

Abstract
AIM: To investigate the hepatoprotective effects and mechanisms of hydrogen-rich water (HRW) in acetaminophen (APAP)-induced liver injury in mice.
METHODS: Male mice were randomly divided into the following four groups: normal saline (NS) control group, mice received equivalent volumes of NS intraperitoneally (ip); HRW control group, mice were given HRW (same volume as the NS group); APAP + NS group, mice received NS ip for 3 d (5 mL/kg body weight, twice a day at 8 am and 5 pm) after APAP injection; APAP + HRW group, mice received HRW for 3 d (same as NS treatment) after APAP challenge. In the first experiment, mice were injected ip with a lethal dose of 750 mg/kg APAP to determine the 5-d survival rates. In the second experiment, mice were injected ip with a sub-lethal dose of 500 mg/kg. Blood and liver samples were collected at 24, 48, and 72 h after APAP injection to determine the degree of liver injury.
RESULTS: Treatment with HRW resulted in a significant increase in the 5-d survival rate compared with the APAP + NS treatment group (60% vs 26.67%, P < 0.05). HRW could significantly decrease the serum alanine aminotransferase level (24 h: 4442 ± 714.3 U/L vs 6909 ± 304.8 U/L, P < 0.01; 48 h: 3782 ± 557.5 U/L vs 5111 ± 404 U/L, P < 0.01; and 3255 ± 337.4 U/L vs 3814 ± 250.2 U/L, P < 0.05, respectively) and aspartate aminotransferase level (24 h: 4683 ± 443.4 U/L vs 5307 ± 408.4 U/L, P < 0.05; 48 h: 3392 ± 377.6 U/L vs 4458 ± 423.6 U/L, P < 0.01; and 3354 ± 399.4 U/L vs 3778 ± 358 U/L, respectively) compared with the APAP treatment group. The alkaline phosphatase, total bilirubin and lactate dehydrogenase levels had the same result. Seventy-two hours after APAP administration, liver samples were collected for pathological examination and serum was collected to detect the cytokine levels. The liver index (5.16% ± 0.26% vs 5.88% ± 0.073%, P < 0.05) and percentage of liver necrosis area (27.73% ± 0.58% vs 36.87% ± 0.49%, P < 0.01) were significantly lower in the HRW-treated animals. The malonyldialdehyde (MDA) contents were significantly reduced in the HRW pretreatment group, but they were increased in the APAP-treated group (10.44 ± 1.339 nmol/mg protein vs 16.70 ± 1.646 nmol/mg protein, P < 0.05). A decrease in superoxide dismutase (SOD) activity in the APAP treatment group and an increase of SOD in the HRW treatment group were also detected (9.74 ± 0.46 U/mg protein vs 12.1 ± 0.67 U/mg protein, P < 0.05). Furthermore, HRW could significantly increase the glutathione (GSH) contents (878.7 ± 76.73 mg/g protein vs 499.2 ± 48.87 mg/g protein) compared with the APAP treatment group. Meanwhile, HRW could reduce the inflammation level (serum TNF-α: 399.3 ± 45.50 pg/L vs 542.8 ± 22.38 pg/L, P < 0.05; and serum IL-6: 1056 ± 77.01 pg/L vs 1565 ± 42.11 pg/L, P < 0.01, respectively). In addition, HRW could inhibit 4-HNE, nitrotyrosine formation, JNK phosphorylation, connexin 32 and cytochrome P4502E expression. Simultaneously, HRW could facilitate hepatocyte mitosis to promote liver regeneration.
CONCLUSION: HRW has significant therapeutic potential in APAP-induced hepatotoxicity by inhibiting oxidative stress and inflammation and promoting liver regeneration.
Keywords: Hydrogen, Liver regeneration, Reactive oxygen species, Acetaminophen, Connexin 32
Core tip: Acetaminophen (APAP)-induced liver injury is a devastating and fatal disease. Hydrogen is a newly-developed antioxidant that has an obvious effect of selectively reducing the strongest oxidants, such as hydroxyl radicals and peroxynitrite. We launched a research study to evaluate the protective role of hydrogen-rich water on APAP-induced hepatotoxicity in mice. We found that hydrogen-rich water treatment was effective in counteracting APAP-induced hepatic damage, oxidative stress and cellular necrosis. It could also promote hepatocyte proliferation and inhibit the expression of connexin 32, cytochrome P4502E and JNK phosphorylation after APAP administration. These results provide a potential therapy for APAP-induced liver injury.
INTRODUCTION
Acetaminophen (N-acetyl-p-aminophenol, APAP) is a widely used analgesic and antipyretic drug in the clinic. APAP is believed to be safe within therapeutic doses, but overdose causes centrilobular hepatic necrosis that leads to acute liver failure (ALF)[1]. Surveys have shown that APAP poisoning accounts for approximately one-half of ALF in the US today, which costs as much as $87 million dollars to treat annually[2].
The severity of APAP-induced liver injury has been the focus of many research studies, and a variety of mechanisms of toxicity both in animals and humans have been elucidated[3,4]. According to pharmacological research, overdoses of APAP can promote the generation of the toxic metabolite N-acetyl-p-benzoquinone imine (NAPQI), which is immediately conjugated with glutathione (GSH) to form the nontoxic metabolite cysteine[5,6]. However, when the GSH is exhausted, NAPQI turns to covalently bind with other proteins to form the protein adducts that directly lead to cell death[7]. Some studies have reported that oxidative stress plays an important role in APAP hepatotoxicity[8]. Both the intracellular (mitochondria) and extracellular (inflammatory cells) sources of reactive oxygen species (ROS) contribute to liver injury[9-11]. Many antioxidant agents have been studied in experimental and clinical studies to reduce or prevent APAP-induced hepatotoxicity. Meanwhile, there is some evidence that APAP administration leads to an increase in the pro-inflammatory cytokines, and treatment of APAP-intoxicated mice with either anti-tumor necrosis factor (TNF)-α or anti-interleukin (IL)-1β can prevent hepatotoxicity[12]. Some proteins or enzymes, such as cytochrome P4502E (CYP2E1), inducible nitric oxide synthase (i-NOS) and c-Jun-NH2-terminal protein kinase (JNK), play important roles in the pathological process of APAP-liver injury[6,13-16].
More recently, the role of gap junctions, which represent an elegant mechanism for direct communication between neighboring cells, has been studied in drug-induced hepatic injury[17]. In liver, connexin 32 (Cx32), the predominant gap junction protein expressed in the liver, has been demonstrated to aggravate drug-induced hepatic injury by enabling direct intercellular communication between coupled cells and the amplification of liver inflammation[18]. Patel et al[19] found that mice deficient in Cx32 were protected against thioacetamide (TAA)-induced liver damage. Inhibition of Cx32 by a pharmaceutical strategy can also decrease the TAA or APAP toxicity[19].
Molecular hydrogen, the lightest and most abundant chemical element in nature, has therapeutic efficacy in many diseases through its efficient anti-oxidant, anti-inflammatory, anti-apoptotic, and anti-allergy effects[20,21]. Although the protective effects of hydrogen on liver diseases including ischemia reperfusion injury, concanavalin-A-induced hepatitis, schistosomiasis-associated liver injury, and nonalcoholic steatohepatitis[22-24] have been confirmed, the effect of hydrogen on APAP-induced liver injury has not been studied. Hydrogen-rich water (HRW) is an effective, convenient way to deliver molecular hydrogen, which has the same effectiveness as inhalation of hydrogen gas and is more suitable for application. Therefore, the main aim of our study was to assess the protective role and potential mechanisms of HRW in APAP-induced hepatic injury in mice.
MATERIALS AND METHODS
Experimental animals and preparation of HRW
This study was conducted using male C57Bl/6 mice (4-5 wk old, 21-26 g) (Animal Feeding Center of Xi’an Jiaotong University Medical School). The animals were acclimatized to laboratory conditions (23 °C, 12 h/12 h light/dark, 50% humidity, and ad libitum access to food and water) for one week prior to experimentation. All mice were housed (5 per cage) in clear, pathogen-free polycarbonate cages in the animal care facility, and they were fed a standard animal diet and water ad libitum under controlled temperature conditions with 12-h light-dark cycles. They were cared for in accordance with the Ethical Committee, Xi’an Jiaotong University Health Science Center. The study was reviewed and approved by the Xi’an Jiaotong University Health Science Center Institutional Review Board. All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of the Xi’an Jiaotong University Health Science Center (IACUC protocol number: NO.XJTULAC2014-207). The animal protocol was designed to minimize pain and discomfort to the animals. All animals were euthanized by isoflurane gas for tissue collection. The HRW was produced by Naturally Plus Japan International Co, Ltd, and was stored under atmospheric pressure at 4 °C in an aluminum bag with no dead volume. Gas chromatography was used to confirm the content of hydrogen by the method described by Ohsawa et al[20] (hydrogen concentration of the HRW used in this study: 0.83-0.91 mmol/L).
Study design
Mice in the present study were divided into the following three groups: (1) normal saline (NS) control group, mice received equivalent volumes of NS intraperitoneally (ip); (2) hydrogen-rich water control group, mice received HRW (same volume as NS); (3) APAP + NS group, mice received NS ip (5 mL/kg body weight, twice per day at 8 am and 5 pm) after APAP injection; and (4) APAP + HRW group, mice received HRW (same volume as NS treatment) after APAP challenge.
In the first experiment, mice (NS control and HRW control groups, n = 5; APAP + NS and APAP + HRW groups, n = 15) were randomly divided as described above and received a lethal dose of 750 mg/kg APAP, administered ip, at 8 am on the first day, and they were monitored for mortality over the next 5 d.
In the second experiment, acute liver injury (ALI) was induced by a sub-lethal dose of 500 mg/kg APAP administered ip at 8 am on the first day. Mice were treated ip with NS or HRW (5 mL/kg, twice a day at 8 am and 5 pm) for 3 d after APAP challenge. Six mice per group were used in this study. Blood samples were collected from all groups by cutting the tail at 24 and 48 h after APAP administration. Mice were sacrificed at 72 h after APAP administration and blood samples were collected from the eyeballs. The serum was separated by centrifugation at 4 °C, 3000× g for 15 min. The liver was removed immediately from each mouse and kept at -80 °C until further analysis.
Measurement of liver function
The serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), total bilirubin and alkaline phosphatase (ALP) activities were determined by an automated procedure in the Department of Inspection, The First Affiliated Hospital of Xi’an Jiaotong University.
Cytokine measurement in murine serum
The levels of serum TNF-α and IL-6 were measured with commercial ELISA kits according to the instructions of the manufacturer (Dakewe, Shenzhen, China).
Measurement of hepatic oxidative stress
The liver tissue was homogenized, and the tissue myeloperoxidase (MPO), malonaldehyde (MDA), superoxide dismutase (SOD), catalase (CAT), glutathione (GSH), glutathione peroxidase (GSH-Px) activities were measured using the activity assay kits from NanJing JianCheng Bioengineering Institute; the methods were previously described[25].
Histological study
Samples from the liver were fixed in 10% formalin solution and embedded in paraffin. Serial sections of 5-μm thickness were obtained and stained with hematoxylin eosin (HE) to evaluate the morphology. Two researchers examined the results in a blinded fashion. For electron microscopy examination, liver tissues were prefixed immediately after harvesting with 1.5% glutaraldehyde and 0.8% paraformaldehyde (0.1 mol/L cacodylate buffer) at room temperature, and they were postfixed in an aqueous solution of 1% OsO4 and 1.5% K4(FeCN)6. Then, the specimens were embedded into Epon by routine procedures. Ultrathin sections (50 nm) were contrasted with lead citrate and uranyl acetate and studied with a CM100 transmission electron microscope.
Immunohistochemistry
Immunohistochemistry (IHC) analysis were performed with 4HNE, nitrotyrosine, bromodeoxyuridine (BrdU), Ki-67, PCNA and Cx32 antibodies (Beijing Biosynthesis Biotechnology Co., Ltd) using previously described methods[26]. Mice given BrdU (0.5 mg/mL) in their drinking water 4 d before APAP administration were analyzed by immunohistochemistry for liver nuclear-labeling indices.
RNA isolation and quantitative reverse transcription-polymerase chain reaction analysis
Total RNA was isolated from liver samples using the RNAfast200 kit (Fastagen Biotech, Shanghai, China). Reverse transcription was performed using the PrimeScript RT reagent kit (TaKaRa Biotechnology, Dalian, China). The mRNA expression was assayed in triplicate and normalized to the 18S mRNA expression. The relative levels were calculated using the Comparative-Ct Method (ΔΔCt method). The primers used in the study are: TNF-α: (Forward 5’-AAGCCTGTAGCCCACGTCGTA-3’ and Reverse 5’-AGGTACAACCCATCGGCTGG-3’); IL-6: (Forward 5’-TCCATCCAGTTGCCTTCTTG-3’ and Reverse 5’-TTCCACGATTTCCCAGAGAAC-3’); Cx32: (Forward 5’-TGAGGCAGGATGAACTGGACAGGT-3’ and Reverse 5’-CACGAAGCAGTCCACTGT-3’); 18S: (Forward 5’-AAACGGCTACCACATCCAAG-3’ and Reverse 5’- CCTCCAATGGATCCTCGTTA -3’).
Western blot analysis
The anti-cyclin D1, PCNA, JNK, phospho-JNK, CYP2E1, and β-actin monoclonal antibodies were purchased from Beijing Biosynthesis Biotechnology CO., Ltd. The protein concentration was determined by the BCA method. Western blot analysis was performed as previously described[27].
Statistical analysis
The survival and mortality rates are expressed as percentages and analyzed using the Kaplan-Meier method. The measurement data are expressed as the mean ± SD. Differences between the experimental and control groups were assessed by either the analysis of variance (ANOVA) or t test, as applicable, using SPSS 18.0 (SPSS, 165 Inc.). A P value of less than 0.05 was considered to be statistically significant.
The statistical methods of this study were reviewed by Dr. Kai Xu from Department of Epidemiology, MD Anderson Cancer Center, University of Texas, United States; and Professor Ya-Feng Dong from University of Kansas School of Medicine, United States.
RESULTS
HRW decreased liver injury in APAP-challenged mice and improved the survival rate
The administration of 500 mg/kg APAP caused a severe illness in the mice, which was characterized by weakness and loss of body weight. After a lethal dose of APAP administration (750 mg/kg), NS treatment resulted in a 73.3% mortality rate in a 5-d observation period, while HRW could improve the 5-d survival rate to 60% (Figure 1A). Three days after a sub-lethal APAP challenge (500 mg/kg), mice were sacrificed with cervical dislocation. Necrosis, dropsy and petechiae could be observed in the liver tissue, which could be obviously mitigated by HRW treatment (Figure 1B-D). HE results also showed that HRW could alleviate the centrilobular necrosis, fatty infiltration and lymphocyte infiltration (Figure 1D). Meanwhile, the liver function detected by the serum ALT, AST, bilirubin, ALP and lactate dehydrogenase (LDH) activities was measured at 24, 48, and 72 h after APAP challenge. Compared with APAP + HRW mice, APAP + NS mice showed a significant increase in the serum ALT, bilirubin, AST, ALP and LDH levels (Figure 1E).
Figure 1.
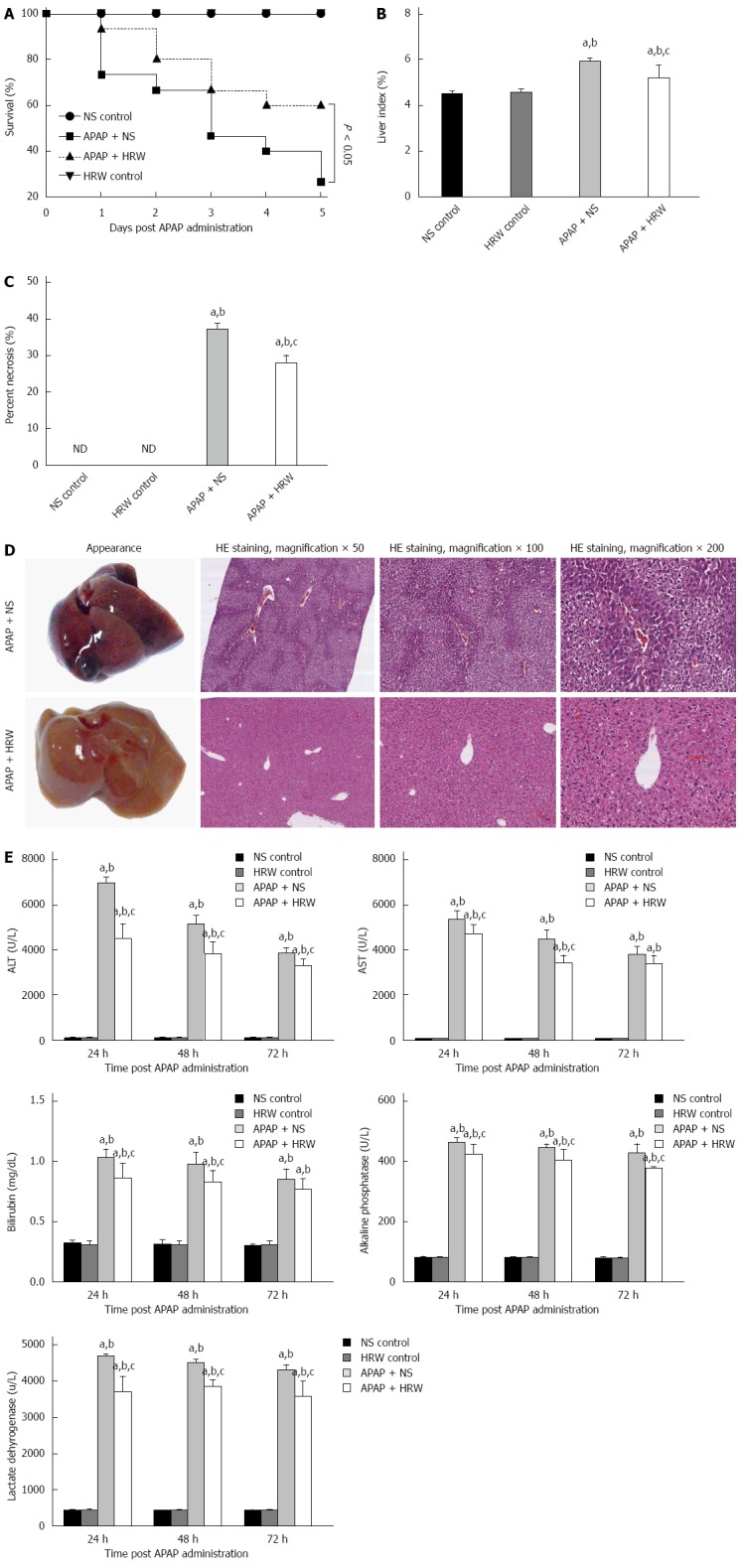
Hydrogen-rich water leads to an obvious enhancement in the survival and decrease in the liver injury after acetaminophen challenge. A: Kaplan-Meier survival curve for mice in three groups in 5 d after a single, lethal dose of 750 mg/kg acetaminophen (APAP) [normal saline (NS) control or hydrogen-rich water (HRW) control groups, n = 5; APAP + NS or APAP + HRW groups, n = 15]; B: Liver tissues were harvested 3 d after a sub-lethal dose of 500 mg/kg APAP. The liver index was used to evaluate the liver injury and it was decreased by HRW administration (n = 6, mean ± SD, aP < 0.05 vs NS control group, bP < 0.05 vs HRW control group, and cP < 0.05 vs APAP + NS group; C: Percent necrosis of mice in different groups (n = 6, mean ± SD, aP < 0.05 vs NS control group, bP < 0.05 vs HRW control group, and cP < 0.05, vs APAP + NS group); D: The representative appearance of livers and HE staining of murine liver sections. Decreased liver hemorrhaging, necrosis, and acute inflammation were observed in the HRW treated mice; E: After a sub-lethal dose of 500 mg/kg APAP administration, blood samples were harvested by tail cutting at 24 and 48 h and eyeball removal at 72 h. Serum was separated by centrifugation from blood to evaluate the liver function. Total bilirubin, alkaline phosphatase (ALP) and lactate dehydrogenase (LDH) at 24, 48 and 72 h were reduced with HRW treatment (n = 6, mean ± SD, aP < 0.05 vs NS control group; bP < 0.05 vs HRW control group; and cP < 0.05 vs APAP + NS group). ALT: Alanine aminotransferase; AST: Aspartate aminotransferase.
HRW inhibited oxidative stress, inflammation and peroxynitrite formation in the liver
Oxidative stress and inflammation could be induced by APAP administration, which played an initial, augmented role in the development of APAP hepatotoxicity. Three days after 500 mg/kg APAP administration, liver samples were removed to assess the oxidative stress in mice. The oxidative stress parameters, including MDA and MPO, in the liver were significantly increased in the APAP + NS group compared with the HRW-treatment group. The protective indicator, SOD, significantly increased with the use of HRW. HRW could also reverse the depletion of GSH and increase the GSH-Px caused by APAP (Figure 2A). Peroxynitrite (NT) formation and 4-HNE expression were also inhibited by HRW treatment (Figure 2B and C). Meanwhile, we also tested the effect of HRW on inflammatory cytokines in the blood and liver tissues 3 d after APAP challenge. The levels of IL-1β and TNF-α in the peripheral blood were markedly increased in the APAP + NS group compared with the normal control and APAP + HRW groups. Meanwhile, HRW could decrease the IL-1β and TNF-α mRNA levels in the liver (Figure 3).
Figure 2.
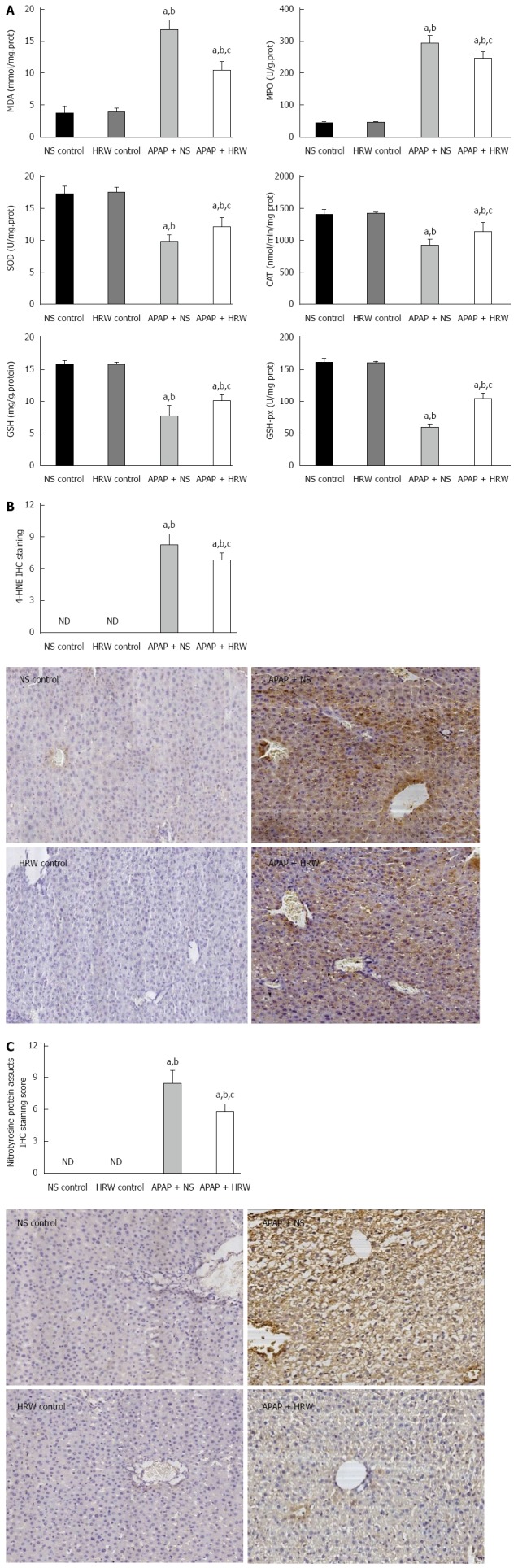
Hydrogen-rich water decreases the oxidative stress and nitrotyrosine formation in the liver after acetaminophen administration. Three days after a sub-lethal dose of 500 mg/kg acetaminophen (APAP) challenge, blood samples and liver tissues were harvested to evaluate the oxidative stress, inflammation and nitrotyrosine formation. A: Hydrogen-rich water (HRW) protected against APAP-induced elevated malonyldialdehyde (MDA) and myeloperoxidase (MPO) levels, decreased superoxide dismutase (SOD), catalase (CAT), glutathione (GSH) and glutathione peroxidase (GSH-px) activities; B: HRW inhibited the expression of 4-HNE in the liver; C: HRW inhibited the nitrotyrosine (NT) protein adduct formation in the liver (n = 6, mean ± SD, aP < 0.05 vs NS control group; bP < 0.05 vs HRW control group; and cP < 0.05 vs APAP + NS group).
Figure 3.
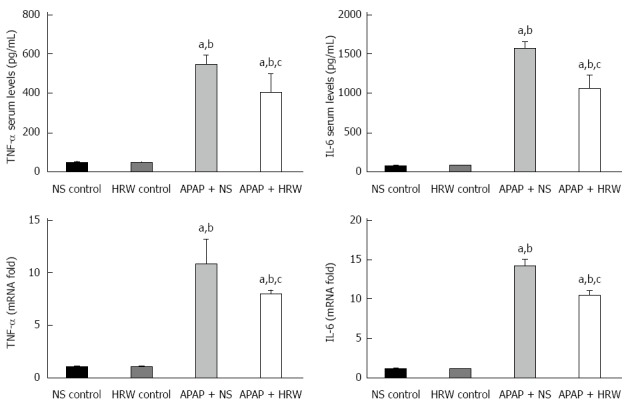
Hydrogen-rich water decreases the inflammation in the liver after acetaminophen administration. Hydrogen-rich water (HRW) reduced the serum TNF-α and IL-6 concentrations and decreased the transcriptional levels of TNF-α and IL-6 in the liver (n = 6, mean ± SD, aP < 0.05 vs NS control group; bP < 0.05 vs HRW control group; and cP < 0.05 vs APAP + NS group). NS: Normal saline.
HRW protected the endoplasmic reticulum and mitochondria in the liver
Three days after APAP administration, mice were sacrificed and the liver tissue was obtained to assess the hepatic subcellular structure injury. Electron microscopy revealed that APAP induced endoplasmic reticulum distension, hyperplasia and breakdown. It could also induce megamitochondria, mitochondria pyknosis, distension and flocculent degeneration. These injuries could be alleviated by different degrees of HRW treatment (Figure 4).
Figure 4.
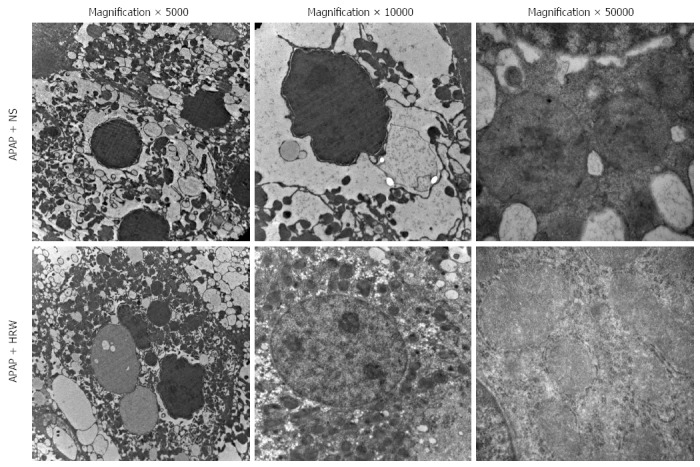
Hydrogen-rich water protects the integrity and stability of organelles after acetaminophen challenge. Representative electron microscopy of endoplasmic reticulum distension, hyperplasia and dissolution as well as hepatocyte megamitochondria in the acetaminophen (APAP) challenged mice. Hydrogen-rich water (HRW) treatment can alleviate the damage to a relatively normal level. NS: Normal saline.
HRW promoted hepatocyte proliferation in vivo after APAP challenge
Hepatocyte proliferation was a key step in recovery from liver injury. Therefore, we hypothesized that HRW might also increase hepatocyte proliferation after APAP overdose. To test this hypothesis, mice were killed at 72 h after APAP administration, and livers were harvested to determine the BrdU, Ki67 and PCNA staining. The tissue from the APAP-challenged mice displayed a small increase in the number of positive staining hepatocytes, which were confined to the centrilobular areas, but HRW significantly enhanced the BrdU, Ki67 and PCNA staining compared with NS-treated APAP-challenged mice (Figure 5A). The induction of cyclin D1 is the most reliable marker for cell cycle (G1 phase) progression in hepatocytes. Western blot was performed using whole-cell extracts prepared from liver tissue to assess the expression of cyclin D1 and PCNA in mice subjected to ALI or the control procedure. As observed in Figure 5B, the cyclin D1 and PCNA expression in the control group was minimal. In contrast, cyclin D1 and PCNA expression was clearly observed in HRW-treated animals at 72 h after APAP administration (Figure 5B).
Figure 5.
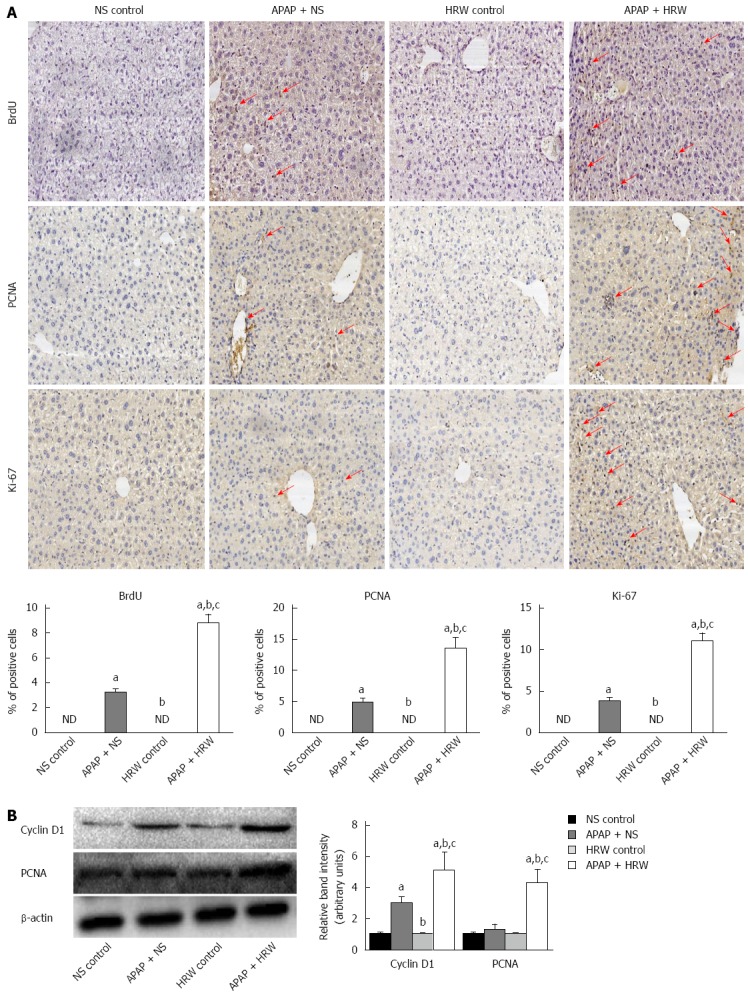
Hydrogen-rich water facilitates hepatocyte proliferation after acetaminophen challenge. A: Representative immunohistochemistry staining of BrdU, Ki67 and PCNA in the saline plus acetaminophen (APAP) as well as hydrogen-rich water (HRW) plus APAP treatment mice. The quantification of hepatocyte proliferation was assessed by calculating the positive cells observed by microscopy (magnification × 200); B: Western blot analysis for the protein content of cyclin D1 and PCNA proteins in response to APAP and HRW treatment in the liver. β-actin was used as an internal control (n = 6, mean ± SD, aP < 0.05 vs NS control group, bP < 0.05 vs APAP+NS group, and cP < 0.05 vs HRW control group). NS: Normal saline.
HRW inhibited Cx32 expression to alleviate liver injury
Three days after APAP administration, mice were sacrificed, the liver tissue was obtained and immunohistochemical analysis was performed to ascertain the localization and expression of Cx32 in liver tissue. Mice treated with APAP displayed significant immunoreactivity for Cx32 around the central veins in the liver. In contrast, the group of mice treated with HRW had lower Cx32 transcription and protein levels (Figure 6A-C).
Figure 6.
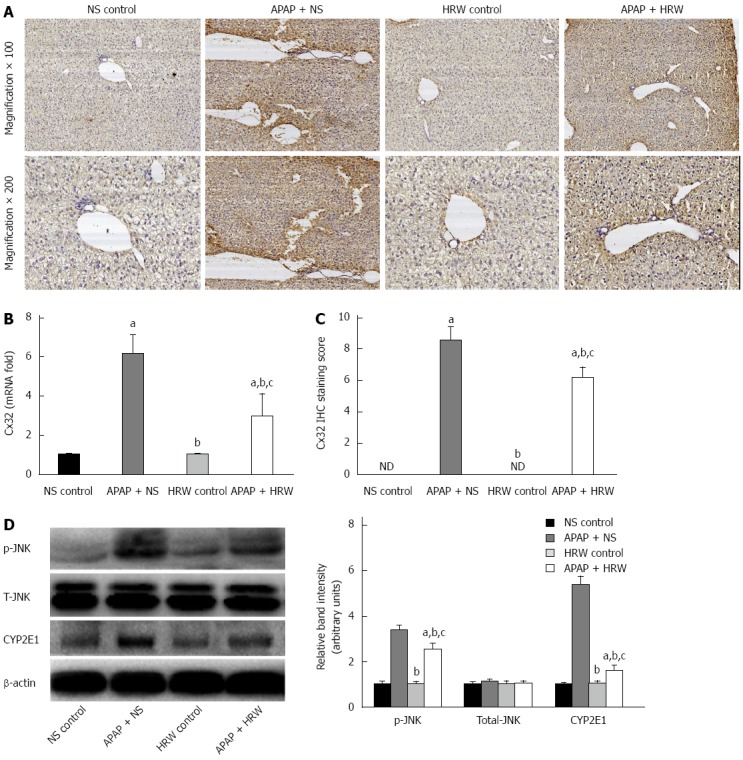
Hydrogen-rich water inhibited connexin 32 expression in the liver tissue. A: Representative photographs of connexin 32 (Cx32) expression in the liver. The acetaminophen (APAP)-challenged mice had more Cx32 immunopositive staining, as indicated by the brown color, while treatment with hydrogen-rich water (HRW) can significantly reduce Cx32 immunostaining; B: The Cx32 IHC staining score indicated that hydrogen therapy could significantly lower the staining score and reduce its activation and expression; C: The relative Cx32 mRNA levels in the three groups; D: Western blot analysis for the protein content of JNK and cytochrome P4502E (CYP2E1) proteins in response to APAP and HRW treatment in the liver. β-actin was used as an internal control (n = 6, mean ± SD, aP < 0.05 vs NS control group; bP < 0.05 vs APAP + NS group, and cP < 0.05 vs HRW control group). NS: Normal saline.
HRW inhibited APAP-induced phosphorylation of JNK and activation of CYP2E1
JNK activation is mediated by oxidative stress and plays pathogenic roles in a diverse array of cellular programs, including cell differentiation, movement, proliferation and death. Three days after APAP administration, mice were sacrificed and the livers were collected to investigate the mechanism for APAP-induced necrotic hepatocyte death; we studied the JNK activation involved in the signal transduction in the hepatic tissues. Western blot analyses showed a significant enhancement in the expression of phospho-JNK after APAP challenge, which could be reduced by the administration of HRW. No significant changes were found in the expression of total JNK. Furthermore, we investigated the effect of APAP on the CYP2E1 protein level and the result showed that the CYP2E1 protein level was much higher in the APAP + NS group compared with the APAP + HRW group (Figure 6D).
DISCUSSION
Progress has been achieved in the research of hydrogen therapy in diseases such as metabolism disorders, cancer, tissue ischemia reperfusion injury and more. Hydrogen has antioxidant, anti-inflammatory, anti-apoptotic and other protective effects, and it selectively quenches detrimental ROS, such as hydroxyl radicals and peroxynitrite, but it does not have an effect on physiological ROS, such as superoxide anion radical, hydrogen peroxide, and nitric oxide[20]. Because of its advantageous distribution characteristics, hydrogen can penetrate biomembranes and diffuse into the cytosol, mitochondria, and nucleus, successfully targeting organelles[28]. In this study, we found that hydrogen could reduce APAP-related liver injury by reducing hepatic necrosis, improving survival, reducing oxidative stress and inflammatory reaction in the liver, facilitating liver regeneration, protecting the integrity and stability of the hepatocyte organelles and inhibiting the expression of Cx32, CYP2E1 and phospho-JNK (Figure 7).
Figure 7.
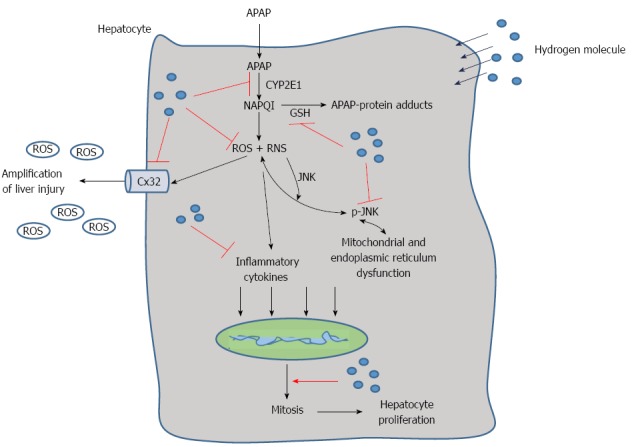
Schematic representation of the proposed effect of the hydrogen molecule in acetaminophen-induced hepatic injury. After acetaminophen (APAP) administration into the hepatocytes, APAP is first metabolized through the cytochrome P4502E (CYP2E1) and generates N-acetyl-p-benzoquinone imine (NAPQI), which binds with glutathione (GSH) and increases reactive oxygen species (ROS) and RNS formation as well as the level of inflammatory cytokines. It can also cause JNK phosphorylation that damages the mitochondria and endoplasmic reticulum. Meanwhile, connexin 32 (Cx32) can be induced with oxidative stress, which can promote the amplification of liver injury. The hepatic self-repair function is also activated after APAP administration, which can promote the cell mitosis to facilitate liver regeneration. In the present study, we found that hydrogen can reduce APAP hepatotoxicity by inhibiting APAP metabolism, ROS and RNS formation, Cx32 expression and promote hepatocyte proliferation.
APAP is a widely used, safe, effective analgesic when used within therapeutic doses. However, long-term and overdose use of APAP can induce severe hepatotoxicity and nephrotoxicity in both animals and humans[29]. The transport function and membrane permeability are impaired by APAP-induced hepatocyte injury, leading to leakage of enzymes from these cells. Therefore, the marked elevated levels of serum transaminases, bilirubin, ALP and LDH activities could be used to detect severe damage to hepatic tissue membranes during APAP-induced hepatotoxicity. However, treatment with HRW effectively reduced these alterations and improved the 5-d survival, demonstrating its hepatoprotective effects. The histopathological analysis of liver sections indicated moderate centrilobular necrosis, fatty infiltration and lymphocytic infiltration in the HRW + APAP treated mice with respect to the NS + APAP treated mice.
CYP2E1 is the major catalyst involved in the metabolism of drugs, and APAP is mainly metabolized by CYP2E1 to form an electrophilic metabolite, NAPQI, which is primarily inactivated by conjugation with GSH and posterior binding with other proteins to form protein adducts[30,31]. The accumulation of the intermediate metabolites and depletion of GSH are key mechanisms of APAP-hepatotoxicity that directly cause liver damage[6]. To detect whether the protective effects of HRW on the liver are associated with the inhibition of CYP2E1, we investigated the expression of CYP2E1 protein levels in different groups. The results suggest that HRW could obviously reduce APAP-induced CYP2E1 expression, which reduces NAPQI formation associated with APAP hepatotoxicity and effectively protects the liver against that pathophysiology.
A number of pieces of evidence implicate the roles of ROS and inflammation in the development of APAP-induced liver injury. For instance, excess depletion of GSH beyond a critical level leads to oxidative stress and exacerbates the hepatotoxicity. The masses of the metabolites produced by APAP are also found to generate ROS in biological systems[32,33]. Therefore, we measured the intracellular ROS production, inflammatory cytokines levels, GSH contents, lipid peroxidation, activities of the antioxidant enzymes and oxidation products (SOD, CAT, MDA, and 4-HNE) in hepatic tissues. APAP intoxication significantly increased the intracellular ROS production, inflammation levels and lipid peroxidation as well as decreased the GSH content. APAP intoxication also decreased the activities of the antioxidant enzymes SOD and CAT. However, the effect of HRW compared to the APAP + NS treatment, which was consistent with previous studies, indicates that hydrogen has a powerful anti-inflammatory and antioxidant effect. ROS are not only the direct damaging factors to the liver, they are also important signaling molecules which can activate JNK[34]. JNK activation is a pivotal regulator of mitochondrial permeabilization and plays a key role in the development of APAP-induced hepatotoxicity[35]. ROS may activate JNK through the oxidation of kinase inhibitors, which can inhibit JNK or upstream ASK1[36]. It can also lead to the redox inactivation of JNK phosphatase, which sustains JNK activation[37]. To check whether HRW could inhibit APAP-induced JNK phosphorylation, we tested the JNK phosphorylation by Western blot analysis. The results of our study showed that HRW could greatly attenuate the expression of phospho-JNK in the liver tissue to alleviate liver injury. It is also well established that APAP overdose induces peroxynitrite formation, as indicated by the appearance of nitrotyrosine protein adducts in centrilobular hepatocytes. Also, we found that hydrogen could inhibit the APAP-induced peroxynitrite formation, which might partly explain its protective mechanism.
To determine the deeper mechanism of the protective effect of HRW against APAP-induced hepatotoxicity, we focused on the effect of hydrogen on the hepatocyte proliferation, maintaining the stability of subcellular fraction and Cx32 expression. Liver regeneration is a compensatory process after a toxic insult, which guarantees the replacement of necrotic cells and full recovery of organ function[38]. The exposure of hepatocytes to growth factor leads to the expression of cell cycle proteins. Cyclin D1 is the most reliable marker for cell cycle (G1 phase) progression in hepatocytes. Once hepatocytes express cyclin D1, they have passed the G1 restriction point and are committed to DNA replication[39]. Notably, in the current investigation, we found that HRW could promote BrdU, ki-67 and PCNA expression in the APAP-challenged liver, and these are the iconic markers for liver regeneration. Meanwhile, the Western blot data showed that HRW markedly increased the level of cyclin D1 in the APAP-challenged liver tissue. These changes in cyclin D1 expression were associated with decreased serum ALT/AST levels and improvement of liver regeneration in HRW-treated mice that received APAP, suggesting that HRW facilitates the activation of cyclin D1-mediated regeneration pathways.
The endoplasmic reticulum (ER) is the major cellular site of protein folding and modification. ER stress occurs when the level of APAP-induced protein adducts entering the ER exceeds its folding capacity[40,41]. Meanwhile, APAP metabolites not only damage the structural integrity of mitochondria, resulting in membrane fracture and electronic leak, they also lead to dysfunction of the respiratory chain and energy generation[42]. Mitochondrial dysfunction is believed to be the propagating event of APAP toxicity, resulting in loss of ATP production, mitochondrial swelling, generation of ROS, formation of the mitochondrial permeability transition pore and the release of mitochondrial contents, all of which ultimately result in hepatic necrosis[30,43]. In this study, treatment with HRW could protect the integrity and function of the ER and mitochondria.
Gap junctions are plasma membrane spatial microdomains constituted by assemblies of channel proteins called connexins, which provide direct intercellular communication pathways, allowing cell-to-cell rapid exchange of ions and metabolites[44]. Cx32 is the major gap junction protein in the liver, and previous studies have shown that interfering with Cx32 greatly reduces liver damage due to several toxic agents, including carbon tetrachloride, D-galactosamine, TAA and APAP[19,45]. In this study, we found that the expression of Cx32 was obviously elevated in the APAP-challenged mice and HRW + APAP administration could significantly reduce the expression compared with the APAP + NS treated group.
In conclusion, the results of the present study demonstrate that HRW has a prophylactic as well as a therapeutic role in preventing APAP-induced hepatotoxicity, most likely due to its unique cytoprotective properties such as antioxidant and anti-inflammation activities. Most importantly, HRW can maintain the stability of the cellular structure, promote hepatocyte regeneration and inhibit the expression of the Cx32 gap junction, JNK phosphorylation and CYP2E1 after APAP challenge. All of these findings indicate that HRW can be a potential therapy for preventing liver injury caused by APAP overdose.
ACKNOWLEDGMENTS
We thank UNIVA Guangzhou Trading Co., Ltd for their assistance in providing hydrogen-rich water.
COMMENTS
Background
Acetaminophen (N-acetyl-p-aminophenol, APAP) is a widely used analgesic and antipyretic drug in the clinic. APAP is believed to be safe within therapeutic doses, but overdose usage causes a centrilobular hepatic necrosis that leads to acute liver failure. Overdoses of APAP can promote the generation of the toxic metabolite N-acetyl-quinoneimine (NAPQI), which is immediately conjugated with glutathione (GSH) to form the nontoxic metabolites cysteine. However, when the GSH is exhausted, NAPQI covalently binds with other proteins to form protein adducts, directly leading to cell death.
Research frontiers
Hydrogen therapy is a new medical approach that has recently become increasingly appreciated. Hydrogen has anti-oxidant, anti-inflammatory, anti-apoptotic, anti-allergy, and anti-cancer effects. Several methods invented to deliver hydrogen, including inhalation, drinking hydrogen-rich water (HRW) and injection with hydrogen-saturated saline, are valid and reliable. In the area of prevention of APAP-induced liver injury, a research hotspot is to search for more effective and convenient methods that more people will accept. Meanwhile, the mechanism of a new medicine is another hotspot.
Innovations and breakthroughs
The authors investigated the effects of HRW on APAP-induced liver injury in mice. The present study concluded that HRW can significantly prevent the APAP-induced acute hepatotoxicity by enhancing the hepatic antioxidant activity, reducing inflammation, protecting the hepatic subcellular structure, promoting liver regeneration, and inhibiting CX32, CYP2E1 and phospho-JNK activation.
Applications
Hydrogen therapy might be safe and effective for preventing liver injury derived from APAP application.
Terminology
Hydrogen is the lightest gas in nature, but it has anti-oxidant and anti-inflammatory effects. It has been proven effective in treating many diseases. HRW is produced by pressurizing the hydrogen gas into the water by a specific device under high pressure.
Peer-review
The manuscript is well-written and interesting because it investigates the hepatoprotective effects and mechanisms of HRW in APAP-induced liver injury in mice. The authors first generated a murine model of APAP-induced liver injury; then, HRW was administered intraperitoneally for 3 d to explore whether HRW has a hepatoprotective effect. They then go on to detect the change in the liver injury index and several cytokines. Meanwhile, they found that HRW promoted hepatocyte proliferation and liver regeneration after APAP administration, indicating that HRW is expected to be a potent hepatoprotective agent in the future and the hepatoprotective effect of HRW is worth studying.
Footnotes
Ethics approval: The study was reviewed and approved by The First Affiliated Hospital of Xi’an Jiaotong University College of Medicine Institutional Review Board.
Institutional animal care and use committee: All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of The First Affiliated Hospital of Xi’an Jiaotong University College (IACUC protocol number: XITULAC2014-207).
Conflict-of-interest: Authors declare no competing financial interests.
Data sharing: Technical appendix, statistical code, and dataset available from the corresponding author at liuchangdoctor@163.com. Participants gave informed consent for data sharing.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: October 18, 2014
First decision: December 2, 2014
Article in press: February 13, 2015
P- Reviewer: Kucera O, Xu CS S- Editor: Yu J L- Editor: Logan S E- Editor: Ma S
References
- 1.Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis. 2007;11:525–48, vi. doi: 10.1016/j.cld.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiødt FV, Ostapowicz G, Shakil AO, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42:1364–1372. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- 3.Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 4.McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 2012;122:1574–1583. doi: 10.1172/JCI59755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahlin DC, Miwa GT, Lu AY, Nelson SD. N-acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc Natl Acad Sci USA. 1984;81:1327–1331. doi: 10.1073/pnas.81.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hinson JA, Roberts DW, James LP. Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol. 2010;(196):369–405. doi: 10.1007/978-3-642-00663-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muldrew KL, James LP, Coop L, McCullough SS, Hendrickson HP, Hinson JA, Mayeux PR. Determination of acetaminophen-protein adducts in mouse liver and serum and human serum after hepatotoxic doses of acetaminophen using high-performance liquid chromatography with electrochemical detection. Drug Metab Dispos. 2002;30:446–451. doi: 10.1124/dmd.30.4.446. [DOI] [PubMed] [Google Scholar]
- 8.Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev. 2012;44:88–106. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agarwal R, MacMillan-Crow LA, Rafferty TM, Saba H, Roberts DW, Fifer EK, James LP, Hinson JA. Acetaminophen-induced hepatotoxicity in mice occurs with inhibition of activity and nitration of mitochondrial manganese superoxide dismutase. J Pharmacol Exp Ther. 2011;337:110–116. doi: 10.1124/jpet.110.176321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cover C, Liu J, Farhood A, Malle E, Waalkes MP, Bajt ML, Jaeschke H. Pathophysiological role of the acute inflammatory response during acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 2006;216:98–107. doi: 10.1016/j.taap.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 11.Bajt ML, Ramachandran A, Yan HM, Lebofsky M, Farhood A, Lemasters JJ, Jaeschke H. Apoptosis-inducing factor modulates mitochondrial oxidant stress in acetaminophen hepatotoxicity. Toxicol Sci. 2011;122:598–605. doi: 10.1093/toxsci/kfr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blazka ME, Wilmer JL, Holladay SD, Wilson RE, Luster MI. Role of proinflammatory cytokines in acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 1995;133:43–52. doi: 10.1006/taap.1995.1125. [DOI] [PubMed] [Google Scholar]
- 13.James LP, McCullough SS, Knight TR, Jaeschke H, Hinson JA. Acetaminophen toxicity in mice lacking NADPH oxidase activity: role of peroxynitrite formation and mitochondrial oxidant stress. Free Radic Res. 2003;37:1289–1297. doi: 10.1080/10715760310001617776. [DOI] [PubMed] [Google Scholar]
- 14.Das J, Ghosh J, Manna P, Sil PC. Acetaminophen induced acute liver failure via oxidative stress and JNK activation: protective role of taurine by the suppression of cytochrome P450 2E1. Free Radic Res. 2010;44:340–355. doi: 10.3109/10715760903513017. [DOI] [PubMed] [Google Scholar]
- 15.Hinson JA, Bucci TJ, Irwin LK, Michael SL, Mayeux PR. Effect of inhibitors of nitric oxide synthase on acetaminophen-induced hepatotoxicity in mice. Nitric Oxide. 2002;6:160–167. doi: 10.1006/niox.2001.0404. [DOI] [PubMed] [Google Scholar]
- 16.Bae MA, Pie JE, Song BJ. Acetaminophen induces apoptosis of C6 glioma cells by activating the c-Jun NH(2)-terminal protein kinase-related cell death pathway. Mol Pharmacol. 2001;60:847–856. [PubMed] [Google Scholar]
- 17.Segretain D, Falk MM. Regulation of connexin biosynthesis, assembly, gap junction formation, and removal. Biochim Biophys Acta. 2004;1662:3–21. doi: 10.1016/j.bbamem.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 18.Patel SJ, King KR, Casali M, Yarmush ML. DNA-triggered innate immune responses are propagated by gap junction communication. Proc Natl Acad Sci USA. 2009;106:12867–12872. doi: 10.1073/pnas.0809292106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel SJ, Milwid JM, King KR, Bohr S, Iracheta-Velle A, Li M, Vitalo A, Parekkadan B, Jindal R, Yarmush ML. Gap junction inhibition prevents drug-induced liver toxicity and fulminant hepatic failure. Nat Biotechnol. 2012;30:179–183. doi: 10.1038/nbt.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, Katsura K, Katayama Y, Asoh S, Ohta S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med. 2007;13:688–694. doi: 10.1038/nm1577. [DOI] [PubMed] [Google Scholar]
- 21.Zhang JY, Liu C, Zhou L, Qu K, Wang R, Tai MH, Lei Lei JC, Wu QF, Wang ZX. A review of hydrogen as a new medical therapy. Hepatogastroenterology. 2012;59:1026–1032. doi: 10.5754/hge11883. [DOI] [PubMed] [Google Scholar]
- 22.Sun H, Chen L, Zhou W, Hu L, Li L, Tu Q, Chang Y, Liu Q, Sun X, Wu M, et al. The protective role of hydrogen-rich saline in experimental liver injury in mice. J Hepatol. 2011;54:471–480. doi: 10.1016/j.jhep.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 23.Kawai D, Takaki A, Nakatsuka A, Wada J, Tamaki N, Yasunaka T, Koike K, Tsuzaki R, Matsumoto K, Miyake Y, et al. Hydrogen-rich water prevents progression of nonalcoholic steatohepatitis and accompanying hepatocarcinogenesis in mice. Hepatology. 2012;56:912–921. doi: 10.1002/hep.25782. [DOI] [PubMed] [Google Scholar]
- 24.Ohta S. Molecular hydrogen as a preventive and therapeutic medical gas: initiation, development and potential of hydrogen medicine. Pharmacol Ther. 2014;144:1–11. doi: 10.1016/j.pharmthera.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 25.Zhang J, Wu Q, Song S, Wan Y, Zhang R, Tai M, Liu C. Effect of hydrogen-rich water on acute peritonitis of rat models. Int Immunopharmacol. 2014;21:94–101. doi: 10.1016/j.intimp.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 26.Zhang JY, Wu QF, Wan Y, Song SD, Xu J, Xu XS, Chang HL, Tai MH, Dong YF, Liu C. Protective role of hydrogen-rich water on aspirin-induced gastric mucosal damage in rats. World J Gastroenterol. 2014;20:1614–1622. doi: 10.3748/wjg.v20.i6.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qu K, Xu X, Liu C, Wu Q, Wei J, Meng F, Zhou L, Wang Z, Lei L, Liu P. Negative regulation of transcription factor FoxM1 by p53 enhances oxaliplatin-induced senescence in hepatocellular carcinoma. Cancer Lett. 2013;331:105–114. doi: 10.1016/j.canlet.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 28.James AM, Cochemé HM, Murphy MP. Mitochondria-targeted redox probes as tools in the study of oxidative damage and ageing. Mech Ageing Dev. 2005;126:982–986. doi: 10.1016/j.mad.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 29.Lee WM, Ostapowicz G. Acetaminophen: pathology and clinical presentation of hepatotoxicity. Drug induced liver disease. 2007:389–405. [Google Scholar]
- 30.Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 2004;40:1170–1179. doi: 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- 31.Lee SS, Buters JT, Pineau T, Fernandez-Salguero P, Gonzalez FJ. Role of CYP2E1 in the hepatotoxicity of acetaminophen. J Biol Chem. 1996;271:12063–12067. doi: 10.1074/jbc.271.20.12063. [DOI] [PubMed] [Google Scholar]
- 32.Yan HM, Ramachandran A, Bajt ML, Lemasters JJ, Jaeschke H. The oxygen tension modulates acetaminophen-induced mitochondrial oxidant stress and cell injury in cultured hepatocytes. Toxicol Sci. 2010;117:515–523. doi: 10.1093/toxsci/kfq208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park BK, Laverty H, Srivastava A, Antoine DJ, Naisbitt D, Williams DP. Drug bioactivation and protein adduct formation in the pathogenesis of drug-induced toxicity. Chem Biol Interact. 2011;192:30–36. doi: 10.1016/j.cbi.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 34.Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 2008;283:13565–13577. doi: 10.1074/jbc.M708916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Latchoumycandane C, Goh CW, Ong MM, Boelsterli UA. Mitochondrial protection by the JNK inhibitor leflunomide rescues mice from acetaminophen-induced liver injury. Hepatology. 2007;45:412–421. doi: 10.1002/hep.21475. [DOI] [PubMed] [Google Scholar]
- 36.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40:928–939. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 38.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45–S53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 39.Yang R, Zhang S, Cotoia A, Oksala N, Zhu S, Tenhunen J. High mobility group B1 impairs hepatocyte regeneration in acetaminophen hepatotoxicity. BMC Gastroenterol. 2012;12:45. doi: 10.1186/1471-230X-12-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 41.Uzi D, Barda L, Scaiewicz V, Mills M, Mueller T, Gonzalez-Rodriguez A, Valverde AM, Iwawaki T, Nahmias Y, Xavier R, et al. CHOP is a critical regulator of acetaminophen-induced hepatotoxicity. J Hepatol. 2013;59:495–503. doi: 10.1016/j.jhep.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 42.Burke AS, MacMillan-Crow LA, Hinson JA. Reactive nitrogen species in acetaminophen-induced mitochondrial damage and toxicity in mouse hepatocytes. Chem Res Toxicol. 2010;23:1286–1292. doi: 10.1021/tx1001755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cover C, Mansouri A, Knight TR, Bajt ML, Lemasters JJ, Pessayre D, Jaeschke H. Peroxynitrite-induced mitochondrial and endonuclease-mediated nuclear DNA damage in acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2005;315:879–887. doi: 10.1124/jpet.105.088898. [DOI] [PubMed] [Google Scholar]
- 44.Maurel M, Rosenbaum J. Closing the gap on drug-induced liver injury. Hepatology. 2012;56:781–783. doi: 10.1002/hep.25779. [DOI] [PubMed] [Google Scholar]
- 45.Asamoto M, Hokaiwado N, Murasaki T, Shirai T. Connexin 32 dominant-negative mutant transgenic rats are resistant to hepatic damage by chemicals. Hepatology. 2004;40:205–210. doi: 10.1002/hep.20256. [DOI] [PubMed] [Google Scholar]