

Summary
Hyperpolarization-activated cyclic nucleotide-gated (HCN) ion channels underlie the cationic Ih current present in many neurons. The direct binding of cAMP to HCN channels increases the rate and extent of channel opening and results in a depolarizing shift in the voltage dependence of activation. TRIP8b is an accessory protein that regulates the cell surface expression and dendritic localization of HCN channels and reduces the cyclic nucleotide dependence of these channels. Here we use electron paramagnetic resonance (EPR) to show that TRIP8b binds to the apo state of the cyclic nucleotide-binding domain (CNBD) of HCN2 channels without changing the overall domain structure. With EPR and nuclear magnetic resonance (NMR), we locate TRIP8b relative to the HCN channel and identify the binding interface on the CNBD. These data provide a structural framework for understanding how TRIP8b regulates the cyclic nucleotide dependence of HCN channels.
Introduction
In the brain, hyperpolarization-activated cyclic nucleotide-gated (HCN) ion channels contribute to rhythmic firing, dendritic excitability, and resting membrane potential in many different types of neurons (Robinson and Siegelbaum, 2003). These cellular functions play critical roles in learning, sleep, and depression (He et al., 2014; Lörincz et al., 2002; Nolan et al., 2003). The expression and regulation of HCN channels varies dramatically among different neuronal subtypes. In pyramidal and neocortical neurons, they show a large gradient of expression that can be as much as 60-fold higher in the distal dendrites than in the soma (Lörincz et al., 2002). In many pathological conditions, including epilepsy and Parkinson’s disease, disruption of the gating, regulation, or localization of HCN channels plays a crucial role in neuronal dysfunction (Chan et al., 2011; He et al., 2014; Lörincz et al., 2002; Nolan et al., 2003; Reid et al., 2012).
Two features differentiate HCN channels from other members of the voltage-activated channel family: 1) they open in response to membrane hyperpolarization as opposed to depolarization, and 2) they are allosterically regulated by the direct binding of cyclic 3’,5’-cyclic adenosine monophosphate (cAMP) (Craven and Zagotta, 2006; Robinson and Siegelbaum, 2003). Binding of cAMP to HCN channels increases the rate and extent of activation and shifts the voltage dependence of activation to more depolarized voltages (Craven and Zagotta, 2006). The cyclic nucleotide dependence of HCN channels arises from a series of conformational changes in a pair of intracellular domains known as the cyclic nucleotide-binding domain (CNBD) and the C-linker (Figure 1A). The C-linker connects the CNBD to the channel pore and is critical for coupling the conformational changes associated with binding of ligand in the CNBD to the opening of the pore (Craven et al., 2008; Puljung and Zagotta, 2013; Puljung et al., 2014; Taraska and Zagotta, 2007). The CNBD and C-linker are characteristic features of the cyclic nucleotide-regulated family of channels, which consists of HCN, cyclic nucleotide-gated (CNG), and KCNH channels (Craven and Zagotta, 2006).
Figure 1. TRIP8bcore binds to and regulated HCN2 channels.

A) Cartoon showing the interactions between HCN channels and full length TRIP8b. The intracellular C-linker and the CNBD of HCN2 are labeled. Only two of the four subunits are shown. The variable, core, and TPR domains of TRIP8b are highlighted (see also Figure S1). B) Representative current traces elicited by hyperpolarizing voltage steps from inside-out patches of oocytes expressing HCN channels with no ligand (left), in the presence of 1 μM cAMP (center), and in the presence of both 1 μM cAMP and 10 μM TRIP8bcore (right). (c) Normalized conductance-voltage relationships of HCN alone (cyan), in the presence of 1μM cAMP (red), and in the presence of both 1 μM cAMP and 10 μM TRIP8bcore (black). Data are represented as mean ± SEM.
In addition to its activation by membrane hyperpolarization and cAMP, HCN channels are also regulated by a cytoplasmic accessory protein called TRIP8b (tetratricopeptide repeat-containing Rab8b-interacting protein) (Santoro et al., 2004). TRIP8b is a highly alternatively spliced protein that regulates HCN channel cell surface expression in an isoform-dependent manner (Lewis et al., 2009; Santoro et al., 2009; Zolles et al., 2009). TRIP8b has been shown to co-localize with HCN1 channels in the distal dendrites of hippocampal pyramidal neurons, and TRIP8b knockout mice show a striking disruption in this expression pattern (Lewis et al., 2011; Piskorowski et al., 2011).
Structurally, TRIP8b is known to interact at two sites on the carboxy-terminal region of HCN channels (Bankston et al., 2012; Han et al., 2011; Lewis et al., 2009; Santoro et al., 2011) (Figure 1A). A series of tetratricopeptide repeats (TPRs) on TRIP8b interact with the terminal tripeptide sequence of HCN channels with high affinity. This site is thought to anchor TRIP8b onto HCN channels. The structure of this C-terminal interaction has been determined at atomic resolution (Bankston et al., 2012). The second site involves an interaction between a segment of TRIP8b containing residues 223-303, termed TRIP8bcore (Figure 1A and Figure S1A), and the CNBD. This interaction dramatically reduces the cAMP regulation of HCN channels (Lewis et al., 2009; Santoro et al., 2009; Zolles et al., 2009). Biochemical data have suggested that TRIP8b reduces cAMP binding by direct competition with the cAMP binding site (Han et al., 2011), whereas electrophysiological evidence suggests that TRIP8b alters the effect of cAMP by inhibiting the conformational change in the CNBD associated with cAMP binding (Hu et al., 2013; Zolles et al., 2009). A nuclear magnetic resonance study by Saponaro et al. suggests that TRIP8b does not interact with the cAMP binding site of HCN2 and therefore does not directly compete with cAMP for binding. (Saponaro et al., 2014). However, the structure of the CNBD-TRIP8b complex and the mechanism of how TRIP8b regulates cAMP dependence are currently unknown.
In this study, we determine structural details of the CNBD-TRIP8b complex. We use continuous-wave electron paramagnetic resonance (CW EPR) and double electron-electron resonance (DEER) spectroscopy to examine the conformational transition in the CNBD of HCN2 channels in response to cAMP and TRIP8b binding. DEER is a pulse EPR technique capable of measuring absolute distances and distance distributions between two paramagnetic spin labels attached to a protein and separated by 15 to 80 Å (Martin et al., 1998; Milov et al., 1984; Pannier et al., 2000). Among distance measurement techniques, a strength of DEER is that it reports a distribution of distances, not merely an average distance. Information on conformational heterogeneity not accessible in crystal structures is fully captured and resolved in DEER distributions from frozen solutions. From DEER measurements between two residues on HCN2, we show that TRIP8b binds to the CNBD in the apo state without significantly altering the overall structure of the domain. Using inter-protein DEER measurements between the CNBD and TRIP8b, we spatially localize two TRIP8b positions relative to the CNBD. With nuclear magnetic resonance (NMR), we identify CNBD residues involved in TRIP8b binding. These data reveal a binding interface on the CNBD and the topology of the CNBD-TRIP8b complex. Together, they provide mechanistic insights into how TRIP8b causes a decrease in the cAMP dependence of HCN channels.
Results
TRIP8bcore modulates HCN2 channels
The TRIP8bcore fragment has been shown to be sufficient to bind to the CNBD of HCN2 channels and antagonize the cyclic nucleotide dependence of these channels (Hu et al., 2013). It contains a central portion of TRIP8b that is highly conserved across species (Figure S1A). To demonstrate that human TRIP8bcore purified from E. coli can bind to and regulate full-length HCN2 channels, we directly applied TRIP8bcore to inside-out patches excised from Xenopus laevis oocytes expressing HCN2 channels. Figure 1B shows HCN2 currents in response to a series of voltage steps to potentials between −70 mV and −140 mV. In control solutions, HCN2 channels opened slowly with a half activation voltage of −130 ± 2 mV. Addition of 1 μM cAMP dramatically increased the rate and extent of HCN2 channel activation and shifted the voltage dependence of activation in the depolarizing direction by 14 mV to −116 ± 3 mV (Figure 1C). Simultaneous perfusion with 10 μM TRIP8bcore and 1 μM cAMP resulted in an almost complete inhibition of the effects of cAMP, shifting the voltage dependence nearly back to control levels (−127 ± 2 mV, Figure 1C). These data are consistent with previous reports and show that this small fragment of TRIP8b is sufficient to bind to and regulate full-length HCN channels.
Intra-CNBD DEER probes TRIP8b binding to the CNBD
The CNBD is composed of an eight-stranded β-roll, followed by two alpha-helices (Figure 2a) (Zagotta et al., 2003). Initial ligand binding is thought to take place at the phosphate-binding cassette (PBC), which is comprised of the region after β6, the P-helix, and a small part of β7 (Figure S1B). After ligand binding, the last helix in the domain, the C-helix, moves closer to the β-roll. This motion is thought to be coupled to channel opening (Flynn et al., 2007; Zhou and Siegelbaum, 2007).
Figure 2. TRIP8bcore and TRIP8b(1a-4) alter the separation of spin labels attached to HCN2-CNBDxt at V537C and R635C.

A) Structure of the HCN2 C-terminal fragment (HCN2-CNBDxt) used in this study (based on accession number 3ETQ) consisting of the cyclic nucleotide binding domain (cyan) and a fraction of the C-linker (gray). B) HCN2-CNBDxt with predicted spin label rotamers highlighted in color. The colored spheres indicated the predicted positions of the midpoints of the N-O bond in MTSL. C) DEER time traces of HCN2-CNBDxt V537C,R635C in the absence (cyan) and presence of TRIP8b(1a-4) (black). D) DEER time traces of HCN2-CNBDxt V537C,R635C in the absence (cyan) and presence of TRIP8bcore (black). E) Distance distributions of HCN2-CNBDxt V537C,R635C calculated from the time traces in (A). F) Distance distributions of HCN2-CNBDxt V537C,R635C calculated from the time traces in (d) (see also Figure S2).
We have previously shown that a cysteine-free version of the HCN2 CNBD and C-linker is able to bind cAMP and undergo conformational change (Flynn et al., 2007; Puljung and Zagotta, 2013; Puljung et al., 2014). To examine whether conformational changes in HCN2 channels accompany TRIP8b binding, we performed DEER experiments on a fragment of HCN2 channels, termed HCN2-CNBDxt, which we expressed and purified from bacteria. This fragment contains the CNBD and helices C’ through F’ of the C-linker (residues 488-640, Figure S1B). Compared to the larger cytosolic fragment (CNBD+C-linker), HCN2-CNBDxt has the advantage that it lacks the A’ and B’ helices required for tetramerization and that it exhibits enhanced solubility when bound to TRIP8bcore. These advantages are required for our EPR and NMR experiments. Tetramerization would lead to multispin effects that complicate EPR results. Also, tetramers would be too large for NMR experiments.
We engineered pairs of cysteines into a cysteine-free version of HCN2-CNBDxt and labeled them with the nitroxide spin label MTSL. Cysteines were placed at either of two positions on the C-helix (A624C or R635C) and at one of three positions on the β-roll (V537C, S563C, or K570C). Figure 2B shows the positions of a single pair of cysteines (V537C and R635C) with spin labels attached. For each spin-labeled site, side chain ensembles were predicted using the MMM rotamer library (Polyhach et al., 2011). These surface sites were selected based on MMM predictions of favorable labeling and to provide good coverage of the β-roll and C-helix. The predicted rotamers are shown with spheres indicating the midpoints of the N-O bond on the spin label, corresponding to the positions of the unpaired electron. DEER measures the separation of the unpaired electrons on the N-O group. For DEER experiments, protein samples are snap-frozen, capturing the distribution of backbone and sidechain conformations present at room temperature.
To verify that our MTSL-labelled HCN2-CNBDxt constructs still bound TRIP8bcore, we performed fluorescence size-exclusion chromatography on wild-type, V537C/R635C, and S563C/A624C HCN2-CNBDxt. HCN2-CNBDxt and TRIP8bcore were mixed together and passed through a Superdex 200 column with an inline fluorescence detector set to detect tryptophan fluorescence (ex. 280 nm, em. 350 nm). HCN2-CNBDxt contains no tryptophan residues while TRIP8bcore contains five tryptophans. Each chromatogram shows TRIP8bcore alone, WT HCN2-CNBDxt alone, and the mixture of the two proteins together (Figure S2). TRIP8bcore elutes at 18.3 mL when run alone and 16.6 mL when run in combination with WT HCN2-CNBDxt indicative of a stable complex (Figure S2A). Similar results were obtained for the other MTSL labeled proteins tested (Figure S2B,C).
To test binding of TRIP8bcore compared to full-length TRIP8b(1a-4), we performed DEER experiments on HCN2-CNBDxt labelled at positions 537 and 635, in the presence of 40 μM full length TRIP8b(1a-4), or 200 μM TRIP8bcore. Modulation-depth normalized DEER time traces for apo-(cyan) and TRIP8b-bound HCN2-CNBDxt (gray) are shown in Figures 2C for TRIP8b(1a-4) and 2D for TRIP8bcore. The unnormalized time traces are shown in Figure S3 A and B. In each case, binding of TRIP8b caused a slower oscillatory component of the time trace to emerge. As the oscillation frequency is proportional to the inverse cube of the distance, these data indicate that a fraction of the spin labels at these two positions moved further apart. DEER distance distributions calculated from the time traces have a maximum at 37 Å in the apo configuration, but are bimodal in the presence of TRIP8b(1a-4) (36 Å and 46 Å) or TRIP8bcore (36 Å and 44 Å) (Figure 2E,F). A peak near 55 Å can be observed in the distance distribution for TRIP8bcore. However, the uncertainty associated with this peak is large, as indicated by the shaded band. Therefore, this peak is not significant. The similarity of the effect of TRIP8b(1a-4) and TRIP8bcore binding suggests that both proteins bind HCN2-CNBDxt in a similar manner. We performed the remainder of the experiments using TRIP8bcore, which expresses in substantially larger quantities than TRIP8b(1a-4) and contains no native cysteines.
DEER measurements were made for the remaining pairs of positions on HCN2-CNBDxt in the apo, cAMP bound, and TRIP8bcore bound states. First, upon cAMP addition, residue 624 at the proximal C-helix moved 9 Å closer to the β-roll, and residue 635 at the distal C-helix made a smaller 1 Å movement towards the β-roll (Figure 3A,B,C and Figure S3C,D,E red traces). This is consistent with similar results obtained with an HCN2 channel fragment consisting of the entire C-linker and CNBD (Puljung et al., 2014). No significant cAMP-induced distance changes were seen between a pair of labels on the β-roll (Figure 3D and Figure S3F red traces). These data suggest that the smaller HCN2-CNBDxt fragment behaves similarly to the HCN fragment that contains the full C-linker and CNBD.
Figure 3. TRIP8bcore binding only alters spin label position at distal C-helix.

DEER distance distributions of HCN2-CNBDxt residues in the presence of TRIP8bcore (black traces), cAMP (red traces) and in the apo state (cyan traces). DEER distributions for A) HCN2-CNBDxt S563C,R635C, B) HCN2-CNBDxt V537C,A624C, C) HCN2-CNBDxt S563C,A624C, and D) HCN2-CNBDxt V537C,K570C. The HCN2-CNBDxt structure above each distribution highlights the predicted spin label rotamers (see also Figure S3).
Addition of TRIP8bcore to HCN2-CNBDxt V537C/A624C and S563C/A624C produced distance distributions that more closely resemble the apo rather than the cAMP-bound distributions (Figure 3B,C, black traces). Similar to cAMP, TRIP8bcore binding to HCN2-CNBDxt had no impact on the distance distributions between 537 and 570 on the β-roll (Figure 3D, black trace).
Interestingly, for the separation between residue 635 (distal C-helix) and residue 563 (β-roll), the addition of TRIP8bcore caused a significant sharpening of the distance distribution accompanied by a small shift in the distribution maximum (30 Å to 33 Å, Figure 3A). In both HCN2-CNBDxt mutants spin labeled at residue 635 (V537C/R635C, S563C/R635C), TRIP8b binding shifted the separation of the spin labels towards longer distances (Figure 2F and 3A). This shift could reflect movement of the distal C-helix away from the β-roll in a fraction of the HCN2-CNBDxt population. However, if TRIP8bcore binds near residue 635, it is also possible that the shift is due to changes in the 635 rotameric distribution induced by steric clashes between TRIP8bcore and the MTSL at position 635.
TRIP8bcore binds near CNBD residue 635
To determine if TRIP8b binding affects the mobility of the spin label at 635, we measured the room temperature continuous wave (CW) EPR spectrum of each spin-labeled position of HCN2-CNBDxt in the presence and absence of 200μM TRIP8bcore.
The addition of TRIP8bcore to HCN2-CNBDxt did not result in observable changes in spin label mobility at any sites except R635C (Figure 4). The R635C spectrum, however, showed significant broadening upon addition of TRIP8bcore, indicating a reduction in MTSL mobility at that position. TRIP8bcore addition increased the rotational correlation time of the spin label from 0.8 ns to 3.0 ns. This spectral broadening could result from a change in the mobility of the distal C-helix or from a significant change in the environment around the spin label at R635C that alters the rotameric distribution of MTSL. In either case, the data suggest that TRIP8bcore binds near R635C on the distal C-helix.
Figure 4. TRIP8bcore binding decreased the mobility of a spin label at the distal end of the C-helix.

CW EPR spectra for spin labels attached to indicated residues on HCN in the absence (cyan) and presence of TRIP8bcore (black). Significant line broadening of the R635C spectrum is seen upon addition of TRIP8bcore.
Inter-protein DEER measurements localize the position of TRIP8bcore bound to HCN2-CNBDxt
To determine the geometry of the HCN2-CNBDxt-TRIP8b complex, we measured DEER distributions between singly spin labeled HCN2-CNBDxt and singly spin labeled TRIP8bcore. Seven residues on HCN, shown in Figure 5A, and two residues on TRIP8bcore (A248C and A261C) were selected. Both TRIP8bcore variants bound to each labeled HCN2-CNBDxt protein and generated DEER oscillations (Figure S5), with the exception of TRIP8bcore A248C and HCN2-CNBDxt A624C, which showed no oscillations when the two proteins were combined. The resulting DEER distance distributions (Figure 5B) feature main peaks at distances between 20 and 45 Å and suggests that TRIP8bcore is localized near the C-helix.
Figure 5. Inter-protein DEER measurements between HCN and TRIP8bcore.

A) Structure of HCN2-CNBDxt with the positions of predicted spin label rotamers highlighted in color. B) Distance distributions obtained from DEER measurements between the indicated residues of singly labeled 150 μM HCN and 40 μM TRIP8bcore. No oscillations were observed in the time trace of A261C of TRIP8bcore and A624C of HCN2 indicating the absence of binding (see also Figure S4).
Using the set of inter-protein DEER data, we proceeded to determine more quantitatively the position of TRIP8b relative to HCN2-CNBDxt. We developed a novel probabilistic trilateration algorithm for locating the spin label on a protein relative to a binding partner with a known structure. We generated 3-dimensional spatial probability distributions for spin labels attached to TRIP8bcore by combining the DEER distributions with the known structure of the HCN2 CNBD. Briefly, rotamer-weighted radial probability distributions were created from the DEER data for every pair of sites. We based our HCN spin label positions on a model of the apo state of the CNBD determined from previous DEER experiments(Puljung et al., 2014). For each TRIP8bcore site, an overall spatial probability density function was then calculated by multiplying the individual radial probability distributions. Since we do not have accurate structural information about the distal C-helix due to its disorder in the apo state, we excluded 635 from our calculation.
Using this probabilistic method, we located the spin labels attached to TRIP8bcore, A248C and A261C, relative to HCN2-CNBDxt. Isosurfaces representing regions of constant probability density where the spin labels are likely to be found are shown in Figure 6. The label attached to TRIP8bcore A248C resides near the proximal portion of the C-helix near residue A624 on HCN2-CNBDxt (Figure 6, yellow surface). This result is consistent with the possibility that this labeled construct did not bind to the MTSL labeled HCN2-CNBDxt A624C due to steric hindrance. Position A261C is localized near the β4-β5 loop and the distal end of the C-helix (Figure 6, green surface). A binding site near the distal C-helix is consistent with our CW-EPR observation of restricted spin label mobility at residue 635 of HCN2-CNBDxt in the presence of TRIP8bcore. A decrease in spin label mobility upon TRIP8bcore binding would also account for the changes we observed in the DEER distribution between residue 635 of HCN2-CNBDxt and residues on the β-roll of HCN2-CNBDxt. Recalculating the isosurfaces using a recent solution state NMR structure of the apo HCN2-CNBD also showed similar probability densities for these residues on TRIP8bcore along the C-helix (data not shown). In addition, including the data from the R635C distributions did not dramatically alter the predicted surfaces. We calculated expected distance distributions from the isosurface-bounded volumes in Figure 6 and compared them with our experimental distance distributions (Figure S6). The good agreement confirmed the robustness of our probabilistic trilateration analysis.
Figure 6. Probability density distributions for the locus of TRIB8bcore residues 248 and 261 relative to HCN2-CNBDxt.
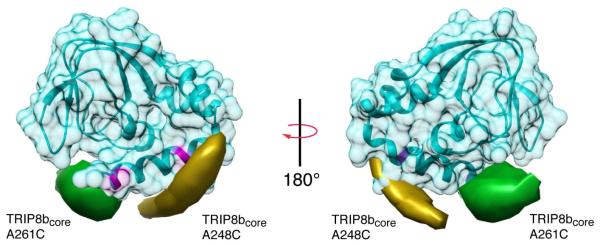
Two views of a structural model of the apo state of HCN2-CNBDxt with calculated trilateration isosurfaces for A248C of TRIP8bcore (yellow) and A261C of TRIP8bcore (green), contoured at a probability density of 1.1×10-4 Å-3. Residues 624 and 635 on the C-helix are highlighted in magenta (see also Figure S5).
NMR identifies a TRIP8bcore binding surface on the CNBD
To further examine the interface between the core domain of TRIP8b and the CNBD of HCN channels, we used NMR spectroscopy to map the TRIP8b binding site on HCN4-CNBDxt, a human HCN4 CNBD construct similar to the one we used for the EPR experiments (Figure S1B). Resonance assignments for a similar human HCN4 CNBD construct and the closely related human HCN2 CNBD have recently been published (Akimoto et al., 2014; Saponaro et al., 2014). We confirmed backbone assignments for the HCN4-CNBDxt under our experimental conditions with a TROSY-HNCA experiment recorded on 15N,13C-labeled HCN4-CNBDxt. We were able to obtain assignments for most (79%) of the HCN4 backbone resonances. Though observed, we were unable to confirm assignment of the disordered N- and C-terminal ends of the protein construct. We were also unable to confirm assignments previously reported for three residues (660-662) corresponding to the P-helix (Saponaro et al., 2014).
We collected a series of 1H,15N-HSQC spectra of 100 μM 15N-labeled HCN4-CNBDxt in the presence of increasing concentrations of unlabeled TRIP8bcore. Significant resonance perturbations, primarily broadenings, were observed at low concentrations of TRIP8bcore (0.25 molar equivalents), with substantial loss of signal at 0.5 molar equivalents of TRIP8bcore, and new resonances beginning to appear at higher concentrations (Figure S7). These observations suggest the interaction between HCN4-CNBDxt and TRIP8bcore is in the intermediate to slow exchange regime on the NMR time scale under these experimental conditions. In the intermediate exchange regime, the observed line width for a peak will be the sum of the population-weighted average of the line widths of the free and bound species and an additional chemical exchange contribution that scales as fffbΔωf 2, (ff and fb refer to the mole fractions of the free and bound species and Δωfb is the difference in chemical shift, in Hz, between the free and bound species). Thus, resonances are predicted to broaden and disappear from the spectrum as a function of mole fraction of bound species. Resonances in the binding interface usually undergo a notable change in chemical shift and will broaden and disappear earlier in the titration due to the quadratic dependence on the chemical shift difference.
To get the most accurate representation of the TRIP8b binding site on the CNBD of HCN4, we focused our analysis on the 1:0.25 complex of HCN4-CNBDxt to TRIB8bcore (Figure 7A). Using these data, we plotted the ratio of peak intensities of the TRIP8b-bound spectrum versus the free HCN4CNBD spectrum for each residue and scored the peaks by determining standard z-scores (Figure 7B). The most strongly affected residues are highlighted in red and are located in the β4-β5 loop (645, 648, 649), the PBC (659, 663, 664), the C-helix (698-699, 703, 705, 707,710) and in the distal C-terminus (712-715, 720, 721, 724).
Figure 7. NMR residues on the CNBD perturbed by TRIP8b binding.

A) 1H, 15N HSQC spectrum of 100 μM labeled HCN4-CNBDxt either unbound (black) or in the presence of 25 μM TRIP8bcore (red). Selected residues from the C-terminus (Ile720, top right), the B4-B5 loop (Lys645, Glu649, middle right), and both regions (Lys648, Ile714) are highlighted. Unlabeled resonances in the highlighted portions of the spectra were not significantly affected by the addition of TRIP8bcore. B) Change in peak intensities for residues in HCN4-CNBDxt upon addition of TRIP8b, measured as a ratio of the intensities of peaks in the bound state over their corresponding intensities in the free state. Unassigned residues are omitted. Peaks with z-scores less than −1 are highlighted in red (see also Figure S6).
Examining these residues on a model of the CNBD in its apo state based on the known crystal structure reveals a putative binding surface for the core domain of TRIP8b that spans the C-helix, PBC, and β4-β5 loop near the cAMP binding site (Figure 8A). Interestingly, the region determined by the NMR data corresponds very well to the DEER-based localization of TRIP8b. Together, they indicate the regions where these two proteins interact. These regions on the CNBD are critical for both cAMP binding and the conformational transition associated with gating. This suggests that TRIP8b could be altering either the binding affinity of cAMP, or the ligand-induced conformational changes, or possibly both.
Figure 8. NMR and DEER localization of a TRIB8b binding site along the C-helix of the CNBD.

A) Two views of a surface representation of the CNBD of HCN2. Residues that were affected by 25 μM TRIP8b binding as measured by NMR are shown in red. Blue residues were assigned but unaffected in the NMR experiments. White residues were not assigned. B) Cartoon summary that shows a tetrameric CNBD (from accession number 1Q5O) with the residues affected by TRIP8b binding colored in red. TRIP8bcore 248C (yellow) and 261C (green) trilateration isosurfaces are shown on the tetramer of the HCN2 CNBD to demonstrate that the localized positions do not clash with the neighboring subunits. The TPR domain of TRIP8b bound to the front subunit is colored in green and shown binding to the distal c-terminus of the HCN channel.
Discussion
TRIP8b has been shown to reduce the cAMP dependence of HCN channels. Interactions between a small central region of TRIP8b and the CNBD of HCN are critical for this gating effect (Hu et al., 2013). The location of the binding site for TRIP8b on the CNBD and the mechanism through which TRIP8b exerts its regulatory effect on HCN channels are still not resolved. To answer these questions, we examined how TRIP8b binding affected the overall structure of the CNBD using intramolecular DEER on HCN2-CNBDxt. In addition, using inter-protein DEER on TRIP8bcore and HCN2-CNBDxt we localized two positions on TRIP8b relative to the CNBD. Lastly, using NMR we found a set of CNBD residues that are at the binding surface for TRIP8b.
The location of the binding surface suggests interesting mechanistic possibilities for how TRIP8b reduces the cAMP dependence of these channels. A model for activation of HCN channels by cAMP has been developed using structural, biochemical, and electrophysiological data (Akimoto et al., 2014; Flynn et al., 2007; Matulef et al., 1999; Saponaro et al., 2014; Tibbs et al., 1998; Zhou and Siegelbaum, 2007). In this model, cAMP initially interacts with residues on the β-roll and P-helix that compose the phosphate binding cassette. Mutations in this region, including residues in β5 and the PBC, have been shown to reduce cyclic nucleotide affinity (Matulef et al., 1999; Tibbs et al., 1998; Xu et al., 2010; Zhou and Siegelbaum, 2007). After initial ligand binding, the C-helix is believed to move toward the binding pocket and form more contacts with the ligand. Mutations in the C-helix have been shown to alter ligand affinity and to affect the ability of the ligand to promote channel opening (Matulef et al., 1999; Zhou and Siegelbaum, 2007).
Previous work has proposed two mechanisms for how TRIP8b could reduce cAMP dependence of HCN channels. Han et al. proposed that TRIP8b could be competing with cAMP for the binding site thus directly reducing its affinity (Han et al., 2011). Hu et al. and Saponaro et al. have suggested that TRIP8b allosterically reduces the effect of cAMP by stabilizing the apo conformation of the HCN channel CNBD, possibly by restricting the motion of the C-helix (Hu et al., 2013; Saponaro et al., 2014). Our results suggest that TRIP8b could be both altering the binding site for the ligand as well as reducing the ability of the CNBD to undergo the conformational transition associated with channel activation. These data could explain why the physiological effects of TRIP8b could not be fit by a simple allosteric model but required a TRIP8b specific effect on cAMP binding (Hu et al., 2013). NMR titrations show that the resonances most perturbed by addition of TRIP8b map to the C-helix, the PBC, and the β4-β5 loop of HCN4-CNBDxt (Figure 7). This is in excellent agreement with the model generated from the DEER and EPR data, which localizes residue 248 of TRIP8b near the proximal C-helix and residue 261 near the distal C-helix and the β4-β5 loop. This binding interface and the trilateration-based interaction topology are compatible with the known 4:4 HCN:TRIP8b stoichiometry in the tetrameric HCN channel (Bankston et al., 2012) (Figure 8B).
The specific NMR resonances on the CNBD of HCN4 altered by TRIP8b binding include amino acids that have been previously shown to affect both the binding of cAMP and the conformational changes associated with channel gating. Many residues across the C-helix, a region that undergoes conformational changes associated with cAMP binding, are affected. For example, an isoleucine in the C-helix (636 in HCN2-CNBDxt and 714 in HCN4-CNBDxt) that is important for ligand selectivity and affinity is impacted by TRIP8b binding (Flynn et al., 2007; Zagotta et al., 2003; Zhou and Siegelbaum, 2007). Additionally, an arginine (632 in HCN2-CNBDxt and 710 in HCN4-CNBDxt) that has been shown to form contacts with the ligand and the core of the β-roll and to stabilize the open conformation of the channel is affected (Zhou and Siegelbaum, 2007). These results are similar to previous data from the homologous CNBD of human HCN2 (Saponaro et al., 2014), which also show extensive changes in C-helix resonances upon addition of TRIP8b. However, our data suggest that TRIP8b binding also affects CNBD regions involved in initial cAMP binding. Specifically, we identify resonance changes in three residues in the PBC and three more in the β4-β5 loop. Each of these regions contain residues that form direct contacts with the cyclic nucleotide (Xu et al., 2010; Zagotta et al., 2003). By affecting both the binding site for cAMP as well as the C-helix, TRIP8b could reduce the cyclic-nucleotide dependence of HCN channels via two mechanisms. It could disrupt ligand affinity and interfere with the ability of the C-helix to couple ligand binding to channel opening.
The binding interface between the CNBD and TRIP8b points to a possible role of electrostatics in the interaction. TRIB8bcore is highly negatively charged (predicted pI 4.88), with three negatively charged residues near position 261 (EWEE) and another four negatively charged residues between position 248 and 261. The distal portion of the C-helix contains a number of positive charges including two arginines (632 and 635 in HCN2-CNBDxt, and 710 and 713 in HCN4-CNBDxt) whose resonances changed in our NMR experiments. Previous results show that charge-inverting mutation of two lysines after the C-helix to glutamates (positions equivalent to 638 and 639 in HCN2-CNBDxt and 716 and 717 in HCN4-CNBDxt) resulted in significantly decreased TRIP8bcore binding (Saponaro et al., 2014). In addition, the β4-β5 loop contains two positively charged lysines that are also perturbed in our NMR data. The cAMP-binding pocket is highly positively charged and is likely necessary for the binding of the negatively charged cAMP. It is possible that the negatively charged TRIP8b could also lower cAMP affinity by altering the electropositive nature of the binding pocket.
The DEER trilateration and NMR perturbation results in this study provide new data on the interaction topology of the complex formed between the CNBD and TRIP8b. In addition, the intra-CNBD DEER data show that TRIP8b binds to a conformation that is largely similar to the apo state. The binding interface is composed of regions on the CNBD that are critical for ligand binding as well as regions critical for the ability of the CNBD to couple binding to channel activation. These data provide a structural basis for understanding how TRIP8b interacts with HCN channels. They suggest that TRIP8b might be altering the cAMP dependence of HCN channels by both restricting ligand binding and interfering with the conformational transition necessary to couple cAMP binding to channel activation.
Experimental Procedures
Molecular Biology
All constructs used in this study were subcloned into the pETM11 vector. The proteins were separated from the histidine tag by a Tobacco Etch Virus (TEV) cleavable linker. TRIP8bcore was derived from the TRIP8b(1a-4) isoform and contains residues 223-303 of that protein. Murine HCN2-CNBDxt contains residues 488-640 in a cysteine free background (Taraska et al., 2009). Human HCN4-CNDBxt is composed of wild-type residues 563-727. Cysteine mutations were engineered into the constructs at the indicated positions using standard PCR-based methods. All constructs were confirmed with fluorescence-based automated sequencing. The cDNA encoding the full length murine HCN2 channel in the pGHE vectors, were kindly provided by Steven Siegelbaum and Bina Santoro (Columbia University).
Electrophysiology
The cRNA for HCN2 channels was transcribed using the mMessage Machine T7 transcription kit (Ambion) and expressed in Xenopus laevis oocytes that were defolliculated and injected with the cRNA as previously described (Zagotta et al., 1989). The vitelline membranes were manually removed, and currents were recorded in the inside-out patch clamp configuration (Hamill et al., 1981) with an EPC-10 patch clamp amplifier (HEKA Elektronik). HCN2 channels are known to run-down upon patch excision due to phosphatidylinositol 4,5-bisphosphate depletion (Pian et al., 2006). Thus, we allowed 20-30 minutes for the HCN current in level from the patch to stabilize before starting the experiment. Patch pipettes were pulled from borosilicate glass and had resistances of 0.40–0.6 MΩ after fire polishing. The solutions for HCN2 recordings were as follows: pipette and bath solution, 130 mM KCl, 3 mM HEPES, and 0.2 mM EDTA, pH 7.2. TRIP8bcore and cAMP were added to these solutions at the concentrations indicated. The solutions were perfused onto the patches using a μflow Low Volume Perfusion System (ALA Scientific Instruments, Farmingdale NY). Patches were held at 0 mV and HCN2 currents were elicited by applying a series of 2-s voltage steps ranging from −70 to −140 mV followed by a 1-s voltage pulse to −40 mV. Data were analyzed using Igor (Wavemetrics, Portland, OR) and MATLAB (MathWorks, Natick, MA).
To determine the conductance-voltage relationships, peak tail current amplitudes measured at −40 mV were normalized to the peak tail current measured in cAMP following steps up to −140 mV. A Boltzmann function,
was then fitted to the data. V is the test voltage, V0.5 is the half-maximal activation voltage, and k is the slope factor.
Protein Expression, Purification, and Spin Labeling
For each protein expressed, the construct was transfected into BL21(DE3) cells and grown at 37 °C to an optical density of 0.6-0.8 . The cells were then induced with 1 mM isopropyl β-D-1-thiogalactopyranoside and grown overnight at 18 °C. For NMR experiments, the bacteria were grown in MOPS minimal media supplemented with 15NH4Cl (1 g/L) and 13C6-glucose (2 g/L). After growth and expression, 1-2 liter cultures of cells were pelleted by centrifugation at 4,000 × g at 4 °C for 10 min and resuspended. For EPR experim ents, cells were resuspended in 150 mM KCl and 30 mM HEPES at pH 7.4 (for HCN2) or pH 8.5 (for TRIP8bcore). DNase at a final concentration of 5 μg/mL and two tablets of protease inhibitors (cOmplete EDTA-free; Roche) were added to the buffer. The resuspended cells were lysed by an Emulsiflex-C3 homogenizer (Avestin) and clarified by centrifugation at 186,000 × g at 4°C for 45 min. The lysate was then purified on a Ni2+ affinity resin column (HisTrap HP, GE Healthcare). The octahistidine tag was removed by TEV protease cleavage overnight at 4°C. The protein (10–50 μM) was then spin-labeled with 100 μM S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate (MTSL; Toronto Research Chemicals) per cysteine mutation for 1 h at room temperature or 6-16 h at 4 °C. To remove the TEV protease and f urther purify the samples, the proteins were passed through ion exchange columns. For HCN2 and HCN4 proteins, a cation exchange column was used (HiTrap SP FF, GE Healthcare). For TRIP8b(1a-4) and TRIP8bcore, an anion exchange column (HiTrap Q HP, GE Healthcare) was used. The proteins were eluted with a KCl gradient from 15 mM to 1 M. Fractions with protein were pooled and concentrated using a 3 kD molecular weight cutoff centrifugal filter (Vivaspin; General Electric). For NMR, HCN4-CNBDxt was additionally purified by size exclusion chromatography on a Superdex 200 column (Amersham Biosciences) and used within 5 days of purification.
EPR Sample Preparation
For CW-EPR and DEER experiments, the protein was buffer exchanged into D2O with 150 mM KCl, 30 mM Tris, and 10% glycerol, pH 8.4 using a PD-10 column (GE Healthcare). Doubly-spin labeled HCN protein for intra-protein DEER measurements was diluted to approximately 50 μM. 1 mM cAMP, 200 μM TRIP8bcore or 40 μM full-length TRIP8b were added as indicated in the text. For inter-protein DEER measurements, 150 μM singly-spin labeled HCN was combined with 40 μM singly labeled TRIP8bcore. For DEER, 50 μL of each protein sample was inserted into a 1.65 mm o.d. quartz tube (Sutter, Q165-115-10) and flash frozen in liquid nitrogen. For CW-EPR, protein was loaded into 1 mm o.d. quartz capillaries (Sutter, Q100-50-7.5) at concentrations of 25 μM HCN2-CNBDxt and 200 μM TRIP8bcore immediately prior to the experiment.
EPR Data Acquisition
Continuous wave (CW) EPR spectra were recorded on a 9-10 GHz Bruker EMX spectrometer with a dielectric resonator (Bruker ER4123D, 9.78 GHz, Q-factor 2000-4000). Experiments were performed at room temperature with 0.2 mW incident power, 2 G modulation amplitude and 100 kHz modulation frequency. DEER data were acquired on a 33-35 GHz Bruker EleXsys E580 spectrometer with an overcoupled dielectric resonator (Bruker EN5107D2, 34.1 GHz, Q-factor 300-700). Experiments were performed at 60 K using a liquid helium cooling system (Oxford). The four pulse, dead-time free DEER sequence (Π/2)probe − T1 − (Π)probe − T1+t − (Π)pump − (T2 − t) − (Π)probe – T2 was used with 22 ns probe pulses and a 44 ns pump pulse. Pulse delays were 120 ns for T1 and 1800 ns for T2,. The delay t was varied from −60 ns to between 1800 and 4000 ns, depending on the experiments, in 10 ns increments. The pump frequency matched the nitroxide spectral maximum. The probe frequency was centered in the resonator dip and was 62 MHz lower than the pump frequency. An eight-step phase cycling protocol combined with extensive averaging at a repetition time of 2 ms was used to collect data. The measurement time for each sample was 10-16 h.
Data Analysis
Rotational correlation times were extracted from CW-EPR data via spectral simulation using EasySpin (Stoll and Schweiger, 2006). Probability densities for spin labels attached to TRIP8bcore were calculated in Matlab. Rotameric models of MTSL attached to HCN2-CNBDxt were obtained using MMM(Polyhach et al., 2011). DEER distance distributions were obtained using DeerAnalysis2013 (Jeschke et al., 2006). A homogeneous three-dimensional background was used for background correction. Time traces were converted to distance distributions using Tikhonov regularization, a model-free least-squares approach. The regularization parameter was optimized separately for each data set according to the L-curve criterion. To estimate errors associated with our measurement, the noise in the time domain traces was linearly transformed to the distance domain. The shaded error bands shown in the distance distributions correspond to two standard deviations of the time-domain noise. Molecular graphics and analyses were performed with the UCSF Chimera and Pymol (Meng et al., 2006; Pettersen et al., 2004; Schrodinger, 2010).
Trilateration
For each TRIP8bcore mutation, every DEER distance distribution between HCN2-CNBDxt and TRIP8bcore was transformed into a radial probability distribution in 3-D space with the origin centered at the MMM predicted N-O bond midpoint of MTSL. For each HCN2-CNBDxt mutation, a weighted average of these distributions was computed using the rotameric populations predicted by MMM as weights. The total probability densities for each TRIP8bcore residue were calculated by multiplying the weighted densities computed for each HCN site.
NMR
NMR spectra were recorded on a Bruker Avance III spectrometer equipped with a cryoprobe and operating at 600 MHz. All spectra were recorded at 25 °C in 30 mM HEPES, 150 mM KCl, 10% D2O, pH 7.6. The HNCA spectrum was collected on samples containing 300 μM of 13C,15N-labeled HCN4-CNBDxt. HSQC spectra were collected on samples containing a constant concentration (100 μM) of 13C,15N-labeled HCN4-CNBDxt from the same preparation in the presence of increasing concentrations of TRIP8bcore (0 μM, 25 μM, 50 μM, 100 μM). NMR data were processed using NMRPipe/NMRDraw (Delaglio et al., 1995) and visualized using NMRView (Johnson and Blevins, 1994). Resonance perturbation analysis examined changes in peak intensities by determining the ratios of a peak in the TRIP8b-bound spectrum to its corresponding peak in the free HCN4CNBD spectrum. We then scored each peak for its standard (z) score using the following formula: z = (X − μ)/(J, where σ is the peak intensity ratio, μ is the mean intensity ratio for all peaks, and σ is the standard deviation of peak intensities. Peaks with z-scores below −1 were considered significantly perturbed.
Supplementary Material
Highlights.
DEER and EPR show TRIP8b binds near the C-helix on the CNBD of HCN channels
NMR shows TRIP8b binding affects the C-helix and the cAMP binding site
TRIP8b likely alters both cAMP binding and CNBD conformational changes
Acknowledgements
We thank Stacey Camp for technical assistance, Ellen Hayes for help with the EPR and DEER experiments, and Yoni Haitin for helpful discussions. This work was supported by NIH grants NS074545 (to J.R.B.), EY010329 (to W.N.Z. and S.S.), 5T32CA080416-15 (to J.C.R) R01 GM098501 (to P.S.B.), American Heart Association grants 14IRG18770000 (to W.N.Z.) and 14CSA20380095 (to W.N.Z. and S.S.), the University of Washington (S.S.), and by a fellowship from the Sackler Scholars Program in Integrative Biophysics (to H.A.D.). Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akimoto M, Zhang Z, Boulton S, Selvaratnam R, VanSchouwen B, Gloyd M, Accili EA, Lange OF, Melacini G. A Mechanism for the Auto-Inhibition of Hyperpolarization-Activated Cyclic Nucleotide-Gated (HCN) Channel Opening and its Relief by cAMP. J. Biol. Chem. 2014 doi: 10.1074/jbc.M114.572164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankston JR, Camp SS, DiMaio F, Lewis AS, Chetkovich DM, Zagotta WN. Structure and stoichiometry of an accessory subunit TRIP8b interaction with hyperpolarization-activated cyclic nucleotide-gated channels. Proc. Natl. Acad. Sci. U. S. A. 2012;109:7899–7904. doi: 10.1073/pnas.1201997109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Glajch KE, Gertler TS, Guzman JN, Mercer JN, Lewis AS, Goldberg AB, Tkatch T, Shigemoto R, Fleming SM, et al. HCN channelopathy in external globus pallidus neurons in models of Parkinson’s disease. Nat. Neurosci. 2011;14:85–92. doi: 10.1038/nn.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven KB, Zagotta WN. CNG and HCN channels: two peas, one pod. Annu. Rev. Physiol. 2006;68:375–401. doi: 10.1146/annurev.physiol.68.040104.134728. [DOI] [PubMed] [Google Scholar]
- Craven KB, Olivier NB, Zagotta WN. C-terminal movement during gating in cyclic nucleotide-modulated channels. J. Biol. Chem. 2008;283:14728–14738. doi: 10.1074/jbc.M710463200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Flynn GE, Black KD, Islas LD, Sankaran B, Zagotta WN. Structure and Rearrangements in the Carboxy-Terminal Region of SpIH Channels. Structure. 2007;15:671–682. doi: 10.1016/j.str.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Han Y, Noam Y, Lewis AS, Gallagher JJ, Wadman WJ, Baram TZ, Chetkovich DM. Trafficking and Gating of Hyperpolarization-activated Cyclic Nucleotide-gated Channels Are Regulated by Interaction with Tetratricopeptide Repeat-containing Rab8b-interacting Protein (TRIP8b) and Cyclic AMP at Distinct Sites. J. Biol. Chem. 2011;286:20823–20834. doi: 10.1074/jbc.M111.236125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Chen F, Li B, Hu Z. Neurophysiology of HCN channels: from cellular functions to multiple regulations. Prog. Neurobiol. 2014;112:1–23. doi: 10.1016/j.pneurobio.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Hu L, Santoro B, Saponaro A, Liu H, Moroni A, Siegelbaum S. Binding of the auxiliary subunit TRIP8b to HCN channels shifts the mode of action of cAMP. J. Gen. Physiol. 2013;142:599–612. doi: 10.1085/jgp.201311013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke G, Chechik V, Ionita P, Godt A. DeerAnalysis2006−a comprehensive software package for analyzing pulsed ELDOR data. Appl. Magn. 2006 [Google Scholar]
- Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Lewis AS, Schwartz E, Chan CS, Noam Y, Shin M, Wadman WJ, Surmeier DJ, Baram TZ, Macdonald RL, Chetkovich DM. Alternatively Spliced Isoforms of TRIP8b Differentially Control h Channel Trafficking and Function. J. Neurosci. 2009;29:6250–6265. doi: 10.1523/JNEUROSCI.0856-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AS, Vaidya SP, Blaiss CA, Liu Z, Stoub TR, Brager DH, Chen X, Bender RA, Estep CM, Popov AB, et al. Deletion of the Hyperpolarization-Activated Cyclic Nucleotide-Gated Channel Auxiliary Subunit TRIP8b Impairs Hippocampal Ih Localization and Function and Promotes Antidepressant Behavior in Mice. J. Neurosci. 2011;31:7424–7440. doi: 10.1523/JNEUROSCI.0936-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lörincz A, Notomi T, Tamás G, Shigemoto R, Nusser Z. Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat. Neurosci. 2002;5:1185–1193. doi: 10.1038/nn962. [DOI] [PubMed] [Google Scholar]
- Martin RE, Pannier M, Diederich F. Determination of Enditoi End Distances in a Series of TEMPO Diradicals of up to 2.8 nm Length with a New FouriPulse Double Electron Electron Resonance …. Angew. Chemie. 1998 doi: 10.1002/(SICI)1521-3773(19981102)37:20<2833::AID-ANIE2833>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Matulef K, Flynn GE, Zagotta WN. Molecular rearrangements in the ligand-binding domain of cyclic nucleotide-gated channels. Neuron. 1999;24:443–452. doi: 10.1016/s0896-6273(00)80857-0. [DOI] [PubMed] [Google Scholar]
- Meng EC, Pettersen EF, Couch GS, Huang CC, Ferrin TE. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics. 2006;7:339. doi: 10.1186/1471-2105-7-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milov AD, Ponomarev AB, Tsvetkov YD. Modulation beats of signal of double electron-electron resonance in spin echo for biradical systems. J. Struct. Chem. 1984 [Google Scholar]
- Nolan MF, Malleret G, Lee KH, Gibbs E, Dudman JT, Santoro B, Yin D, Thompson RF, Siegelbaum SA, Kandel ER, et al. The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell. 2003;115:551–564. doi: 10.1016/s0092-8674(03)00884-5. [DOI] [PubMed] [Google Scholar]
- Pannier M, Veit S, Godt A, Jeschke G, Spiess HW. Dead-Time Free Measurement of Dipole-Dipole Interactions between Electron Spins. J. Magn. Reson. 2000;142:331–340. doi: 10.1006/jmre.1999.1944. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pian P, Bucchi A, Robinson RB, Siegelbaum SA. Regulation of gating and rundown of HCN hyperpolarization-activated channels by exogenous and endogenous PIP2. J. Gen. Physiol. 2006;128:593–604. doi: 10.1085/jgp.200609648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskorowski R, Santoro B, Siegelbaum SA. TRIP8b Splice Forms Act in Concert to Regulate the Localization and Expression of HCN1 Channels in CA1 Pyramidal Neurons. Neuron. 2011;70:495–509. doi: 10.1016/j.neuron.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyhach Y, Bordignon E, Jeschke G. Rotamer libraries of spin labelled cysteines for protein studies . Phys. Chem. Chem. Phys. 2011;13:2356–2366. doi: 10.1039/c0cp01865a. [DOI] [PubMed] [Google Scholar]
- Puljung MC, Zagotta WN. A secondary structural transition in the C-helix promotes gating of cyclic nucleotide-regulated ion channels. J. Biol. Chem. 2013;288:12944–12956. doi: 10.1074/jbc.M113.464123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puljung MC, DeBerg HA, Zagotta WN, Stoll S. Double electron-electron resonance reveals cAMP-induced conformational change in HCN channels. Proc. Natl. Acad. Sci. U. S. A. 2014;111:9816–9821. doi: 10.1073/pnas.1405371111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA, Phillips AM, Petrou S. HCN channelopathies: pathophysiology in genetic epilepsy and therapeutic implications. Br. J. Pharmacol. 2012;165:49–56. doi: 10.1111/j.1476-5381.2011.01507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu. Rev. Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- Santoro B, Wainger BJ, Siegelbaum SA. Regulation of HCN Channel Surface Expression by a Novel C-Terminal Protein-Protein Interaction. J. Neurosci. 2004;24:10750–10762. doi: 10.1523/JNEUROSCI.3300-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Piskorowski RA, Pian P, Hu L, Liu H, Siegelbaum SA. TRIP8b Splice Variants Form a Family of Auxiliary Subunits that Regulate Gating and Trafficking of HCN Channels in the Brain. Neuron. 2009;62:802–813. doi: 10.1016/j.neuron.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Hu L, Liu H, Saponaro A, Pian P, Piskorowski RA, Moroni A, Siegelbaum SA. TRIP8b Regulates HCN1 Channel Trafficking and Gating through Two Distinct C-Terminal Interaction Sites. J. Neurosci. 2011;31:4074–4086. doi: 10.1523/JNEUROSCI.5707-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saponaro A, Pauleta SR, Cantini F, Matzapetakis M, Hammann C, Donadoni C, Hu L, Thiel G, Banci L, Santoro B, et al. Structural basis for the mutual antagonism of cAMP and TRIP8b in regulating HCN channel function. Proc. Natl. Acad. Sci. U. S. A. 2014 doi: 10.1073/pnas.1410389111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger LLC. The PyMOL Molecular Graphics System. 2010 Version~1.3r1. [Google Scholar]
- Stoll S, Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Taraska JW, Zagotta WN. Structural dynamics in the gating ring of cyclic nucleotide-gated ion channels. Nat. Struct. Mol. Biol. 2007;14:854–860. doi: 10.1038/nsmb1281. [DOI] [PubMed] [Google Scholar]
- Taraska JW, Puljung MC, Olivier NB, Flynn GE, Zagotta WN. Mapping the structure and conformational movements of proteins with transition metal ion FRET. Nat. Methods. 2009;6:532–537. doi: 10.1038/nmeth.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbs GR, Liu DT, Leypold BG, Siegelbaum SA. A state-independent interaction between ligand and a conserved arginine residue in cyclic nucleotide-gated channels reveals a functional polarity of the cyclic nucleotide binding site. J. Biol. Chem. 1998;273:4497–4505. doi: 10.1074/jbc.273.8.4497. [DOI] [PubMed] [Google Scholar]
- Xu X, Vysotskaya Z. V, Liu, Q., Zhou L. Structural basis for the cAMP-dependent gating in the human HCN4 channel. J. Biol. Chem. 2010;285:37082–37091. doi: 10.1074/jbc.M110.152033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Gating of single Shaker potassium channels in Drosophila muscle and in Xenopus oocytes injected with Shaker mRNA. Proc. Natl. Acad. Sci. U. S. A. 1989;86:7243–7247. doi: 10.1073/pnas.86.18.7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- Zhou L, Siegelbaum SA. Gating of HCN Channels by Cyclic Nucleotides: Residue Contacts that Underlie Ligand Binding, Selectivity, and Efficacy. Structure. 2007;15:655–670. doi: 10.1016/j.str.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolles G, Wenzel D, Bildl W, Schulte U, Hofmann A, Müller CS, Thumfart J-O, Vlachos A, Deller T, Pfeifer A, et al. Association with the Auxiliary Subunit PEX5R/Trip8b Controls Responsiveness of HCN Channels to cAMP and Adrenergic Stimulation. Neuron. 2009;62:814–825. doi: 10.1016/j.neuron.2009.05.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.