

SUMMARY
Vascular endothelial growth factor-C (VEGF-C) is a potent lymphangiogenic cytokine that signals via the coordinated action of two cell surface receptors, Neuropilin-2 (Nrp2) and VEGFR-3. Diseases associated with both loss and gain of VEGF-C function, lymphedema and cancer, respectively, motivate studies of VEGF-C/Nrp2 binding and inhibition. Here we demonstrate that VEGF-C binding to Nrp2 is regulated by C-terminal proteolytic maturation. The structure of the VEGF-C C-terminus in complex with the ligand-binding domains of Nrp2 demonstrates that a cryptic Nrp2 binding motif is released upon proteolysis, allowing specific engagement with the b1 domain of Nrp2. Based on the identified structural requirements for Nrp2 binding to VEGF-C, we hypothesized that the endogenous secreted splice form of Nrp2, s9Nrp2, may function as a selective inhibitor of VEGF-C. We find that s9Nrp2 forms a stable dimer that potently inhibits VEGF-C/Nrp2 binding and cellular signaling. These data provide critical insight into VEGF-C/Nrp2 binding and inhibition.
INTRODUCTION
The Vascular Endothelial Growth Factor (VEGF) family of cytokines are critical regulators of endothelial cell function. There are five VEGF family members: VEGF-A, -B, -C, -D and placental growth factor (PlGF). Of these five, VEGF-C and VEGF-D selectively control lymphangiogenesis. While they show partially overlapping biological activity and physical properties, VEGF-C is essential for viability while VEGF-D is not (Baldwin et al., 2005; Karkkainen et al., 2004). Endothelial cells of homozygous VEGF-C knockout mice do not sprout to form lymphatic vessels, which results in an alymphatic embryo and embryonic lethality (Karkkainen et al., 2004). Overexpression of VEGF-C results in selective induction of lymphatic but not vascular endothelial cell proliferation and lymphatic vessel enlargement (Jeltsch et al., 1997). In addition to its critical physiological role, VEGF-C signaling is also important for pathological lymphangiogenesis that is associated with both aberrant loss of function in lymphedema (Saaristo et al., 2002) or gain of function in tumorigenesis and metastasis (Caunt et al., 2008; Ellis, 2006; Stacker et al., 2002).
VEGF-C signals via the coordinated activity of two families of endothelial cell surface receptors, the VEGF-receptor (VEGFR) family of receptor tyrosine kinases (RTK) (rev. in (Stuttfeld and Ballmer-Hofer, 2009)) and the Neuropilin (Nrp) family of co-receptors (rev. in (Parker et al., 2012a)). VEGF-C function is specifically mediated through VEGFR-2/3 (Joukov et al., 1996; Kukk et al., 1996; Lymboussaki et al., 1999) and Nrp2 (Karkkainen et al., 2001; Xu et al., 2010), with VEGF-C capable of simultaneously engaging both families of receptors (Favier et al., 2006). VEGFR-2/3 have dual functionality in both angiogenesis and lymphangiogenesis (rev. in (Lohela et al., 2009)). In contrast, Nrp2 knockout mice display normal angiogenesis but abnormal lymphatic vessel development (Yuan et al., 2002), similar to the tissue specific function observed in the VEGF-C knockout (Karkkainen et al., 2004). Intriguingly, it has also been demonstrated that Nrp2 can function in VEGF-C signaling independent of its role as a co-receptor for VEGFR (Caunt et al., 2008).
Each member of the VEGF family of ligands is produced in multiple forms by either alternative splicing (e.g. VEGF-A, -B, and PlGF) or proteolytic processing (e.g. VEGF-C and -D) (Holmes and Zachary, 2005). In all cases, an invariant core cystine-knot domain, which specifically interacts with VEGFR, is combined with a variable C-terminal domain. VEGF-C is synthesized as a pro-protein with N- and C-terminal domains flanking the central core cystine-knot domain. Prior to secretion, the C-terminal propeptide is cleaved followed by extracellular cleavage of the N-terminus (Joukov et al., 1997). These processing events critically alter both the physiological and pathological bioactivity of VEGF-C (Siegfried et al., 2003). The mature dual processed VEGF-C shows dramatically enhanced stimulatory activity in situ (McColl et al., 2003) and loss of C-terminal processing ablates function in vivo (Khatib et al., 2010). However, the physical basis for the enhanced activity of the mature form of VEGF-C remains unclear and has been connected to different properties including differential receptor binding and interactions with heparin/extracellular matrix (ECM) (Harris et al., 2013; Joukov et al., 1997; Karpanen et al., 2006). The role of VEGF-C proteolytic maturation in regulating Nrp2 binding is unknown.
The structural basis for VEGF-C binding to VEGFR-2/3 has recently been elucidated and was shown to involve the invariant cystine-knot domain of VEGF-C binding to the N-terminal domains of VEGFR-2 and VEGFR-3 (Leppanen et al., 2010; Leppanen et al., 2013). However, the structural basis for VEGF-C binding to Nrp2 remains to be determined. Alternative splicing and proteolysis modify the C-terminal variable region of VEGF and regulate Nrp binding (Makinen et al., 1999; Parker et al., 2012c; Soker et al., 1998). It has been demonstrated that Nrp1 binds the C-terminal basic domain of the Semaphorin-3 (Sema3) and VEGF family of ligands (He and Tessier-Lavigne, 1997; Soker et al., 1996) utilizing a binding pocket for ligands that contain a C-terminal arginine (Parker et al., 2010; Parker et al., 2012c; Vander Kooi et al., 2007; von Wronski et al., 2006). Importantly, the Sema3 family of ligands undergoes furin-dependent proteolytic maturation within their C-terminal domain, a process that liberates an extended basic sequence and directly regulates bioactivity and Nrp binding (Adams et al., 1997; Parker et al., 2010; Parker et al., 2013).
Nrp2-dependent VEGF-C signaling is important in a variety of tumors and overexpression of these factors is correlated with advanced stage disease and poor prognosis (Ellis, 2006; Stacker et al., 2002). Thus, specific Nrp2/VEGF-C inhibitors are of clinical interest. Soluble receptor fragments are common endogenous inhibitors (Albuquerque et al., 2009; Ambati et al., 2006; Kendall and Thomas, 1993; Rose-John and Heinrich, 1994). A soluble Nrp1 isoform was first identified as an endogenous inhibitor of prostate cancer in vivo (Gagnon et al., 2000). Soluble extracellular domain fragments can also be engineered for use clinically, including VEGF-trap (Aflibercept), a chimeric VEGFR-1/2-Fc fusion, which is an inhibitor of VEGF-A (Holash et al., 2002). A soluble splice form of Nrp2, s9Nrp2, has been identified at the transcript level (Rossignol et al., 2000). s9Nrp2 is produced by intron inclusion, which contains an in-frame stop codon. This stop codon is located prior to the transmembrane domain, and is thus predicted to produce a secreted form of Nrp2. Interestingly, the insertion occurs in the middle of the second coagulation factor domain (b2), rather than in an interdomain region. The two Nrp2 coagulation factor domains (b1b2) form an integral unit (Appleton et al., 2007) and thus the nature of the production and function of s9Nrp2 is unclear. Further, domains b1b2 of Nrp2 have been demonstrated to bind VEGF-C (Karpanen et al., 2006), bringing into question whether this soluble splice form contains the structural requirements necessary to bind and sequester its ligands.
Here we demonstrate that removal of the VEGF-C C-terminal propeptide directly regulates binding to Nrp2. The structure of the mature VEGF-C C-terminus in complex with Nrp2 demonstrates that a cryptic Nrp2-binding motif is liberated upon C-terminal processing. This offers the first structural insight into the physical basis for VEGF-C binding to Nrp2, showing that the proteolytically liberated C-terminal arginine of VEGF-C directly binds the Nrp2 b1 domain. Mutagenesis of both VEGF-C and Nrp2 confirms the critical nature of the VEGF-C C-terminal sequence in Nrp2-b1 binding. Understanding the physical interactions underlying VEGF-C/Nrp2 binding led us to consider mechanisms for VEGF-C inhibition. The secreted Nrp2 splice form, s9Nrp2, contains an intact Nrp2 b1 domain but a subsequent stop codon, and we assessed its function as a pathway specific inhibitor. Strikingly, this soluble receptor forms a disulfide-linked dimer with two tightly integrated b1 domains and functions as a potent inhibitor of VEGF-C binding to Nrp2.
RESULTS
Structural basis for proteolytic-dependent VEGF-C binding to Nrp2
VEGF-C is synthesized as a pro-protein with N and C-terminal pro-peptides. Removal of the VEGF-C C-terminal propeptide critically regulates its bioactivity. C-terminal processing of VEGF-C liberates a polypeptide stretch rich in basic amino acids that terminates with a di-arginine sequence (Figure 1A), a structural motif conserved across the VEGF and Sema3 family of ligands and known to be important for Nrp1 binding. Thus, we hypothesized that processing of VEGF-C may directly regulate a physical interaction with Nrp2. To test this hypothesis, we produced peptides corresponding to the unprocessed (215-RQVHSIIRRSLPA-227) and processed (215-RQVHSIIRR-223) VEGF-C C-terminus and measured the ability of each peptide to bind Nrp2 domains b1b2 using a differential scanning fluorimetry (DSF) thermal shift assay (Figure 1B). Processed VEGF-C significantly stabilized Nrp2-b1b2 (Tm 48.8°C ± 0.06°C to 50.3°C ± 0.05°C), while unprocessed VEGF-C showed no effect (Tm 48.4°C ± 0.04°C). Further, the processed VEGF-C pep tide showed dose-dependent saturable binding to Nrp2-b1b2 with an apparent dissociation constant Kd = 199 μM ± 71 μM (Figure 1C). These data demonstrate that C-terminal proteolytic maturation directly regulates VEGF-C binding to Nrp2.
Figure 1. Crystal structure of the VEGF-C/Nrp2 complex reveals the basis for proteolytic-dependent binding.
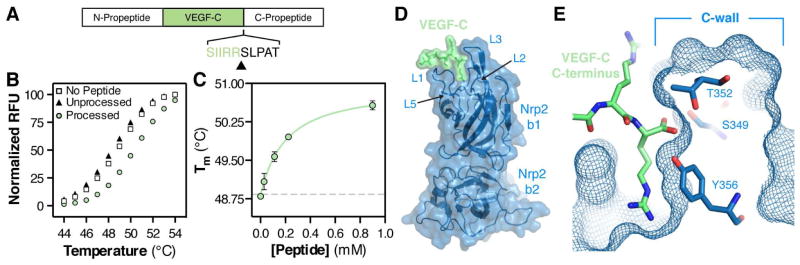
(A) Organization of the VEGF-C pro-protein and site of C-terminal processing (black arrow). (B) Peptides corresponding to processed (green circle) and unprocessed (black triangle) VEGF-C were assayed for the ability to bind Nrp2-b1b2 as measured by DSF thermal shift assay. Peptides were added to Nrp2-b1b2 to a final concentration of 0.5 mM and melting was monitored between 20 and 90°C. All samples were measured in triplicate and a representative melting curve is shown for each. (C) Processed VEGF-C dose-dependently enhances the Nrp2-b1b2 Tm. Error bars indicate the standard deviation (SD) of the three measurements. (D) Structure of Nrp2-b1b2 (blue) in complex with the C-terminus of VEGF-C (green). (E) Cross-section of the Nrp2 binding pocket demonstrates that the free carboxy terminus of VEGF-C is buried against the Nrp2 C-wall, which is formed by the third coagulation factor loop.
To define the physical basis for proteolytic-dependent binding of VEGF-C to Nrp2, we determined the crystal structure of the processed VEGF-C C-terminus in complex with Nrp2 domains b1b2. The C-terminal five amino acids of mature VEGF-C (219-SIIRR-223), which are strictly conserved across species and also with VEGF-D, were fused to the C-terminus of human Nrp2 domains b1b2 (residues 276–595). The fusion protein was expressed in E. coli, purified, and crystallized. The structure was solved by molecular replacement and was refined to a resolution of 1.9 Å (Figure 1D, Table 1). There were two molecules in the asymmetric unit oriented in an anti-parallel fashion (Figure S1A). Both molecules demonstrated specific binding of the VEGF-C encoded residues via an intermolecular interaction with a symmetry-related molecule.
Table 1.
Data collection and refinement statistics
| Construct: | Nrp2-VEGF-C | Nrp2-T319R | s9Nrp2B |
|---|---|---|---|
| Data Collection | |||
| Beamline | APS 22-ID | APS 22-BM | APS 22-ID |
| Wavelength | 1.0000 | 1.0000 | 1.0000 |
| Space group | P21 | P212121 | P21212 |
| Cell dimensions (Å) | 41.05, 120.81, 69.84 | 34.90, 70.76, 122.97 | 69.36, 91.39, 67.33 |
| Cell dimensions (°) | 90.0, 103.29, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Unique reflections | 44,081 | 12,223 | 16,303 |
| Completeness (%) | 90.6(82.0) | 96.4(83.2) | 94.1(79.8) |
| Resolution (Å) | 1.95(2.02–1.95) | 2.40(2.49–2.40) | 2.40(2.49–2.40) |
| Rmerge (%) | 9.9(46.6) | 8.0(29.2) | 9.9(32.7) |
| Redundancy | 5.1(4.2) | 6.8(5.9) | 4.4(4.1) |
| I/σ(I) | 13.1(3.0) | 29.4(5.1) | 12.3(3.2) |
| Refinement | |||
| Resolution limits (Å) | 20.00(1.95) | 20.00(2.40) | 20.00(2.40) |
| No. reflections/no. to compute Rfree | 41,511/2140 | 11,490/586 | 15,439/821 |
| R(Rfree) | 21.0(24.1) | 20.1(25.5) | 21.0(26.4) |
| No. protein residues | 632 | 313 | 361 |
| No. solvent/ion molecules | 333 | 123 | 107 |
| RMSD Bond, Å | 0.006 | 0.008 | 0.006 |
| RMSD Angle, ° | 1.11 | 1.19 | 1.04 |
| Protein geometry | |||
| Ramachandran outlier/favored (%) | 0/96.7 | 0/96.1 | 0/96.7 |
| Residues with bad bonds/angles | 0/0 | 0/0 | 0/0 |
| Rotamer outliers | 0 | 0 | 0 |
Analysis of the structure reveals that VEGF-C (green) engages a binding pocket formed by the Nrp2-b1 (blue) coagulation factor loops (Figure 1D, S1B and S1C). Indeed, this interloop cleft uniquely accommodates the C-terminal residue of processed VEGF-C (Figure 1E). The free carboxy-terminus of VEGF-C is integrated into the binding pocket through interactions with residues from the third coagulation factor loop (L3) of Nrp2-b1, which form a wall at one side of the binding pocket (“C-wall”). Specifically, an extensive hydrogen bond network forms between the VEGF-C free C-terminal carboxylate and the side chains of the C-wall residues S349, T352, and Y356 (Figure 1E). Importantly, the position of the C-wall would preclude binding of the unprocessed protein, providing a physical mechanism for the observed proteolytic-dependent binding of VEGF-C to Nrp2-b1b2.
Characterization of the VEGF-C/Nrp2 interaction
Clear electron density for the VEGF-C-encoded region was observed, permitted modeling of both the VEGF-C polypeptide and interfacing solvent that bridge the two molecules (Figure 2A). Analysis of the VEGF-C/Nrp2 interface reveals direct interactions between VEGF-C and residues within the L1, L5, and L3 loops of Nrp2-b1 (Figure 2B), the regions that show the largest conformational changes when comparing the bound structure to the previously reported apo structure (Figure S2) (Appleton et al., 2007). In addition to the hydrogen bond network formed between the VEGF-C free carboxy-terminus and the Nrp2 L3 loop, the side chain of the VEGF-C C-terminal arginine, R223, forms extensive interactions with the Nrp2 binding pocket. The guanidinium of VEGF-C R223 forms a salt-bridge with the Nrp2-b1 L5 loop residue D323. Additionally, the aliphatic portion of the R223 side chain displays extensive van der Waals interactions with two tyrosine residues of Nrp2-b1 that demarcate the sides of the binding pocket, Y299 (L1 loop) and Y356 (L3 loop). In addition to interactions mediated by VEGF-C R223, there is a hydrogen bond between the backbone carbonyl of I221 and the aromatic hydroxyl of Nrp2 Y299.
Figure 2. Mechanism of VEGF-C binding to Nrp2.
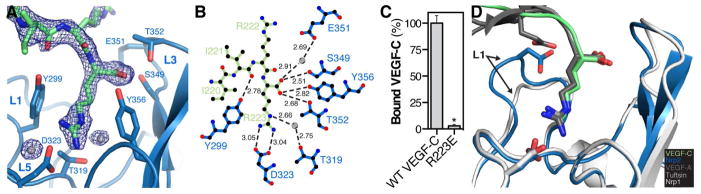
(A) Zoom of the intermolecular interface between Nrp2 (blue) and VEGF-C (green) with the 2Fo-Fc electron density map for VEGF-C contoured at 1.0σ. Interfacing water is shown as grey spheres. (B) Ligplot+ generated representation of the interaction between VEGF-C (green) and Nrp2 (blue). Bond distances (Å) are labeled in black and water is shown as grey spheres. (C) Nrp2 binding was compared between VEGF-C and VEGF-C R223E. Binding was measured in triplicate and is reported as mean ± SD (*p<0.05). (D) Superimposition of the VEGF-A HBD/Nrp1 complex (PDB=4DEQ) and the tuftsin/Nrp1 complex (PDB=2ORZ) onto the structure of the VEGF-C/Nrp2 complex demonstrates the shared and unique modes of engagement within this ligand/receptor family.
While protein-protein binding is primarily mediated by direct interactions between polypeptide chains, interfacing solvent also plays a critical role in stabilizing protein-protein complexes (Janin, 1999; Karplus and Faerman, 1994). Three water molecules, two of which bridge the interaction between VEGF-C and Nrp2, are observed in the binding site. One solvent molecule facilitates a water-mediated hydrogen bond between the side chain hydroxyl of Nrp2 T319, located at the base of the binding pocket, and the side chain guanidinium of VEGF-C R223 (Figure 2B). Likewise, a second solvent molecule bridges the side chain carboxylate of Nrp2 E351 and the free carboxylate of VEGF-C. These solvent-mediated interactions appear to further stabilize the position of the VEGF-C C-terminus within the Nrp2-b1 binding pocket.
To confirm the critical role of the VEGF-C C-terminus, we mutated the C-terminal arginine of VEGF-C to glutamate (R223E) and compared the ability of alkaline phosphatase (AP)-tagged VEGF-C and VEGF-C R223E to bind Nrp2-b1b2 affinity plates (Figure 2C). Robust binding was observed between AP-VEGF-C and Nrp2-b1b2 but R223E binding was reduced by >95%. These data demonstrate that the C-terminal arginine of mature VEGF-C is necessary for high-affinity Nrp2-b1b2 binding, and confirm the importance of C-terminal propeptide processing within VEGF-C to produce a C-terminal arginine that allows avid engagement of Nrp2.
The interaction observed between Nrp2-b1 and VEGF-C buries 374 Å2 surface area of the VEGF-C C-terminus. This is comparable to that observed for the exon 8 encoded residues of VEGF-A (338 Å2 buried surface area) (Figure 2D, dark grey) (Parker et al., 2012c) and tuftsin (328 Å2 buried surface area) (Figure 2D, light grey) (Vander Kooi et al., 2007) which complex with an equivalent binding site on Nrp1-b1. Importantly, these ligands, like VEGF-C, also contain a C-terminal arginine. All three ligands traverse the L1 loop, an orientation that is maintained by the engagement of the carboxy-terminus by the C-wall. Collectively, the shared use of a C-terminal arginine in VEGF-A and VEGF-C explains their ability to bind both Nrp receptors (Karpanen et al., 2006; Parker et al., 2012b), while electrostatic repulsion by the L1 loop and adjacent regions account for receptor selectivity (Figure 2D) (Parker et al., 2012b; Parker et al., 2012c).
Occluding the Nrp2 interloop cleft abolishes binding
The structure of VEGF-C in complex with Nrp2 reveals a critical role for the Nrp2-b1 interloop cleft, which forms the VEGF-C binding pocket. To confirm that the Nrp2 binding pocket is responsible for VEGF-C binding, we carried out site-directed mutagenesis of Nrp2-b1b2 to generate a construct with an occluded binding pocket. Specifically, T319, located at the residue at the base of the Nrp2-b1 interloop cleft, was mutated to arginine (Nrp2-T319R). We determined the crystal structure of Nrp2-T319R to a resolution of 2.4 Å (Figure 3A, Table 1). The R319 side-chain showed clear electron density extending into the interloop cleft between the two binding pocket tyrosines, Y299 and Y356 (Figure 3B). Superposing the VEGF-C/Nrp2 complex onto Nrp2-T319R demonstrates that the binding site occupied by VEGF-C is occluded in the Nrp2 mutant (Figure 3C). The Nrp2-T319R mutant was then used to analyze the contribution of the interloop cleft to VEGF-C binding. We compared the binding of VEGF-C to Nrp2-b1b2 and Nrp2-T319R (Figure 3D). While robust binding was observed between AP-VEGF-C and Nrp2-b1b2, binding to Nrp2-T319R was completely abolished. These data confirm that the interloop cleft, formed by the Nrp2-b1 coagulation factor loops, forms a structure that uniquely accommodates the C-terminus of VEGF-C to mediate binding of the C-terminally processed ligand.
Figure 3. Crystal structure and VEGF-C binding properties of Nrp2-T319R.

(A) Structure of Nrp2-T319R with the stick representation for T319R shown in red. (B) Zoom of the Nrp2-T319R binding pocket. The blue mesh illustrates the 2Fo-Fc electron density map for R319 contoured at 1.0σ. (C) Superimposition of VEGF-C (green) onto the structure of Nrp2-T319R demonstrates that the binding pocket normally occupied by VEGF-C is blocked in the mutant. (D) VEGF-C binding was compared between Nrp2-b1b2 and Nrp2-T319R. Binding was measured in triplicate and is reported as mean ± SD (*p<0.05).
A dimeric soluble Nrp2 splice form
Based on the specific binding of VEGF-C to the Nrp2 b1 domain, we hypothesized that the previously identified splice form of Nrp2, s9Nrp2, could function as a selective inhibitor of VEGF-C. s9Nrp2 is an alternative Nrp2 splice form that arises from intron inclusion in the b2 domain (Figure 4A). An in-frame stop-codon encoded within the intron is predicted to result in termination of translation prior to the transmembrane domain, and thus production of a secreted Nrp2 receptor that contains the first two CUB domains (a1 and a2) and the first coagulation factor domain (b1), but only a portion of the coding sequence for the second coagulation factor domain (b2). Given that the b1 domain of Nrp2 is solely responsible for VEGF-C binding, we hypothesized that s9Nrp2 may be able to effectively sequester VEGF-C, thereby functioning as an inhibitor. However, it is unknown whether the s9Nrp2 transcript produces a functional protein, since s9Nrp2 retains residues coding only a portion of the b2 domain (114 of 159 residues). Indeed, s9Nrp2 lacks the coding region for three of the eight core β-strands that normally integrate to form the distorted jelly-roll fold that typifies the b1 and b2 domains of Nrp. Additionally, it was unknown whether s9Nrp2 could accommodate the loss of the canonical C-terminal capping cysteine of the b2 domain. To investigate the physical and functional activity of s9Nrp2, we tested the ability of this isoform to be secreted from eukaryotic cells. We produced s9Nrp2 and a construct containing solely the ligand-binding coagulation factor domains, s9Nrp2B (Figure 4A), as a Human Growth Hormone (Hgh)-fusion in Chinese Hamster Ovary (CHO) cells. Western blot analysis demonstrated that both constructs were efficiently produced and secreted (Figure 4B). We next produced s9Nrp2B protein in bacteria. Analysis of s9Nrp2B by reducing SDS-PAGE revealed that purified s9Nrp2B, while running with a larger apparent MW than Nrp2-b1 alone, was smaller than expected from its primary sequence (Figure 4C, observed MW = 22 kDa, expected MW = 34 kDa). Mass spectrometry confirmed that s9Nrp2B is an essentially homogeneous single species with MW = 22,775 Da ± 20 Da. These data, together with the observed intact N-terminal His-tag, indicate that s9Nrp2B is cleaved C-terminal to E457 (predicted MW = 22,792 Da). Thus, the proteolyzed s9Nrp2B contains only a single cysteine residue from the b2 domain (C434), which normally forms an intradomain disulfide. Surprisingly, under non-reducing conditions, s9Nrp2B ran with an apparent MW = 38 kDa, indicating the formation of a disulfide-linked intermolecular dimer via the free b2 domain cysteine (Figure 4C). Predominantly disulfide-linked dimeric protein is also observed in s9Nrp2B protein purified from CHO-cell conditioned media (Figure S3A). The difference in oligomeric state was evident from size exclusion chromatography (SEC) (Figure 4D). Nrp2-b1 eluted off SEC with an apparent MW = 16 kDa (grey line), while s9Nrp2B had an apparent MW = 38 kDa (black line), consistent with the SDS-PAGE analysis.
Figure 4. s9Nrp2B forms a disulfide-linked dimer.

(A) Domain organization of Nrp2 and the protein fragment utilized for our studies, s9Nrp2B. (B) Western blot analysis of Hgh-tagged s9Nrp2 and s9Nrp2B expressed in CHO cells. (C) Non-reducing and reducing SDS-PAGE analysis of Nrp2-b1 and s9Nrp2B. (D) The oligomeric state of s9Nrp2B (black line) was analyzed by size-exclusion chromatography. Nrp2-b1 was run as a reference (grey line).
s9Nrp2B is a uniquely potent inhibitor of VEGF-C/Nrp2 binding
To understand the structural arrangement of the s9Nrp2B dimer, we determined the crystal structure of s9Nrp2B to a resolution of 2.4 Å (Figure 5A, Table 1). Continuous electron density was observed from F275 through S453, consistent with the C-terminus defined using mass spectrometry. A single dimer was present in the asymmetric unit, with the base of each b1 domain apposed to the other, thus forming an extended anti-parallel dimer. The orientation of the dimer is stabilized by both the intermolecular disulfide and, unexpectedly, a unique dimeric helical bundle formed by residues from the b1-b2 linker and b2 domain (Figure 5B). The residues that form this unique helix (residues 428–453) display dramatic structural reorganization relative to that observed in the intact b2-domain where they form an extended sheet and loop motif (Figure 5C). The C-terminal helix runs approximately 20° off parallel from the base of the b1 domain, an angle that is maintained by a cluster of hydrophobic residues at the hinge region between the helix and domain b1. The helix both caps the b1 domain and mediates the intermolecular interaction interface with the other monomer of the s9Nrp2B dimer. The intermolecular interface is composed of both helix-helix interactions, which are mostly hydrophobic in nature (Figure 5B), and helix-b1 interactions, which are mostly hydrophilic in nature. Truncation of the helix decreased the amount of dimeric species formed, demonstrating a role for the helix in the formation of a stable disulfide-linked dimer (Figure S3B).
Figure 5. Crystal structure and inhibitory properties of s9Nrp2B.

(A) Crystal structure of the s9Nrp2B dimer (Chain A: light orange; Chain B: dark orange). The intermolecular disulfide is shown in black and the Nrp2-b1 binding pockets are labeled with arrows. (B) Zoom of the dimeric helical bundle with the 2Fo-Fc electron density map contoured at 1σ. (C) The residues of the Nrp2 b1-b2 linker and b2 domain show a dramatic structural reorganization from an extended loop in the b1b2 sequence (blue) to an extended helix in the s9Nrp2B dimer (orange). (D) ATWLPPR (grey), Nrp2-b1 (blue), and s9Nrp2B (orange) were assayed for the ability to inhibit VEGF-C binding to Nrp2. ATWLPPR inhibited binding with an IC50 = 10 μM (log[IC50] = −4.98 ± 0.03), Nrp2-b1 inhibited binding with an IC50 = 1.5 μM (log[IC50] = −5.82 ± 0.09), and s9Nrp2B inhibited binding with an IC50 = 250 nM (log[IC50] = −6.60 ± 0.08). (E) s9Nrp2B was assayed for the ability to alter VEGF-C binding to VEGFR3. Addition of 4μM s9Nrp2B fully inhibited VEGF-C/Nrp2 binding but showed no effect on VEGF-C/VEGFR3 binding. (F) Inhibition of C4-2 cell prostatosphere formation was used to assess the biological activity of s9Nrp2B. Prostatosphere formation was compared in the absence and presence of s9Nrp2B, as well as with C-furSema (positive control) and C-Sema (negative control). (G) Model illustrating the mechanism of action for s9Nrp2B. s9Nrp2B sequesters VEGF-C and prevents activation of the VEGFR3/Nrp2 signaling complex. All inhibition experiments were measured in triplicate and reported as mean ± SD (*p<0.05).
The two binding pockets within the s9Nrp2B dimer are positioned 71 Å apart, suggesting that it could simultaneously engage both subunits of the VEGF-C dimer, which is 68 Å wide (Leppanen et al., 2010). Thus, we hypothesized that co-engagement of both VEGF-C monomers by s9Nrp2B would allow the dimer to function as a uniquely potent inhibitor of VEGF-C/Nrp2 binding. To test this hypothesis, we compared the inhibitory potency of ATWLPPR, an optimized peptide inhibitor of Nrp that functions by competitive binding (Parker and Vander Kooi, 2014; Starzec et al., 2007), with Nrp2-b1 and s9Nrp2B, both of which function as soluble competitors through sequestration of VEGF-C (Figure 5D). ATWLPPR showed dose dependent inhibition of VEGF-C binding to Nrp2 with an IC50 = 10 μM (grey line), consistent with its modest reported potency. Next, we examined the ability of Nrp2-b1 to inhibit binding (blue line). Nrp2-b1 sequestered VEGF-C with improved potency compared to the peptide inhibitor, with an IC50 = 1.5 μM. As expected for a monomeric competitive inhibitor, the Hill slope was approximately −1 (ATWLPPR = −1.08 and Nrp2-b1 = −0.97). These data are consistent with independent engagement of each VEGF-C monomer by a single Nrp2-b1. Next, we measured the inhibitory potency of s9Nrp2B (orange line). Strikingly, s9Nrp2B potently sequestered VEGF-C with an IC50 = 250 nM, a significant improvement in potency from both the peptide inhibitor and Nrp2-b1. Additionally, the Hill slope for s9Nrp2B was −1.5. Thus, the enhanced potency of s9Nrp2B is due to its ability to synergistically sequester the VEGF-C dimer through simultaneous and cooperative engagement of the two VEGF-C monomers.
VEGF-C signaling requires the coordinated action of Nrp2 and the receptor tyrosine kinase VEGFR3 (Xu et al., 2010). Nrp2 enhances signaling via VEGF-C through both a direct interaction with VEGF-C and via coupling with VEGFR3 (Favier et al., 2006). Thus, while s9Nrp2B could inhibit VEGF-C signaling by sequestering VEGF-C ligand from Nrp2, it is also possible that s9Nrp2B could interact with VEGFR3 and actually enhance VEGF-C binding and signaling. Therefore, we tested the effect of s9Nrp2B on VEGF-C binding to VEGFR3. While s9Nrp2B blocked VEGF-C binding to Nrp2, it showed no effect on VEGF-C binding to VEGFR3, indicating that the binding events are independent (Figure 5E).
We extended our studies to assess the efficacy of s9Nrp2B as an inhibitor of Nrp2 signaling in prostate cancer. Nrp2 and VEGF-C expression have both been reported to function in the survival and aggressiveness of prostate cancer (Goel et al., 2012; Muders et al., 2009). We assessed the ability of Nrp inhibition to reduce the formation of prostatospheres by C4-2 cells. Incubation with s9Nrp2B resulted in a significant decrease in prostatosphere formation (Figure 5F). Incubation with the specific Nrp inhibitor C-furSema (Goel et al., 2013; Parker et al., 2010) likewise significantly reduced prostatosphere formation whereas the control C-Sema did not, demonstrating the specific role for Nrp in prostatosphere formation. These data demonstrate that s9Nrp2B can effectively sequester VEGF-C and raises the exciting possibility of using engineered Nrp ectodomains as inhibitors of pathological Nrp-dependent signaling (Figure 5G).
DISCUSSION
Structural characterization of the mechanism for VEGF-C binding to Nrp2 represents an important step for understanding the physiological and pathological activity of VEGF-C. These data also inform the rational design of specific VEGF-C/D antagonists, including s9Nrp2B, which potently inhibits VEGF-C/Nrp2 binding and represents a potential therapeutic avenue. Collectively, these results have important implications for interpreting both the aberrant loss and gain of function in the VEGF-C/Nrp2 signaling axis that critically underlies a number of disease states.
With complementary biochemical and structural approaches we show that VEGF-C C-terminal proteolysis is required for Nrp2 binding. The requirement for proteolytic processing is determined by the position of the Nrp2 C-wall, formed by the L3 coagulation factor loop residues, which specifically engages the VEGF-C free carboxy-terminus, precluding binding of unprocessed protein. These results provide critical insight for interpreting the altered in vitro and in vivo functionality of alternative VEGF-C forms. While both N- and C-terminal processing regulate VEGF-C activity (Joukov et al., 1997; McColl et al., 2003), processing at these sites is not functionally equivalent. Indeed, loss of C-terminal processing is uniquely detrimental, fully ablating VEGF-C function in vivo (Khatib et al., 2010), which we demonstrate blocks Nrp2 binding.
The loss of VEGF-C binding to Nrp2-T319R, a mutant with an occluded binding pocket, demonstrates the use of a C-terminal arginine for ligand engagement. Indeed, the VEGF-C C-terminal arginine side chain and free carboxylate form extensive interactions with the Nrp2-b1 binding pocket. Interestingly, VEGF-C is not the only VEGF family member that, in the absence of post-translational modification, lacks a C-terminal arginine. Of the five VEGF family members, three contain Nrp-binding domains that lack this structural motif (VEGF-C, VEGF-D, and VEGF-B186). VEGF-D, a close structural and functional homologue of VEGF-C, is processed at an equivalent site in its C-terminus to produce a C-terminal arginine (Stacker et al., 1999) and thus likely utilizes a similar binding mode to Nrp2. This observation provides additional functional insight, as loss of VEGF-D C-terminal processing also ablates function in vivo (Harris et al., 2013). There are three VEGF-B isoforms, VEGF-B167, VEGF-B127, and VEGF-B186, all of which differ in their C-terminal domain (Olofsson et al., 1996a; Olofsson et al., 1996b). Characterization of VEGF-B186 demonstrated that it exhibited proteolytic-dependent binding to Nrp1 and identified the site of proteolysis as R227 (Makinen et al., 1999). Thus, the mechanism of proteolytic-dependent VEGF-C binding to Nrp2 has broad explanatory power for understanding Nrp binding across the VEGF family.
Determining the structural basis for VEGF-C signaling via Nrp2 informs ongoing studies to describe the effect of signaling deficiency on human disease. Deficient VEGF-C signaling via Nrp2 has significant implications for both primary and secondary lymphedema. Mutations in both VEGFR-3 (Karkkainen et al., 2000) and VEGF-C (Gordon et al., 2013) have been demonstrated to underlie hereditary lymphedema and Nrp2 has been identified as an additional candidate gene (Ferrell et al., 2008; Karkkainen et al., 2001). Additionally, both VEGF-C and Nrp2 have recently been identified as candidate genes for the development of secondary lymphedema following surgery in breast cancer (Miaskowski et al., 2013). The structural insights gleaned from the VEGF-C/Nrp2 complex also provide an important molecular basis for interpreting emerging exome sequencing data that has identified Nrp2 variants in close proximity to the ligand binding interface. Intriguingly, a stringent examination of exome sequencing data has reported both common and rare Nrp2 variants in human populations (Tennessen et al., 2012). Several of these variants are located in the coagulation factor loops of Nrp2-b1, the region to which VEGF-C binds. Specifically, there are two reported variants in the L5 loop (N321I and L322M) that are located proximal to the critical salt-bridge formed by D323, and two in the L3 loop (Q353H and N354K). The structural data presented here provides a rationale for examining specific coagulation factor loop variants for loss of function on both a physical and functional level.
As opposed to aberrant VEGF-C loss of function in lymphedema, aberrant activation of VEGF-C signaling via Nrp2 is associated with cancer initiation, survival, and progression (Ellis, 2006; Stacker et al., 2002). The Nrp2/VEGF-C signaling axis contributes to tumorigenesis via multiple mechanisms. Mimicking its physiological function, VEGF-C signaling via Nrp2 stimulates lymphatic vessel recruitment to tumors and directly contributes to cancer metastasis (Caunt et al., 2008). Importantly, the role of VEGF-C and Nrp2 in tumorigenesis is not exclusively associated with aberrant lymphangiogenesis. Indeed, in situ studies have demonstrated that autocrine VEGF-C signaling in breast cancer cells stimulates cellular motility (Timoshenko et al., 2007). Further, recent reports indicate that cancer cell survival is enhanced through VEGF-C/Nrp2-dependent autophagy (Stanton et al., 2012) and that autocrine Nrp2 signaling maintains the population of cancer stem cells (Goel et al., 2013). VEGF-C also functions to protect prostate cancer cells from oxidative stress in a Nrp2-dependent fashion (Muders et al., 2009). Thus, selective inhibition of Nrp2 represents a promising, multipronged anti-cancer therapeutic strategy.
Secreted splice forms of angiogenic receptors have essential roles in vivo (Albuquerque et al., 2009; Ambati et al., 2006; Kendall and Thomas, 1993) and have been engineered to serve as therapeutic inhibitors that block aberrant pathway activation by ligand sequestration (Stewart, 2012). Here we demonstrate that the alternative Nrp2 splice form, s9Nrp2B, potently sequesters VEGF-C and inhibits binding to Nrp2. The biological function and localized tissue-specific expression of s9Nrp2 is of significant interest. Indeed, s9Nrp2 may be analogous or complementary to sVEGFR-2, the secreted splice form of VEGFR-2 which functions as an endogenous lymphangiogenesis inhibitor (Albuquerque et al., 2009). VEGF-D also functions in lymphatic angiogenesis and has been shown to have partially overlapping biological function with VEGF-C and important pathological functions (Haiko et al., 2008; Harris et al., 2013; Karpanen et al., 2006). The conservation of Nrp2-interacting residues between VEGF-C and VEGF-D strongly suggest that s9Nrp2B will equivalently sequester both VEGF-C and VEGF-D. In contrast, the heparin-binding domain of VEGF-A contains specificity determinants that limit binding to Nrp2 (Parker et al., 2012b; Parker et al., 2012c). Thus, s9Nrp2B is likely to selectively sequester the lymphangiogenic-specific VEGF family members, VEGF-C and VEGF-D.
The practice of engineering inhibitor multimerization to increase potency is well established for soluble receptor fragments. Most commonly, soluble receptors are dimerized by expression as an Fc fusion protein (e.g. VEGF-trap). s9Nrp2B represents a unique mechanism for generation of a multimeric protein that maintains the benefits of avidity but does not require introduction of an exogenous polypeptide sequence. Additional optimization of s9Nrp2B potency, selectivity, and stability is an important future direction for the development of a therapeutically useful inhibitor.
EXPERIMENTAL PROCEDURES
Protein expression and purification
Human Nrp2-b1b2 (residues 276–595), human Nrp2-b1 (residues 276–430), human Nrp2-T319R (residues 276–595 with T319R mutation), s9Nrp2B (residues 275–555: isoform O60462-6), s9Nrp2B-Δ-helix (residues 276–436) and the Nrp2-b1b2/VEGF-C fusion were expressed in E. coli as His-tag fusion proteins from pET28b (Merck KGaA, Darmstadt, Germany). Proteins were purified via immobilized metal ion affinity chromatography (IMAC) and either heparin affinity or SEC. AP-VEGF-C (residues 108–223) wild-type and mutant and Hgh-tagged proteins were produced by transient transfection of CHO cells (Aricescu et al., 2006). The VEGFR-3 extracellular domain was produced via baculovirus mediated expression (residues 21–776) and purified by IMAC and SEC.
Structure determination
Purified Nrp2-b1b2-VEGF-C fusion, Nrp2-T319R, and s9Nrp2B were concentrated to 2.0 mg/mL, 2.1 mg/mL and 3.5 mg/mL, respectively, and crystals grown by hanging-drop vapor-diffusion experiments. Fusion protein crystals were obtained in two weeks at RT in 0.1 M MES pH 6.5, 0.5 M ammonium sulfate. Nrp2-T319R crystals were obtained in five days at RT in 0.1 M HEPES pH 7, 18% (w/v) PEG 12000. s9Nrp2B crystals were obtained in two week at RT in 10% PEG 1000/10% PEG 8000. Crystals were passed through mother liquor supplemented with 10% glycerol and then flash frozen in liquid nitrogen. Diffraction data were collected at the SER-CAT 22-ID and 22-BM beamlines of the Advanced Photon Source, Argonne National Laboratories, and processed using HKL2000 (Otwinowski and Minor, 1997). Structures were solved by molecular replacement using Nrp2-b1b2 (PDB=2QQJ) followed by iterative modeling building and refinement using COOT (Emsley et al., 2010) and Refmac5 (Murshudov, 1997) to generate a final refined model (Table 1).
Differential scanning fluorimetry (DSF)
Peptides corresponding to processed and unprocessed VEGF-C were produced with an N-terminal tryptophan to allow accurate quantitation by UV280 absorbance (LifeTein LLC, Hillsborough, NJ). Peptides were resuspended and combined with 2 μM of Nrp2-b1b2 and 5x SYPRO Orange Protein Gel Stain (Life Technologies, Grand Island, NY) in PBS. Nrp2-b1b2 melting was monitored on a CFX96 Real-Time PCR system (BioRad, Hercules, CA) from 20 to 90°C at a rate of 1°C/50s with fluorescent readi ngs taken every 1°C.
Binding and inhibition assays
Plate binding and soluble Nrp competition assays were performed by measuring the binding of AP-tagged VEGF-C to Nrp2-b1b2, Nrp2-T319R, or VEGFR3 affinity plates. For direct binding assays, ligand was directly added to Nrp2-affinity plates, incubated for 1 hr at RT, washed, and developed using p-Nitrophenyl Phosphate (pNPP) AP substrate. For competition experiments, ligand was pre-mixed with inhibitor and then added to affinity plates as with binding.
Prostatosphere assays
Prostatosphere cultures utilized C4-2 prostate cancer cells (UroCor, Inc., Oklahoma City, OK) (Cao et al., 2011). 5000 cells/well were cultured in suspension in serum-free DMEM-F12 (Life Technologies), supplemented with B27 (1:50, Life Technology), 20 ng/mL EGF (Peprotech, Rocky Hill, NJ), and 4 μg/mL insulin (Sigma-Aldrich, St. Louis, MO) in 6-well ultra-low attachment plates (Corning, Corning, NY). s9Nrp2B, C-furSema, or C-Sema inhibitors were added at a concentration of 5.0 μM while plating the cells. The prostatospheres were cultured for 6 days and 1 ml of culture medium was added every other day. Spheres larger than 100 μm were counted.
Supplementary Material
HIGHLIGHTS.
C-terminal processing of VEGF-C regulates direct binding to Nrp2
Structure of the VEGF-C/Nrp2 complex demonstrates the basis for ligand binding
Alternative Nrp2 splicing produces a soluble, dimeric receptor with novel structure
Soluble Nrp2 functions as a potent antagonist of VEGF-C/Nrp2 binding
Acknowledgments
We thank Dr. Carol Beach for assistance with mass spectrometry and Dr. Hou-Fu Guo and Xiaobo Li for valuable advice and technical help. This work was supported by National Institutes of Health (NIH) Grants R01GM094155 (C.W.V.K), T32HL072743 (M.W.P.), R01CA168464 (A.M.M., H.L.G.) and P30GM103486 (Core support), and NSF REU DBI-1004931 (A.D.L.).
Footnotes
The authors declare no conflict of interest.
ACCESSION NUMBERS
Coordinates and structure factors have been deposited in the Protein Data Bank (PDB), www.pdb.org, with accession codes 4QDQ, 4QDR, 4QDS for the complex, T319R, and dimer structures, respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams RH, Lohrum M, Klostermann A, Betz H, Puschel AW. The chemorepulsive activity of secreted semaphorins is regulated by furin-dependent proteolytic processing. EMOB J. 1997;16:6077–6086. doi: 10.1093/emboj/16.20.6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque RJ, Hayashi T, Cho WG, Kleinman ME, Dridi S, Takeda A, Baffi JZ, Yamada K, Kaneko H, Green MG, et al. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat Med. 2009;15:1023–1030. doi: 10.1038/nm.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambati BK, Nozaki M, Singh N, Takeda A, Jani PD, Suthar T, Albuquerque RJ, Richter E, Sakurai E, Newcomb MT, et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature. 2006;443:993–997. doi: 10.1038/nature05249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton BA, Wu P, Maloney J, Yin J, Liang WC, Stawicki S, Mortara K, Bowman KK, Elliott JM, Desmarais W, et al. Structural studies of neuropilin/antibody complexes provide insights into semaphorin and VEGF binding. EMBO J. 2007;26:4902–4912. doi: 10.1038/sj.emboj.7601906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aricescu AR, Lu W, Jones EY. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr D, Biol Crystallogr. 2006;62:1243–1250. doi: 10.1107/S0907444906029799. [DOI] [PubMed] [Google Scholar]
- Baldwin ME, Halford MM, Roufail S, Williams RA, Hibbs ML, Grail D, Kubo H, Stacker SA, Achen MG. Vascular endothelial growth factor D is dispensable for development of the lymphatic system. Mol Cell Biol. 2005;25:2441–2449. doi: 10.1128/MCB.25.6.2441-2449.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Mani RS, Ateeq B, Dhanasekaran SM, Asangani IA, Prensner JR, Kim JH, Brenner JC, Jing X, Cao X, et al. Coordinated regulation of polycomb group complexes through microRNAs in cancer. Cancer Cell. 2011;20:187–199. doi: 10.1016/j.ccr.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caunt M, Mak J, Liang WC, Stawicki S, Pan Q, Tong RK, Kowalski J, Ho C, Reslan HB, Ross J, et al. Blocking neuropilin-2 function inhibits tumor cell metastasis. Cancer Cell. 2008;13:331–342. doi: 10.1016/j.ccr.2008.01.029. [DOI] [PubMed] [Google Scholar]
- Ellis LM. The role of neuropilins in cancer. Mol Cancer Ther. 2006;5:1099–1107. doi: 10.1158/1535-7163.MCT-05-0538. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D, Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favier B, Alam A, Barron P, Bonnin J, Laboudie P, Fons P, Mandron M, Herault JP, Neufeld G, Savi P, et al. Neuropilin-2 interacts with VEGFR-2 and VEGFR-3 and promotes human endothelial cell survival and migration. Blood. 2006;108:1243–1250. doi: 10.1182/blood-2005-11-4447. [DOI] [PubMed] [Google Scholar]
- Ferrell RE, Kimak MA, Lawrence EC, Finegold DN. Candidate gene analysis in primary lymphedema. Lymphat Res Biol. 2008;6:69–76. doi: 10.1089/lrb.2007.1022. [DOI] [PubMed] [Google Scholar]
- Gagnon ML, Bielenberg DR, Gechtman Z, Miao HQ, Takashima S, Soker S, Klagsbrun M. Identification of a natural soluble neuropilin-1 that binds vascular endothelial growth factor: In vivo expression and antitumor activity. P Natl Acad Sci USA. 2000;97:2573–2578. doi: 10.1073/pnas.040337597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel HL, Chang C, Pursell B, Leav I, Lyle S, Xi HS, Hsieh CC, Adisetiyo H, Roy-Burman P, Coleman IM, et al. VEGF/neuropilin-2 regulation of Bmi-1 and consequent repression of IGF-IR define a novel mechanism of aggressive prostate cancer. Cancer Disc. 2012;2:906–921. doi: 10.1158/2159-8290.CD-12-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel HL, Pursell B, Chang C, Shaw LM, Mao J, Simin K, Kumar P, Vander Kooi CW, Shultz LD, Greiner DL, et al. GLI1 regulates a novel neuropilin-2/alpha6beta1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO Mol Med. 2013;5:488–508. doi: 10.1002/emmm.201202078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon K, Schulte D, Brice G, Simpson MA, Roukens MG, van Impel A, Connell F, Kalidas K, Jeffery S, Mortimer PS, et al. Mutation in vascular endothelial growth factor-C, a ligand for vascular endothelial growth factor receptor-3, is associated with autosomal dominant milroy-like primary lymphedema. Circ Res. 2013;112:956–960. doi: 10.1161/CIRCRESAHA.113.300350. [DOI] [PubMed] [Google Scholar]
- Haiko P, Makinen T, Keskitalo S, Taipale J, Karkkainen MJ, Baldwin ME, Stacker SA, Achen MG, Alitalo K. Deletion of vascular endothelial growth factor C (VEGF-C) and VEGF-D is not equivalent to VEGF receptor 3 deletion in mouse embryos. Mol Cell Biol. 2008;28:4843–4850. doi: 10.1128/MCB.02214-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris NC, Davydova N, Roufail S, Paquet-Fifield S, Paavonen K, Karnezis T, Zhang YF, Sato T, Rothacker J, Nice EC, et al. The propeptides of VEGF-D determine heparin binding, receptor heterodimerization, and effects on tumor biology. J Biol Chem. 2013;288:8176–8186. doi: 10.1074/jbc.M112.439299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Tessier-Lavigne M. Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell. 1997;90:739–751. doi: 10.1016/s0092-8674(00)80534-6. [DOI] [PubMed] [Google Scholar]
- Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R, Hylton D, Burova E, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. P Natl Acad Sci USA. 2002;99:11393–11398. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes DI, Zachary I. The vascular endothelial growth factor (VEGF) family: angiogenic factors in health and disease. Genome Biol. 2005;6:209. doi: 10.1186/gb-2005-6-2-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin J. Wet and dry interfaces: the role of solvent in protein–protein and protein–DNA recognition. Structure. 1999;7:R277–R279. doi: 10.1016/s0969-2126(00)88333-1. [DOI] [PubMed] [Google Scholar]
- Jeltsch M, Kaipainen A, Joukov V, Meng X, Lakso M, Rauvala H, Swartz M, Fukumura D, Jain RK, Alitalo K. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science. 1997;276:1423–1425. doi: 10.1126/science.276.5317.1423. [DOI] [PubMed] [Google Scholar]
- Joukov V, Pajusola K, Kaipainen A, Chilov D, Lahtinen I, Kukk E, Saksela O, Kalkkinen N, Alitalo K. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996;15:1751. [PMC free article] [PubMed] [Google Scholar]
- Joukov V, Sorsa T, Kumar V, Jeltsch M, Claesson-Welsh L, Cao Y, Saksela O, Kalkkinen N, Alitalo K. Proteolytic processing regulates receptor specificity and activity of VEGF-C. EMBO J. 1997;16:3898–3911. doi: 10.1093/emboj/16.13.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkkainen MJ, Ferrell RE, Lawrence EC, Kimak MA, Levinson KL, McTigue MA, Alitalo K, Finegold DN. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet. 2000;25:153–159. doi: 10.1038/75997. [DOI] [PubMed] [Google Scholar]
- Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, Jeltsch M, Jackson DG, Talikka M, Rauvala H, et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. 2004;5:74–80. doi: 10.1038/ni1013. [DOI] [PubMed] [Google Scholar]
- Karkkainen MJ, Saaristo A, Jussila L, Karila KA, Lawrence EC, Pajusola K, Bueler H, Eichmann A, Kauppinen R, Kettunen MI, et al. A model for gene therapy of human hereditary lymphedema. P Natl Acad Sci USA. 2001;98:12677–12682. doi: 10.1073/pnas.221449198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpanen T, Heckman CA, Keskitalo S, Jeltsch M, Ollila H, Neufeld G, Tamagnone L, Alitalo K. Functional interaction of VEGF-C and VEGF-D with neuropilin receptors. FASEB J. 2006;20:1462–1472. doi: 10.1096/fj.05-5646com. [DOI] [PubMed] [Google Scholar]
- Karplus PA, Faerman C. Ordered water in macromolecular structure. Curr Opin Struc Biol. 1994;4:770–776. [Google Scholar]
- Kendall RL, Thomas KA. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. P Natl Acad Sci USA. 1993;90:10705–10709. doi: 10.1073/pnas.90.22.10705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatib AM, Lahlil R, Scamuffa N, Akimenko MA, Ernest S, Lomri A, Lalou C, Seidah NG, Villoutreix BO, Calvo F, et al. Zebrafish ProVEGF-C expression, proteolytic processing and inhibitory effect of unprocessed ProVEGF-C during fin regeneration. PloS ONE. 2010;5:e11438. doi: 10.1371/journal.pone.0011438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukk E, Lymboussaki A, Taira S, Kaipainen A, Jeltsch M, Joukov V, Alitalo K. VEGF-C receptor binding and pattern of expression with VEGFR–3 suggests a role in lymphatic vascular development. Development. 1996;122:3829–3837. doi: 10.1242/dev.122.12.3829. [DOI] [PubMed] [Google Scholar]
- Leppanen VM, Prota AE, Jeltsch M, Anisimov A, Kalkkinen N, Strandin T, Lankinen H, Goldman A, Ballmer-Hofer K, Alitalo K. Structural determinants of growth factor binding and specificity by VEGF receptor 2. P Natl Acad Sci USA. 2010;107:2425–2430. doi: 10.1073/pnas.0914318107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppanen VM, Tvorogov D, Kisko K, Prota AE, Jeltsch M, Anisimov A, Markovic-Mueller S, Stuttfeld E, Goldie KN, Ballmer-Hofer K, et al. Structural and mechanistic insights into VEGF receptor 3 ligand binding and activation. P Natl Acad Sci USA. 2013;110:12960–12965. doi: 10.1073/pnas.1301415110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohela M, Bry M, Tammela T, Alitalo K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol. 2009;21:154–165. doi: 10.1016/j.ceb.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Lymboussaki A, Olofsson B, Eriksson U, Alitalo K. Vascular endothelial growth factor (VEGF) and VEGF-C show overlapping binding sites in embryonic endothelia and distinct sites in differentiated adult endothelia. Circ Res. 1999;85:992–999. doi: 10.1161/01.res.85.11.992. [DOI] [PubMed] [Google Scholar]
- Makinen T, Olofsson B, Karpanen T, Hellman U, Soker S, Klagsbrun M, Eriksson U, Alitalo K. Differential binding of vascular endothelial growth factor B splice and proteolytic isoforms to neuropilin-1. J Biol Chem. 1999;274:21217–21222. doi: 10.1074/jbc.274.30.21217. [DOI] [PubMed] [Google Scholar]
- McColl BK, Baldwin ME, Roufail S, Freeman C, Moritz RL, Simpson RJ, Alitalo K, Stacker SA, Achen MG. Plasmin activates the lymphangiogenic growth factors VEGF-C and VEGF-D. J Exp Med. 2003;198:863–868. doi: 10.1084/jem.20030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miaskowski C, Dodd M, Paul SM, West C, Hamolsky D, Abrams G, Cooper BA, Elboim C, Neuhaus J, Schmidt BL, et al. Lymphatic and angiogenic candidate genes predict the development of secondary lymphedema following breast cancer surgery. PloS ONE. 2013;8:e60164. doi: 10.1371/journal.pone.0060164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muders MH, Zhang H, Wang E, Tindall DJ, Datta K. Vascular endothelial growth factor-C protects prostate cancer cells from oxidative stress by the activation of mammalian target of rapamycin complex-2 and AKT-1. Cancer Res. 2009;69:6042–6048. doi: 10.1158/0008-5472.CAN-09-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D, Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Olofsson B, Pajusola K, Kaipainen A, von Euler G, Joukov V, Saksela O, Orpana A, Pettersson RF, Alitalo K, Eriksson U. Vascular endothelial growth factor B, a novel growth factor for endothelial cells. P Natl Acad Sci USA. 1996a;93:2576–2581. doi: 10.1073/pnas.93.6.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson B, Pajusola K, von Euler G, Chilov D, Alitalo K, Eriksson U. Genomic organization of the mouse and human genes for vascular endothelial growth factor B (VEGF-B) and characterization of a second splice isoform. J Biol Chem. 1996b;271:19310–19317. doi: 10.1074/jbc.271.32.19310. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Parker MW, Guo HF, Li X, Linkugel AD, Vander Kooi CW. Function of members of the neuropilin family as essential pleiotropic cell surface receptors. Biochemistry. 2012a;51:9437–9446. doi: 10.1021/bi3012143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MW, Hellman LM, Xu P, Fried MG, Vander Kooi CW. Furin processing of semaphorin 3F determines its anti-angiogenic activity by regulating direct binding and competition for neuropilin. Biochemistry. 2010;49:4068–4075. doi: 10.1021/bi100327r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MW, Linkugel AD, Vander Kooi CW. Effect of C-Terminal Sequence on Competitive Semaphorin Binding to Neuropilin-1. J Mol Biol. 2013;425:4405–4414. doi: 10.1016/j.jmb.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MW, Vander Kooi CW. Microplate-based screening for small molecule inhibitors of neuropilin-2/vascular endothelial growth factor-C interactions. Anal Biochem. 2014;453:4–6. doi: 10.1016/j.ab.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MW, Xu P, Guo HF, Vander Kooi CW. Mechanism of Selective VEGF-A Binding by Neuropilin-1 Reveals a Basis for Specific Ligand Inhibition. PloS ONE. 2012;7:e49177. doi: 10.1371/journal.pone.0049177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MW, Xu P, Li X, Vander Kooi CW. Structural basis for selective vascular endothelial growth factor-A (VEGF-A) binding to neuropilin-1. J Biol Chem. 2012;287:11082–11089. doi: 10.1074/jbc.M111.331140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem J. 1994;300(Pt 2):281–290. doi: 10.1042/bj3000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol M, Gagnon ML, Klagsbrun M. Genomic organization of human neuropilin-1 and neuropilin-2 genes: identification and distribution of splice variants and soluble isoforms. Genomics. 2000;70:211–222. doi: 10.1006/geno.2000.6381. [DOI] [PubMed] [Google Scholar]
- Saaristo A, Karkkainen MJ, Alitalo K. Insights into the molecular pathogenesis and targeted treatment of lymphedema. Ann NY Acad Sci. 2002;979:94–110. doi: 10.1111/j.1749-6632.2002.tb04871.x. [DOI] [PubMed] [Google Scholar]
- Siegfried G, Basak A, Cromlish JA, Benjannet S, Marcinkiewicz J, Chretien M, Seidah NG, Khatib AM. The secretory proprotein convertases furin, PC5, and PC7 activate VEGF-C to induce tumorigenesis. J Clin Invest. 2003;111:1723–1732. doi: 10.1172/JCI17220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soker S, Fidder H, Neufeld G, Klagsbrun M. Characterization of novel vascular endothelial growth factor (VEGF) receptors on tumor cells that bind VEGF165 via its exon 7-encoded domain. J Biol Chem. 1996;271:5761–5767. doi: 10.1074/jbc.271.10.5761. [DOI] [PubMed] [Google Scholar]
- Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- Stacker SA, Achen MG, Jussila L, Baldwin ME, Alitalo K. Lymphangiogenesis and cancer metastasis. Nat Rev Cancer. 2002;2:573–583. doi: 10.1038/nrc863. [DOI] [PubMed] [Google Scholar]
- Stacker SA, Stenvers K, Caesar C, Vitali A, Domagala T, Nice E, Roufail S, Simpson RJ, Moritz R, Karpanen T, et al. Biosynthesis of vascular endothelial growth factor-D involves proteolytic processing which generates non-covalent homodimers. J Biol Chem. 1999;274:32127–32136. doi: 10.1074/jbc.274.45.32127. [DOI] [PubMed] [Google Scholar]
- Stanton MJ, Dutta S, Zhang H, Polavaram NS, Leontovich AA, Honscheid P, Sinicrope FA, Tindall DJ, Muders MH, Datta K. Autophagy control by VEGF-C/NRP-2 axis in cancer and its implication for treatment resistance. Cancer Res. 2012;73:160–171. doi: 10.1158/0008-5472.CAN-11-3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starzec A, Ladam P, Vassy R, Badache S, Bouchemal N, Navaza A, du Penhoat CH, Perret GY. Structure-function analysis of the antiangiogenic ATWLPPR peptide inhibiting VEGF(165) binding to neuropilin-1 and molecular dynamics simulations of the ATWLPPR/neuropilin-1 complex. Peptides. 2007;28:2397–2402. doi: 10.1016/j.peptides.2007.09.013. [DOI] [PubMed] [Google Scholar]
- Stewart MW. Aflibercept (VEGF Trap-eye): the newest anti-VEGF drug. Brit J Ophthalmol. 2012;96:1157–1158. doi: 10.1136/bjophthalmol-2011-300654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuttfeld E, Ballmer-Hofer K. Structure and function of VEGF receptors. IUBMB Life. 2009;61:915–922. doi: 10.1002/iub.234. [DOI] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timoshenko AV, Rastogi S, Lala PK. Migration-promoting role of VEGF-C and VEGF-C binding receptors in human breast cancer cells. Brit J Cancer. 2007;97:1090–1098. doi: 10.1038/sj.bjc.6603993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Kooi CW, Jusino MA, Perman B, Neau DB, Bellamy HD, Leahy DJ. Structural basis for ligand and heparin binding to neuropilin B domains. P Natl Acad Sci USA. 2007;104:6152–6157. doi: 10.1073/pnas.0700043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Wronski MA, Raju N, Pillai R, Bogdan NJ, Marinelli ER, Nanjappan P, Ramalingam K, Arunachalam T, Eaton S, Linder KE, et al. Tuftsin binds neuropilin-1 through a sequence similar to that encoded by exon 8 of vascular endothelial growth factor. J Biol Chem. 2006;281:5702–5710. doi: 10.1074/jbc.M511941200. [DOI] [PubMed] [Google Scholar]
- Xu Y, Yuan L, Mak J, Pardanaud L, Caunt M, Kasman I, Larrivee B, Del Toro R, Suchting S, Medvinsky A, et al. Neuropilin-2 mediates VEGF-C-induced lymphatic sprouting together with VEGFR3. J Cell Biol. 2010;188:115–130. doi: 10.1083/jcb.200903137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Moyon D, Pardanaud L, Breant C, Karkkainen MJ, Alitalo K, Eichmann A. Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development. 2002;129:4797–4806. doi: 10.1242/dev.129.20.4797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.