] [Google Scholar]
- 27.Leboyer M, et al. Whole blood serotonin and plasma beta-endorphin in autistic probands and their first-degree relatives. Biological psychiatry. 1999;45(2):158–163. doi: 10.1016/s0006-3223(97)00532-5. [DOI] [PubMed] [Google Scholar]
- 28.Kane MJ, et al. Mice genetically depleted of brain serotonin display social impairments, communication deficits and repetitive behaviors: possible relevance to autism. PloS one. 2012;7(11):e48975. doi: 10.1371/journal.pone.0048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dzirasa K, Kumar S, Sachs BD, Caron MG, Nicolelis MA. Cortical-amygdalar circuit dysfunction in a genetic mouse model of serotonin deficiency. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33(10):4505–4513. doi: 10.1523/JNEUROSCI.4891-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veenstra-VanderWeele J, et al. Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(14):5469–5474. doi: 10.1073/pnas.1112345109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meikle L, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27(21):5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang M, Clarke AM, Crawley JN. Postnatal lesion evidence against a primary role for the corpus callosum in mouse sociability. The European journal of neuroscience. 2009;29(8):1663–1677. doi: 10.1111/j.1460-9568.2009.06714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas A, et al. Marble burying reflects a repetitive and perseverative behavior more than novelty-induced anxiety. Psychopharmacology. 2009;204(2):361–373. doi: 10.1007/s00213-009-1466-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McMahon J, et al. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32(45):15704–15714. doi: 10.1523/JNEUROSCI.2392-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vickers SP, et al. Modulation of 5-HT(2A) receptor-mediated head-twitch behaviour in the rat by 5-HT(2C) receptor agonists. Pharmacology, biochemistry, and behavior. 2001;69(3–4):643–652. doi: 10.1016/s0091-3057(01)00552-4. [DOI] [PubMed] [Google Scholar]
- 36.Darmani NA, Martin BR, Glennon RA. Withdrawal from chronic treatment with (+/−)-DOI causes super-sensitivity to 5-HT2 receptor-induced head-twitch behaviour in mice. European journal of pharmacology. 1990;186(1):115–118. doi: 10.1016/0014-2999(90)94066-7. [DOI] [PubMed] [Google Scholar]
- 37.Huang X, et al. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiology of disease. 2010;40(1):193–199. doi: 10.1016/j.nbd.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buckmaster PS, Ingram EA, Wen X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29(25):8259–8269. doi: 10.1523/JNEUROSCI.4179-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29(21):6964–6972. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yasuhara A. Correlation between EEG abnormalities and symptoms of autism spectrum disorder (ASD) Brain & development. 2010;32(10):791–798. doi: 10.1016/j.braindev.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 41.Talos DM, et al. The interaction between early life epilepsy and autistic-like behavioral consequences: a role for the mammalian target of rapamycin (mTOR) pathway. PloS one. 2012;7(5):e35885. doi: 10.1371/journal.pone.0035885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lippman-Bell JJ, et al. AMPA receptor antagonist NBQX attenuates later-life epileptic seizures and autistic-like social deficits following neonatal seizures. Epilepsia. 2013;54(11):1922–1932. doi: 10.1111/epi.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uhlmann EJ, et al. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Annals of neurology. 2002;52(3):285–296. doi: 10.1002/ana.10283. [DOI] [PubMed] [Google Scholar]
- 44.Carson RP, Van Nielen DL, Winzenburger PA, Ess KC. Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiology of disease. 2012;45(1):369–380. doi: 10.1016/j.nbd.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chevere-Torres I, Maki JM, Santini E, Klann E. Impaired social interactions and motor learning skills in tuberous sclerosis complex model mice expressing a dominant/negative form of tuberin. Neurobiology of disease. 2012;45(1):156–164. doi: 10.1016/j.nbd.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin WH, et al. Seizure-induced 5-HT release and chronic impairment of serotonergic function in rats. Neuroscience letters. 2013;534:1–6. doi: 10.1016/j.neulet.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 47.Huang X, McMahon J, Huang Y. Rapamycin attenuates aggressive behavior in a rat model of pilocarpine-induced epilepsy. Neuroscience. 2012;215:90–97. doi: 10.1016/j.neuroscience.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kanemura H, Sano F, Tando T, Sugita K, Aihara M. Can EEG characteristics predict development of epilepsy in autistic children? European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society. 2013;17(3):232–237. doi: 10.1016/j.ejpn.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 49.van Eeghen AM, et al. Understanding relationships between autism, intelligence, and epilepsy: a cross-disorder approach. Developmental medicine and child neurology. 2013;55(2):146–153. doi: 10.1111/dmcn.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eisermann MM, et al. Infantile spasms in Down syndrome--effects of delayed anticonvulsive treatment. Epilepsy research. 2003;55(1–2):21–27. doi: 10.1016/s0920-1211(03)00088-3. [DOI] [PubMed] [Google Scholar]
- 51.Chugani DC. Serotonin in autism and pediatric epilepsies. Mental retardation and developmental disabilities research reviews. 2004;10(2):112–116. doi: 10.1002/mrdd.20021. [DOI] [PubMed] [Google Scholar]
- 52.Betancur C, et al. Serotonin transporter gene polymorphisms and hyperserotonemia in autistic disorder. Molecular psychiatry. 2002;7(1):67–71. doi: 10.1038/sj.mp.4001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McDougle CJ, et al. Effects of tryptophan depletion in drug-free adults with autistic disorder. Archives of general psychiatry. 1996;53(11):993–1000. doi: 10.1001/archpsyc.1996.01830110029004. [DOI] [PubMed] [Google Scholar]
- 54.Dolen G, Darvishzadeh A, Huang KW, Malenka RC. Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature. 2013;501(7466):179–184. doi: 10.1038/nature12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.West L, Brunssen SH, Waldrop J. Review of the evidence for treatment of children with autism with selective serotonin reuptake inhibitors. Journal for specialists in pediatric nursing : JSPN. 2009;14(3):183–191. doi: 10.1111/j.1744-6155.2009.00196.x. [DOI] [PubMed] [Google Scholar]
- 56.Chadman KK. Fluoxetine but not risperidone increases sociability in the BTBR mouse model of autism. Pharmacology, biochemistry, and behavior. 2011;97(3):586–594. doi: 10.1016/j.pbb.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 57.Hollander E, et al. A double-blind placebo-controlled trial of fluoxetine for repetitive behaviors and global severity in adult autism spectrum disorders. The American journal of psychiatry. 2012;169(3):292–299. doi: 10.1176/appi.ajp.2011.10050764. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.