

Abstract
The search for compounds active against Mycobacterium tuberculosis is reliant upon high throughput screening (HTS) in whole cells. We have used Bayesian machine learning models which can predict anti-tubercular activity to filter an internal library of over 150,000 compounds prior to in vitro testing. We used this to select and test 48 compounds in vitro; 11 were active with MIC values ranging from 0.4 µM to 10.2 µM, giving a high hit rate of 22.9%. Among the hits, we identified several compounds belonging to the same series including five quinolones (including ciprofloxacin), three molecules with long aliphatic linkers and three singletons. This approach represents a rapid method to prioritize compounds for testing that can be used alongside medicinal chemistry insight and other filters to identify active molecules. Such models can significantly increase the hit rate of HTS, above the usual 1% or lower rates seen. In addition, the potential targets for the 11 molecules were predicted using TB Mobile and clustering alongside a set of over 740 molecules with known M. tuberculosis target annotations. These predictions may serve as a mechanism for prioritizing compounds for further optimization.
Keywords: Bayesian models, Collaborative Drug Discovery Tuberculosis database, function class fingerprints, Virtual Screening, Mycobacterium tuberculosis
Introduction
The search for drugs to prevent or treat infectious diseases is an urgent research focus both in academia and across the pharmaceutical industry. In recent years there has been an increase in the efforts around high throughput screening (HTS) for Mycobacterium tuberculosis, in order to find compounds as therapeutics against tuberculosis (TB) 1–6, A recent review of the state of TB research has summarized the limited pipeline of molecules in various drug discovery/development stages 7. Collaborative efforts that coordinate fragmented TB research efforts by individual groups will be critical to improve the chances of success in both identifying new targets and finding new molecules that could target them. Such efforts include the initiatives funded by NIAID, the Bill and Melinda Gates Foundation (BMGF) and the FP7 funded More Medicines For Tuberculosis (MM4TB) project.
The pipeline for TB therapeutics had not produced a new approved drug in over 40 years until the recently FDA-approved Bedaquiline 8–10, although there are several candidates in the clinic 9, 11. Only a tiny fraction of TB targets have been addressed with approved drugs or early leads 12 and recent testing has targeted additional proteins (e.g. MmpL3 13). The relative lack of success with target-based screening compared with whole cell phenotypic screening is a pattern observed for other antibacterial targets, reflecting the difficulty of target-based high-throughput screening for novel antibiotics 14. In pharmaceutical companies, computational approaches are widely used to aid in drug discovery, but these have not been as exhaustively applied or validated for TB research. For example, virtual screening of compound libraries is used as a complement to high-throughput screening in vitro for many diseases 15–21. Computational approaches applied to TB have been generally used by specialists focused on a single target or series of compounds and rarely in combination with other computational tools 22, 23. We recently exhaustively reviewed this topic 22, 24, as computational methods are used in workflows by many pharmaceutical company project teams 18. We found several gaps when we look at how computational methods could be used in TB drug discovery including limited use of filtering for drug-likeness or lead-likeness 25, target deconvolution 26, lack of sequential virtual and biochemical screening and lack of in silico ADME/Tox model use22. A clear disconnect was noted between the generation, utilization, dissemination, sharing and reuse of computational models and the entire drug discovery process 22.
We have proposed using recently retrospectively validated Bayesian machine learning models for M. tuberculosis 25, 27, 28 for prospective compound evaluation. Three recent studies have also explored the optimization of these models by combining bioactivity and cytotoxicity data 29–31 and delivered hit rates in excess of 20%. In the current study we have validated the use of three Bayesian machine learning models by prospectively selecting a small percentage of an in house library for testing. We have identified 11 compounds with in vitro activity and predicted their potential targets using ligand-based computational approaches.
Experimental Methods
Chemicals
Compounds were purchased from ChemBridge (San Diego, CA), ChemDiv (San Diego, CA), Maybridge/Thermo Fisher Scientific Inc. (Waltham, MA) and Sigma - Aldrich (St. Louis, Mo).
CDD Database and SRI datasets
The development of the CDD TB database (Collaborative Drug Discovery Inc. Burlingame, CA) has been previously described 25. The Tuberculosis Antimicrobial Acquisition and Coordinating Facility (TAACF) and Molecular Libraries Small Molecule Repository (MLSMR) screening datasets 2–4 were collected and uploaded in CDD TB from sdf files and mapped to custom protocols 32. All of the public M. tuberculosis datasets are available for free public read-only access and mining upon registration, making them a valuable molecule resource for researchers along with available contextual data on these samples from other non M. tuberculosis assays. These datasets are also publically available in PubChem 33. The IDRI database and screening data used in modeling is proprietary.
Machine learning models for M. tuberculosis
We have previously described the generation and validation of Laplacian-corrected Bayesian classifier models 25, 27, 28 developed with single point screening and dose response data. In this study we have generated Laplacian-corrected Bayesian classifier models using Discovery Studio 2.5.5 34–38 Molecular function class fingerprints of maximum diameter 6 (FCFP_6) 39, AlogP, molecular weight, number of rotatable bonds, number of rings, number of aromatic rings, number of hydrogen bond acceptors, number of hydrogen bond donors, and molecular fractional polar surface area were calculated from input sdf files using the “calculate molecular properties” protocol to distinguish between compounds that are active against M. tuberculosis and those that are inactive in this study. A Bayesian classifier model with the molecular descriptors described above was built using the “create Bayesian model” protocol and IDRI % inhibition at 20 µM for 1106 samples (308 active with >90% inhibition) 40. Each model was validated using leave-one-out cross-validation. Each sample was left out one at a time, and a model built using the results of the samples, and that model used to predict the left-out sample. Once all the samples had predictions, a receiver operator curve (ROC) plot was generated, and the cross validated (XV) ROC area under the curve (AUC) calculated (Table 1). All models generated were additionally evaluated by leaving out 50% of the data and rebuilding the model 100 times using a custom protocol for validation, in order to generate the XV ROC and AUC (Table 1). These models were also used for screening the “Infectious Disease Research Institute (IDRI) library” of 156,719 compounds with M. tuberculosis activity.
Table 1.
Mean (SD) leave one out and leave out 50% × 100 cross validation of M. tuberculosis Bayesian models (ROC = receiver operator characteristic)
| Datasets (number of molecules) |
Leave one out ROC |
Leave out 50% × 100 External ROC Score |
Leave out 50% × 100Internal ROC Score |
Leave out 50% × 100 Concordance |
Leave out 50% × 100 Specificity |
Leave out 50% × 100 Sensitivity |
|---|---|---|---|---|---|---|
| IDRI % inhibition at 20uM (1106) |
0.82 | 0.77 ± 0.02 | 0.78 ± 0.02 | 73.4 ± 2.98 | 77.3 ± 6.28 | 63.4 ± 7.62 |
M. tuberculosis assays for biological activity
Molecules were screened at a single concentration of 20 µM in Middlebrook 7H9 medium plus 10% v/v OADC (oleic acid, albumen, dextrose, catalase) and 0.05 % w/v Tween 80; actives were classified as having ≥90% inhibition of growth of M. tuberculosis H37Rv after 5 d 40. MICs were determined in liquid medium 41; briefly a 10 point serial dilution of compounds was run and % growth of M. tuberculosis determined after 5 days incubation 41. Curves were generated using the Gompertz fit and MICs determined as minimal concentration required to inhibit growth completely.
Target prediction for IDRI compounds
Over 700 compounds with known M. tuberculosis targets were collated from the literature 42 and made available in the mobile application TB Mobile (Collaborative Drug Discovery Inc. Burlingame, CA) which is freely available for iOS and Android platforms 12, 43. This dataset was recently updated to 745 compounds and covers over 70 targets. Molecules representing hits from screening in this study were input as queries in TB Mobile and the similarity of all molecules calculated in the application. The top most structurally similar compounds were used to infer M. tuberculosis targets. In most cases multiple targets are shown were the top 2–3 molecules had different targets. The 745 compounds with known M. tuberculosis targets and the hit compounds from this study were used to generate a Principal Component Analysis (PCA) using the interpretable descriptors used for machine learning model building previously in Discovery Studio (AlogP, molecular weight, number of rotatable bonds, number of rings, number of aromatic rings, number of hydrogen bond acceptors, number of hydrogen bond donors, and molecular fractional polar surface area). 1200 M. tuberculosis screening hits (actives and non-toxic only from the SRI screens 29–31) were used to show how they covered the target-chemistry PCA space alongside the 745 compounds.
The 745 compounds with known M. tuberculosis targets and the hit compounds from this study were also clustered (100 clusters) using MDL fingerprints in Discovery Studio, and the position of the screening hits in specific clusters identified along with the targets of the other molecules in these clusters. In cases when a hit was a singleton, the identity of targets for clusters around a hit was noted. This clustering approach can also be used to infer targets alongside TB Mobile.
Results
IDRI – Bayesian model
A Bayesian model was generated with whole cell M. tuberculosis data for 1106 previously described TAACF and MLSMR actives and inactives [34]. The leave one out ROC was 0.82 and this decreased slightly (0.77) with internal validation with leave out 50% × 100 (Table 2). The concordance (73.4%), specificity (77.3%) and selectivity (63.4%) were in line with the other models described previously (Table 1) 25, 27, 29, 30. Using the FCFP-6 descriptors we can identify those substructure descriptors that contribute to the M. tuberculosis activity in the training set including imidazole, benzothiazole and quinolone, (Figure 1) and those that are not present in active compounds including acetamide, thioether, pyrrole, phenylether and piperazine (Figure 2).
Table 2.
Hits from IDRI library selected with Bayesian models. Promiscuity is calculated from activity of compound in PubChem as this represents a very large database of compounds and bioassays. Tanimoto similarity is reported when compared to the CDD database which contains over 300,000 compounds screened in vitro against Mtb. Target prediction was performed with TB Mobile and clustering as described in the Materials and Methods.
| Compound | Structure | Bayesian Model & Score |
MIC90 (µM) |
Promiscuity | NIH PubChem Notes |
Highest Tanimoto similarity in CDD public TB datasets |
Prediction with TB Mobile |
Prediction with Clustering |
|---|---|---|---|---|---|---|---|---|
| IDR- 0157809 |
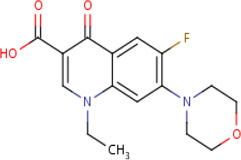 |
MLSMR 20.01 |
5.0 | 0.18 | Active in 3 out of 17 bioassays including against Escherichia coli Pseudomonas aeroginosa and Staphylococcus aureus |
95% | GyrA (Rv0006) GyrB (Rv0005) |
GyrA (Rv0006) GyrB (Rv0005) MurD (Rv2155c) cluster 84 |
| IDR- 018217* |
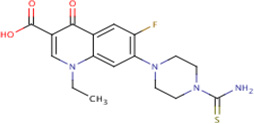 |
MLSMR 21.29 |
4.4 | 0 | No Data | 99% | GyrA (Rv0006) GyrB (Rv0005) |
GyrA (Rv0006) GyrB (Rv0005) MurD (Rv2155c) cluster 84 |
| IDR- 0159662 |
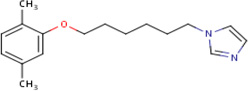 |
IDRI 20.45 |
9.4 | 0 | Tested in 7 assays inactive in all - no bacterial species tested |
97% | InhA (Rv 1484) ThiL |
InhA (Rv 1484) cluster 80 |
| IDR- 0168354 |
IDRI 19.41 |
4.8 | 0 | Tested in 7 assays -inactive in all - no bacterial species tested |
93% | InhA (Rv 1484) ThiL (Rv2977c) |
InhA (Rv 1484) cluster 80 |
|
| IDR- 0171075 |
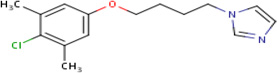 |
IDRI 20.47 |
10.2 | 0 | Tested in 1 assay - inactive in all - no bacterial species tested |
97% | KasA (Rv2245) |
InhA (Rv 1484) cluster 80 |
| IDR- 0173634 |
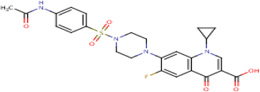 |
MLSMR 17.91 |
2.2 | 0 | No Data | 100%* | InhA (Rv 1484) |
GyrA (Rv0006) cluster 53 |
| IDR- 0229683* |
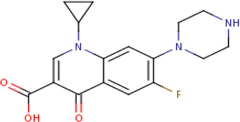 |
MLSMR 27.39 |
0.4 | 0.76 | Ciprofloxacin - active in 6698 out of 8773 bioaasays including against M tuberculosis |
80% | GyrA (Rv0006) |
GyrA (Rv0006) GyrB (Rv0005) murD (Rv2155c) cluster 84 |
| IDR- 0198303 |
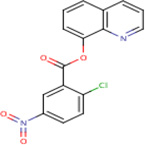 |
IDRI 20.79 |
2.4 | 0 | Tested in 1 bioassay - inactive |
89% | FolP1 (Rv3608c) FolP2 (Rv1207) Rv1885c |
Singleton in cluster 89 Dxs1 (Rv2682c) cluster 88 MbyA (Rv2384) cluster 90 |
| IDR- 0204586 |
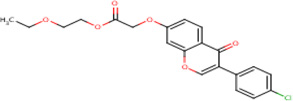 |
MLSMR+ cytotox 38.11 |
6.3 | 0 | No Data | 100%* | PtpA (Rv2234) | Molecule a singleton in cluster 72 RplJ (Rv0651) TlyA (Rv1694) cluster 71 Alr (Rv3423c) Cluster 73 |
| IDR- 0218592 |
 |
MLSMR 21.71 |
9.9 | 0 | No Data | 64% | ThiL (Rv2977c) |
RpoB (Rv0667) cluster 95 |
| IDR- 0236229* |
 |
MLSMR 22.48 |
8.7 | 0.793991 | Perfloxacin - active in 185 out of 233 bioassays including Mycobacteriuim leprae |
100% | GyrA (Rv0006) GyrB (Rv0005) |
GyrA (Rv0006) GyrB (Rv0005) MurD (Rv2155c) cluster 84 |
not cytotoxic based on Vero cell toxicity data in CDD.
Figure 1.

Good features identified in the IDRI Bayesian Model
Figure 2.

Bad features identified in the IDRI Bayesian Model
IDRI - Prospective testing of the Bayesian Models
The previously published MLSMR dose response model 25, MLSMR dose response and cytotoxicity model 29 and the IDRI Bayesian model were used to rank the “IDRI library” of 156,719 compounds for M. tuberculosis activity. This library can be considered leadlike based on the mean Molecular weight (344.5), log P (3.3), hydrogen bond donors (1.0), hydrogen bond acceptors (3.6) and other properties (Supplemental Figure 1). After ranking the library with the Bayesian score derived from each Bayesian model, the top 1000 compounds were selected and analyzed. There was minimal overlap between all three models and compounds in the top scoring 1000 (Figure 3). The MLSMR models overlapped to the greatest degree (over 20% of the top 1000). Forty eight compounds were selected from these ranked lists and tested in vitro; 11 of these were classed as hits (22.9% hit rate) as they possessed anti-tubercular activity with MIC <10 µM (Table 2). To illustrate the diversity of hits (Table 2), this included five quinolones including ciprofloxacin, three azole containing molecules with long aliphatic linkers and three singletons. Six compounds were found with the MLSMR dose response model, four were found with the IDRI model and one with the MLSMR dose response and cytotoxicity model. The Tanimoto similarity of the 11 compounds were compared to all the publically accessible TB related datasets and these ranged from 64–100%.
Figure 3.

Venn diagram showing the overlap of IDRI library compounds selected with the MLSMR dose response model, the MLSMR dose response and cytotoxicity model and the IDRI model for M. tuberculosis whole cell activity.
Target prediction for IDRI compounds
The PCA model of compounds with annotated M. tuberculosis targets represents the target-chemistry space and 88.7% of the variance is explained by the 3 principal components. The 11 hit compounds from this study were also added to this set and show they are clustered in a relatively narrow region (Figure 4A). Similarly, the hits from previous SRI screens only partially cover the target space (Figure 4B). Clustering the 11 hits with the 745 compounds with annotated target information enabled complementary target predictions with those based on molecular similarity performed with TB mobile (Table 2, Supplemental Figure 2). The known gyrase inhibitor class, fluoroquinolones were well predicted by both target inference methods. The remaining compounds had divergent predictions apart from the azoles, which were predicted to be InhA inhibitors.
Figure 4.


Principal Component Analysis of 745 compounds with A. known M. tuberculosis targets (Blue) from TB Mobile and 11 screening hits (yellow) and B. 1200 active and non toxic compounds from SRI screens (yellow)
Discussion
Drug discovery is time consuming and very costly 44, 45 such that any tools that can point out liabilities earlier will have considerable value 21, 46, 47. The need for new anti-tubercular therapies is unquestioned in the face of drug resistance that has progressed to the point of the identification of totally drug resistant strains in India 48 and a call for re-opening the TB sanatoria that were closed more than 60 years ago 49. To address the challenge of drug resistance in TB infection, many groups have turned to HTS campaigns with chemically diverse libraries of small molecules to identify novel starting points for drug discovery 1, 50. The TB community must now ask how to mine efficiently and leverage this growing database to provide new drug candidates, in the face of well known complications such as latency and persistence 51 and the numerous issues associated with typical HTS data 52. To help answer this question, we have identified a significant opportunity for the tuberculosis drug discovery community to harness pharmaceutical industry-tested computational methodologies 22. Subsequently, we turned in part to the cheminformatics methods which occupy an important place in the industrial drug discovery workflow. Ligand- and protein-based methods, for example, have been used as a complement to high-throughput screening in vitro 15. In order to validate the predictions from such methods we are required to test molecules for their whole cell TB activity.
We have developed and utilized machine learning models for M. tuberculosis 25, 27, 28 using large publically accessible HTS data sets 3, 4. During retrospective validation of these models we observed at least 4–10 fold enrichment in identifying TB actives in the top scoring molecules 28. These results indicated that using whole cell screening data from one laboratory for computational models can be used to benefit other laboratories via predictions of their compounds of interest and narrowing down the number of compounds to be tested in vitro 28. We have recently updated our approach to incorporate cytotoxicity data into the models 29–31. These previously published Bayesian models had considerably higher hit rates than random HTS screening 29, 30. One study virtually screened over 82,000 molecules, 550 were tested in vitro and 124 actives were identified in total (22.5% hit rate) 30. A second study virtually screened over 38,000 molecules, tested 106 in vitro and identified 17 actives (22.5% hit rate) 29. In the current study we utilized several previously published models as well as a newly constructed model generated with new data from >1000 previously published active compounds. Three Bayesian models were ultimately used to screen the in house library of 156,719 molecules and 48 (0.03%) of the compounds were tested in vitro resulting in 11 hits. Again this confirmed the hit rates previously observed with a value of 22.9%. In our experience and as a point of contrast, the whole cell HTS hit rate for the IDRI group has varied from 0.6 – 2% depending on the assay (unpublished).
Using several Bayesian models for M. tuberculosis activity to prioritize compounds from a screening library of this size is by far the largest such analysis we have performed to date to our knowledge 29–31. The results obtained further validate the hypothesis that Bayesian models 25, 27–31, 42 identify subsets of compound libraries enriched with active compounds, therefore requiring the testing of far fewer compounds. Future research will involve investigating open source descriptors and algorithms that can enable deploying such models more widely 53, 54. This Bayesian modeling and virtual screening approach is also applicable to other neglected diseases.
This study also further utilized a recently developed mobile application for inferring potential M. tuberculosis targets for the 11 hits (Table 2, Supplemental Figure 2). This application draws together known small molecules and their annotated targets as well as other information relevant to the pathway targeted, essentiality, human ortholog etc. 12, 42. Generating predictions with this application was also complemented by using clustering of the known compounds with targets and assessing which clusters the 11 compounds were in. The fluoroquinolone compounds are not surprisingly predicted as gyrase inhibitors using both target inference methods, apart from IDR-0173634 which is also predicted as a potential inhibitor of InhA. Although azoles are well known cytochrome P450 inhibitors 55, 56 they are predominantly predicted as targeting InhA. These and the remaining 3 singleton compounds with different predicted targets with no concordance with the target inference methods would be worthy of testing in vitro. Our analysis of the 11 hits suggest, as one would expect, that they are covering a very narrow section of chemistry and target space. In particular we have multiple fluoroquinolones and azoles (Table 2) so these may essentially count as a single data point in each case. Our approach (using known compounds with Mtb targets) to infer potential targets for similar compounds is more conservative than methods which would use similarity to compounds known to be active against targets in other organisms. Such target prediction efforts would help us to prioritize targets to test.
In conclusion we have presented an approach using multiple Bayesian models to prioritize compounds for testing which identified active compounds. These in turn were used with TB Mobile 12 and clustering as mechanisms for predicting potential targets for compounds in M. tuberculosis, thereby serving as an approach for further identifying the best compounds for optimization. Such computational workflows leveraging prior knowledge further our aim of optimally using and integrating the data and resources available to us in order to accelerate drug discovery for M. tuberculosis 22.
Supplementary Material
Acknowledgement
S.E. acknowledges colleagues at CDD for developing the software. Dr. Alex Clark and Dr. Malabika Sarker are acknowledged for assistance with TB Mobile. Accelrys are kindly acknowledged for providing Discovery Studio. S.E. acknowledges Dr. Joel Freundlich and Dr. Robert Reynolds for numerous discussions on TB and Bayesian models. We thank Torey Alling, Mai Ann Bailey and Juliane Ollinger for running the MICs at IDRI, Susantha Chandrasekera, Edward Kesicki and Joshua Odingo for assistance with compound structural information and Alfredo Blakeley for technical assistance with compound handling.
Funding
The CDD TB has been developed thanks to funding from the Bill and Melinda Gates Foundation (Grant#49852 “Collaborative drug discovery for TB through a novel database of SAR data optimized to promote data archiving and sharing”). The project described was supported by Award Number R43 LM011152-01 “Biocomputation across distributed private datasets to enhance drug discovery” from the National Library of Medicine. TB Mobile was developed with funding from Award Number 2R42AI088893-02 “Identification of novel therapeutics for tuberculosis combining cheminformatics, diverse databases and logic based pathway analysis” from the National Institutes of Allergy and Infectious Diseases.
R.C.R. and S. G. F. acknowledge the American Reinvestment and Recovery Act Grant 1RC1AI086677-01 (National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases (NIAID)) – “Targeting MDR-TB.” The work at IDRI was funded in part by Eli Lilly and Company in support of the mission of the Lilly TB Drug Discovery Initiative and with Grant #42844 from the Bill and Melinda Gates Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information Available
Supplemental material is available online. The Bayesian models created in Discovery Studio using the previously published data in CDD are available from the authors upon written request.
Conflict of interest statement
Sean Ekins is a consultant for Collaborative Drug Discovery Inc. Barry A. Bunin is the Founder and CEO of Collaborative Drug Discovery Inc.
References
- 1.Ballel L, Field RA, Duncan K, Young RJ. New small-molecule synthetic antimycobacterials. Antimicrobial agents and chemotherapy. 2005;49:2153–2163. doi: 10.1128/AAC.49.6.2153-2163.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reynolds RC, Ananthan S, Faaleolea E, Hobrath JV, Kwong CD, Maddox C, Rasmussen L, Sosa MI, Thammasuvimol E, White EL, Zhang W, Secrist JA., 3rd . Tuberculosis. Scotland: Edinburgh; 2011. High throughput screening of a library based on kinase inhibitor scaffolds against Mycobacterium tuberculosis H37Rv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maddry JA, Ananthan S, Goldman RC, Hobrath JV, Kwong CD, Maddox C, Rasmussen L, Reynolds RC, Secrist JA, 3rd, Sosa MI, White EL, Zhang W. Tuberculosis. Vol. 89. Scotland: Edinburgh; 2009. Antituberculosis activity of the molecular libraries screening center network library; pp. 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ananthan S, Faaleolea ER, Goldman RC, Hobrath JV, Kwong CD, Laughon BE, Maddry JA, Mehta A, Rasmussen L, Reynolds RC, Secrist JA, 3rd, Shindo N, Showe DN, Sosa MI, Suling WJ, White EL. Tuberculosis. Vol. 89. Scotland: Edinburgh; 2009. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv; pp. 334–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mak PA, Rao SP, Ping Tan M, Lin X, Chyba J, Tay J, Ng SH, Tan BH, Cherian J, Duraiswamy J, Bifani P, Lim V, Lee BH, Ling Ma N, Beer D, Thayalan P, Kuhen K, Chatterjee A, Supek F, Glynne R, Zheng J, Boshoff HI, Barry CE, 3rd, Dick T, Pethe K, Camacho LR. A High-Throughput Screen To Identify Inhibitors of ATP Homeostasis in Non-replicating Mycobacterium tuberculosis. ACS Chem Biol. 2012;7:1190–1197. doi: 10.1021/cb2004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stanley SA, Grant SS, Kawate T, Iwase N, Shimizu M, Wivagg C, Silvis M, Kazyanskaya E, Aquadro J, Golas A, Fitzgerald M, Dai H, Zhang L, Hung DT. Identification of Novel Inhibitors of M. tuberculosis Growth Using Whole Cell Based High-Throughput Screening. ACS Chem Biol. 2012;7:1377–1384. doi: 10.1021/cb300151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thayer A, Taking down TB. Chem Eng News. 2007 Sep 24;:21–32. [Google Scholar]
- 8.Voelker R. MDR-TB has new drug foe after fast-track approval. Jama. 2013;309:430. doi: 10.1001/jama.2013.94. [DOI] [PubMed] [Google Scholar]
- 9.Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. The challenge of new drug discovery for tuberculosis. Nature. 2011;469:483–490. doi: 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- 10.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. Science. Vol. 307. New York, NY: 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis; pp. 223–227. [DOI] [PubMed] [Google Scholar]
- 11.Kaneko T, Cooper C, Mdluli K. Challenges and opportunities in developing novel drugs for TB. Future Med Chem. 2011;3:1373–1400. doi: 10.4155/fmc.11.115. [DOI] [PubMed] [Google Scholar]
- 12.Ekins S, Clark AM, Sarker M. TB Mobile: A Mobile App for Anti-tuberculosis Molecules with Known Targets. J Cheminform. 2013;5:13. doi: 10.1186/1758-2946-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tahlan K, Wilson R, Kastrinsky DB, Arora K, Nair V, Fischer E, Barnes SW, Walker JR, Alland D, Barry CE, 3rd, Boshoff HI. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrobial agents and chemotherapy. 2012;56:1797–1809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Payne DA, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Disc. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 15.Schneider G. Virtual screening: an endless staircase? Nature reviews. 2010;9:273–276. doi: 10.1038/nrd3139. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Fourches D, Sedykh A, Zhu H, Golbraikh A, Ekins S, Clark J, Connelly MC, Sigal M, Hodges D, Guiguemde A, Guy RK, Tropsha A. Discovery of Novel Antimalarial Compounds Enabled by QSAR-Based Virtual Screening. Journal of chemical information and modeling. 2013;53:475–492. doi: 10.1021/ci300421n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scior T, Bender A, Tresadern G, Medina-Franco JL, Martinez-Mayorga K, Langer T, Cuanalo-Contreras K, Agrafiotis DK. Recognizing Pitfalls in Virtual Screening: A Critical Review. Journal of chemical information and modeling. 2012 doi: 10.1021/ci200528d. [DOI] [PubMed] [Google Scholar]
- 18.Duffy BC, Zhu L, Decornez H, Kitchen DB. Early phase drug discovery: cheminformatics and computational techniques in identifying lead series. Bioorganic & medicinal chemistry. 2012;20:5324–5342. doi: 10.1016/j.bmc.2012.04.062. [DOI] [PubMed] [Google Scholar]
- 19.Krueger BA, Weil T, Schneider G. Comparative virtual screening and novelty detection for NMDA-GlycineB antagonists. Journal of computer-aided molecular design. 2009;23:869–881. doi: 10.1007/s10822-009-9304-1. [DOI] [PubMed] [Google Scholar]
- 20.Noeske T, Jirgensons A, Starchenkovs I, Renner S, Jaunzeme I, Trifanova D, Hechenberger M, Bauer T, Kauss V, Parsons CG, Schneider G, Weil T. Virtual Screening for Selective Allosteric mGluR1 Antagonists and Structure-Activity Relationship Investigations for Coumarine Derivatives. ChemMedChem. 2007;2:1763–1773. doi: 10.1002/cmdc.200700151. [DOI] [PubMed] [Google Scholar]
- 21.Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol. 2007;152:9–20. doi: 10.1038/sj.bjp.0707305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ekins S, Freundlich JS, Choi I, Sarker M, Talcott C. Computational Databases, Pathway and Cheminformatics Tools for Tuberculosis Drug Discovery. Trends in microbiology. 2011;19:65–74. doi: 10.1016/j.tim.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ballester PJ, Mangold M, Howard NI, Robinson RL, Abell C, Blumberger J, Mitchell JB. Hierarchical virtual screening for the discovery of new molecular scaffolds in antibacterial hit identification. J R Soc Interface. 2012;9:3196–3207. doi: 10.1098/rsif.2012.0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ekins S, Freundlich JS. Methods in molecular biology. Vol. 993. Clifton, NJ: 2013. Computational models for tuberculosis drug discovery; pp. 245–262. [DOI] [PubMed] [Google Scholar]
- 25.Ekins S, Bradford J, Dole K, Spektor A, Gregory K, Blondeau D, Hohman M, Bunin B. A Collaborative Database And Computational Models For Tuberculosis Drug Discovery. Mol BioSystems. 2010;6:840–851. doi: 10.1039/b917766c. [DOI] [PubMed] [Google Scholar]
- 26.Prathipati P, Ma NL, Manjunatha UH, Bender A. Fishing the target of antitubercular compounds: in silico target deconvolution model development and validation. Journal of proteome research. 2009;8:2788–2798. doi: 10.1021/pr8010843. [DOI] [PubMed] [Google Scholar]
- 27.Ekins S, Kaneko T, Lipinksi CA, Bradford J, Dole K, Spektor A, Gregory K, Blondeau D, Ernst S, Yang J, Goncharoff N, Hohman M, Bunin B. Analysis and hit filtering of a very large library of compounds screened against Mycobacterium tuberculosis. Molecular bioSystems. 2010;6:2316–2324. doi: 10.1039/c0mb00104j. [DOI] [PubMed] [Google Scholar]
- 28.Ekins S, Freundlich JS. Validating new tuberculosis computational models with public whole cell screening aerobic activity datasets. Pharm Res. 2011;28:1859–1869. doi: 10.1007/s11095-011-0413-x. [DOI] [PubMed] [Google Scholar]
- 29.Ekins S, Reynolds R, Kim H, Koo M-S, Ekonomidis M, Talaue M, Paget SD, Woolhiser LK, Lenaerts AJ, Bunin BA, Connell N, Freundlich JS. Bayesian Models Leveraging Bioactivity and Cytotoxicity Information for Drug Discovery. Chem Biol. 2013;20:370–378. doi: 10.1016/j.chembiol.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ekins S, Reynolds RC, Franzblau SG, Wan B, Freundlich JS, Bunin BA. Enhancing Hit Identification in Mycobacterium tuberculosis Drug Discovery Using Validated Dual-Event Bayesian Models. PLOSONE. 2013;8:e63240. doi: 10.1371/journal.pone.0063240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ekins S, Freundlich JS, Hobrath JV, White EL, Reynolds RC. Combining Computational Methods for Hit to Lead Optimization in Mycobacterium tuberculosis Drug Discovery. Pharm Res. 2013 doi: 10.1007/s11095-013-1172-7. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anon. Collaborative Drug Discovery, Inc. http://www.collaborativedrug.com/register.
- 33.Anon. The PubChem Database. http://pubchem.ncbi.nlm.nih.gov/
- 34.Prathipati P, Ma NL, Keller TH. Global Bayesian models for the prioritization of antitubercular agents. Journal of chemical information and modeling. 2008;48:2362–2370. doi: 10.1021/ci800143n. [DOI] [PubMed] [Google Scholar]
- 35.Bender A, Scheiber J, Glick M, Davies JW, Azzaoui K, Hamon J, Urban L, Whitebread S, Jenkins JL. Analysis of Pharmacology Data and the Prediction of Adverse Drug Reactions and Off-Target Effects from Chemical Structure. ChemMedChem. 2007;2:861–873. doi: 10.1002/cmdc.200700026. [DOI] [PubMed] [Google Scholar]
- 36.Klon AE, Lowrie JF, Diller DJ. Improved naive Bayesian modeling of numerical data for absorption, distribution, metabolism and excretion (ADME) property prediction. Journal of chemical information and modeling. 2006;46:1945–1956. doi: 10.1021/ci0601315. [DOI] [PubMed] [Google Scholar]
- 37.Hassan M, Brown RD, Varma-O'brien S, Rogers D. Cheminformatics analysis and learning in a data pipelining environment. Mol Divers. 2006;10:283–299. doi: 10.1007/s11030-006-9041-5. [DOI] [PubMed] [Google Scholar]
- 38.Rogers D, Brown RD, Hahn M. Using extended-connectivity fingerprints with Laplacian-modified Bayesian analysis in high-throughput screening follow-up. J Biomol Screen. 2005;10:682–686. doi: 10.1177/1087057105281365. [DOI] [PubMed] [Google Scholar]
- 39.Jones DR, Ekins S, Li L, Hall SD. Computational approaches that predict metabolic intermediate complex formation with CYP3A4 (+b5) Drug Metab Dispos. 2007;35:1466–1475. doi: 10.1124/dmd.106.014613. [DOI] [PubMed] [Google Scholar]
- 40.Roberts D, Ollinger J, Bailey M, Casey A, Parish T. Development of a whole cell high-throughput screen to identify inhibitors of Mycobacterium tuberculosis. Unpublished. 2012 doi: 10.1371/journal.pone.0205479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ollinger J, Bailey MA, Moraski GC, Casey A, Florio S, Alling T, Miller MJ, Parish T. A dual read-out assay to evaluate the potency of compounds active against Mycobacterium tuberculosis. PloS one. 2013;8:e60531. doi: 10.1371/journal.pone.0060531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarker M, Talcott C, Madrid P, Chopra S, Bunin BA, Lamichhane G, Freundlich JS, Ekins S. Combining cheminformatics methods and pathway analysis to identify molecules with whole-cell activity against Mycobacterium tuberculosis. Pharm Res. 2012;29:2115–2127. doi: 10.1007/s11095-012-0741-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.TB Mobile. https://itunes.apple.com/us/app/tb-mobile/id567461644?mt=8.
- 44.Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nature reviews. 2010;9:203–214. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- 45.Munos B. Lessons from 60 years of pharmaceutical innovation. Nature reviews. 2009;8:959–968. doi: 10.1038/nrd2961. [DOI] [PubMed] [Google Scholar]
- 46.Ekins S, Waller CL, Swaan PW, Cruciani G, Wrighton SA, Wikel JH. Progress in predicting human ADME parameters in silico. J Pharmacol Toxicol Methods. 2000;44:251–272. doi: 10.1016/s1056-8719(00)00109-x. [DOI] [PubMed] [Google Scholar]
- 47.Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: applications to targets and beyond. Br J Pharmacol. 2007;152:21–37. doi: 10.1038/sj.bjp.0707306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Udwadia ZF, Amale RA, Ajbani KK, Rodrigues C. Totally drug-resistant tuberculosis in India. Clin Infect Dis. 2012;54:579–581. doi: 10.1093/cid/cir889. [DOI] [PubMed] [Google Scholar]
- 49.Dheda K, Migliori GB. The global rise of extensively drug-resistant tuberculosis: is the time to bring back sanatoria now overdue? Lancet. 2012;379:773–775. doi: 10.1016/S0140-6736(11)61062-3. [DOI] [PubMed] [Google Scholar]
- 50.Sacchettini JC, Rubin EJ, Freundlich JS. Drugs versus bugs: in pursuit of the persistent predator Mycobacterium tuberculosis. Nat Rev Microbiol. 2008;6:41–52. doi: 10.1038/nrmicro1816. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y. The magic bullets and tuberculosis drug targets. Annu Rev Pharmacol Toxicol. 2005;45:529–564. doi: 10.1146/annurev.pharmtox.45.120403.100120. [DOI] [PubMed] [Google Scholar]
- 52.Malo N, Hanley JA, Cerquozzi S, Pelletier J, Nadon R. Statistical practice in high-throughput screening data analysis. Nature biotechnology. 2006;24:167–175. doi: 10.1038/nbt1186. [DOI] [PubMed] [Google Scholar]
- 53.Gupta RR, Gifford EM, Liston T, Waller CL, Bunin B, Ekins S. Using open source computational tools for predicting human metabolic stability and additional ADME/TOX properties. Drug metabolism and disposition: the biological fate of chemicals. 2010;38:2083–2090. doi: 10.1124/dmd.110.034918. [DOI] [PubMed] [Google Scholar]
- 54.Ekins S, Gupta RR, Gifford E, Bunin BA, Waller CL. Chemical space: missing pieces in cheminformatics. Pharm Res. 2010;27:2035–2039. doi: 10.1007/s11095-010-0229-0. [DOI] [PubMed] [Google Scholar]
- 55.Rupp B, Raub S, Marian C, Holtje HD. Molecular design of two sterol 14alpha-demethylase homology models and their interactions with the azole antifungals ketoconazole and bifonazole. Journal of computer-aided molecular design. 2005;19:149–163. doi: 10.1007/s10822-005-3692-7. [DOI] [PubMed] [Google Scholar]
- 56.Hitchcock CA, Dickinson K, Brown SB, Evans EG, Adams DJ. Interaction of azole antifungal antibiotics with cytochrome P-450-dependent 14 alpha-sterol demethylase purified from Candida albicans. Biochem J. 1990;266 doi: 10.1042/bj2660475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.