

Abstract
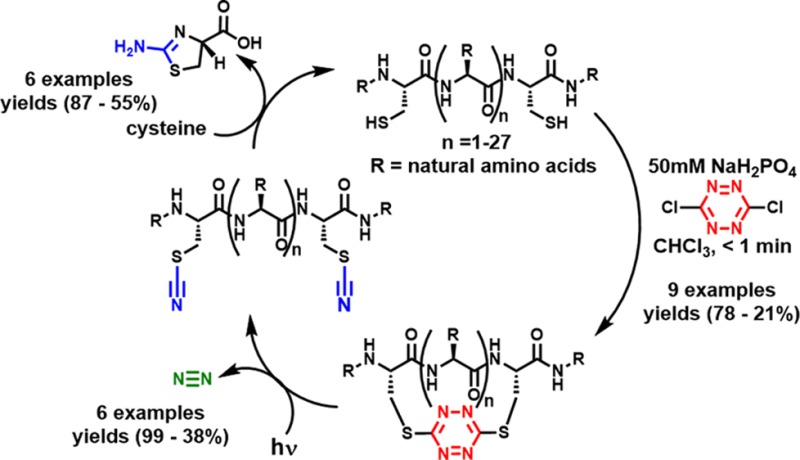
Protocols have been achieved that permit facile introduction of s-tetrazine into unprotected peptides and the protein, thioredoxin, between two cysteine sulfhydryl groups (i.e., staple), followed by photochemical release (i.e., unstaple) and regeneration of the peptide/protein upon removal of the cyano groups from the derived bisthiocyanate. The S,S-tetrazine macrocycles in turn provide a convenient handle for probe introduction by exploiting the inverse electron demand Diels–Alder reactivity of the tetrazine.
Modulation of peptide and protein conformation comprises one of the principal determinates of biofunction.1 Toward this end, macrocyclization has become an effective tactic to increase proteolytic stability, vary polarity, and/or improve drugability.2−5 While a number of methods5−18 have been reported to construct macrocyclic peptides, our recent focus has been on the construction of peptide/protein macrocycles via side-chain–side-chain tethering, a tactic known as stapling, useful for both helical5,6 and non-helical scaffolds.7−12
Development of the all-hydrocarbon stapling tactic13 has provided a number of bioactive peptides,5 locking their active conformation to target extra- and intracellular protein–protein interactions. Examples of helix stabilization via heteroatom side-chain tethering of peptides/proteins with amides,14 triazoles,15 thioethers,14 oximes,17 and perfluoroaryl linkages18 have also been developed. To date however, to the best of our knowledge, there has been only one example of the removal of an inserted staple to regenerate the native peptide/protein without severely disrupting the structure.16
In a recent biophysical program to define peptide folding dynamics, we, in collaboration with Hochstrasser, introduced the s-tetrazine moiety as an ultrafast photochemical trigger to release peptide/protein conformation on the picosecond time scale.19−23 In that venture, we designed and synthesized a series of S,S-tetrazine peptide linchpins by bridging two cysteines with the reagent dichlorotetrazine, employing solid-phase peptide synthesis (SPPS).20 The linchpins were then reacted with helices available via SPPS in a solution-phase fragment union strategy to construct larger peptides with known conformations.21Upon irradiation of the S,S-tetrazine chromophore with a biocompatible light flash (355 or 410 nm), the restricted conformation was released on the ps time scale, generating two thiocyanates and molecular nitrogen.22 This tactic played an important role in the ultrafast photochemical release of a kinked helix when employed with the Hochstrasser–Zanni pump/probe 2D-IR technique,24 enabling the collection of structural snapshots and relaxation kinetics of the local bond conformational reorganization, previously unobservable, on the ps time scale.23
Here we describe a new general approach for the stapling and unstapling of peptides comprising a facile biphasic protocol that enables rapid, highly efficient incorporation of the s-tetrazine chromophore into unprotected peptides that possess two proximal cysteines, and a monophasic protocol for the protein thioredoxin. Central to both protocols is the high chemoselective nucleophilicity of the sulfhydryl groups. Importantly, the S,S-tetrazine stapled macrocycles can be readily unstapled by photolysis to form the peptide/protein possessing two thiocyanates, which can be removed by reaction with cysteine to provide the disulfhydryl peptide or protein (vide infra).
We initiated this investigation by determining suitable phase-transfer conditions for incorporation of the s-tetrazine chromophore into unprotected peptides employing bis-sulfhydryl model peptide 1a (Scheme 1), prepared by Fmoc-based SPPS. 1a was designed to possess many of the reactive side chains commonly found within bioactive peptides and proteins. We next examined a series of solvents, denaturants, and pH levels for the conversion of 1a to 2a. Separation of the phases (i.e., phase transfer) containing the two reactants permitted excellent chemoselective control for sulfhydryl addition to the highly electrophilic dichlorotetrazine. Mild acidic conditions (pH 5) were slightly favored in the reaction, with the rate of tetrazine insertion somewhat slower at low pH values, while at pH >9 the product became unstable. Several denaturants were investigated with the idea to decrease aggregation; addition of guanidine hydrochloride did not noticeably affect the yield. To prevent disulfide formation, we also added a reducing agent to the reaction solution; tris(2-carboxyethyl)phosphine (TCEP) was found to react with dichlorotetrazine during the course of the reaction and to reduce the s-tetrazine motif once inserted. However, in the event that disulfide bond formation is a problem, immobilized TCEP can be employed to reduce the disulfide bond and then removed by filtration prior to addition of the dichlorotetrazine. The best phase-transfer conditions comprise addition of 1a in a 50 mM aqueous solution of monosodium phosphate, with the “red” dichlorotetrazine dissolved in chloroform as a separate phase. Vigorous mixing of the two phases rapidly (<1 min) furnishes 2a (Scheme 1), qualitatively indicated by the red color now present in the aqueous phase. Minimal side products were formed during the reaction. Pleasingly, purification by reverse-phase chromatography yielded the red peptide 2a (78%). Notably, the s-tetrazine adduct 2a is stable in a variety of buffers (pH 3–8) and organic solvents, in the temperature range of 0–100 °C, and without exclusion of ambient light. Subsequent steady-state irradiation in a Rayonet photoreactor equipped with UV-A lamps (λmax = 365 nm)20 led, on preparative scale (15 mg), to the dithiocyanate peptide 3a in near-quantitative yield.
Scheme 1. Stapling and Unstapling of Peptides.
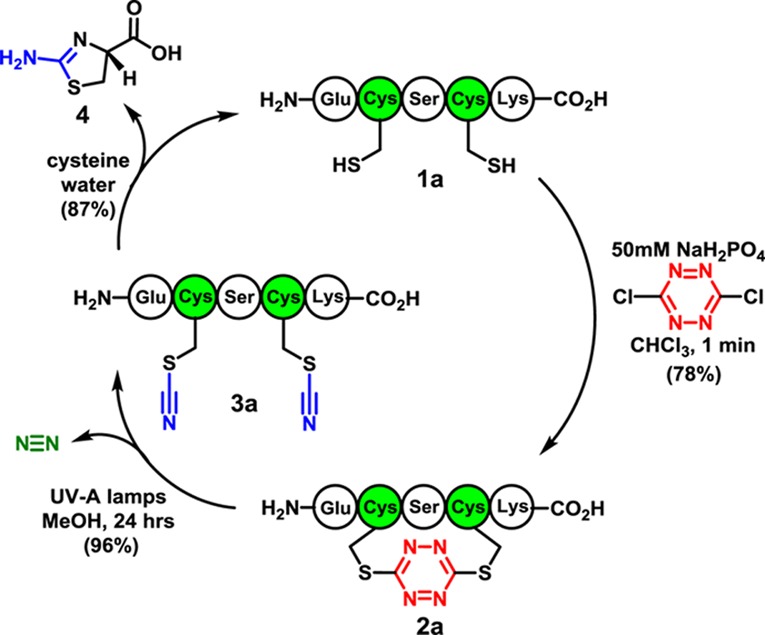
We next developed a mild protocol to remove the nitriles from the thiocyanate groups in 3a to regenerate the native disulfhydryl peptide (Scheme 1). Early studies25,26 suggested that the cyanyl groups could be removed from the thiocyanates with cysteine. To this end, peptide 2a was treated with cysteine in water, giving 1a in 87% yield along with thiazolamine 4. The mechanism for nitrile removal (Figure 1) is proposed to be analogous to native chemical ligation.27
Figure 1.
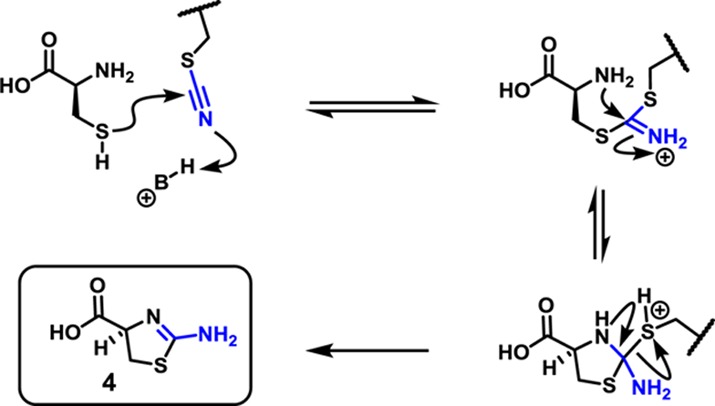
Proposed mechanism for thiocyanate removal.
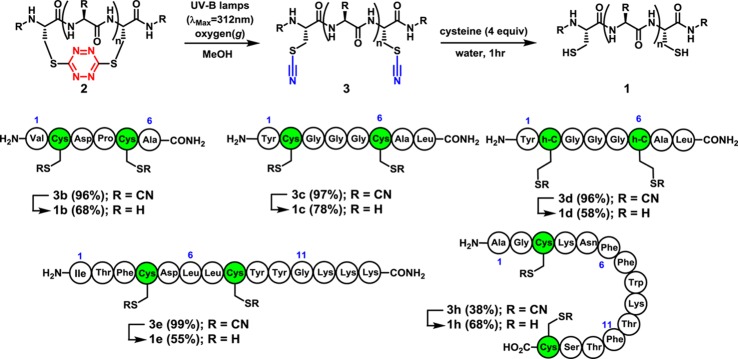
Encouraged by the results of the model study, we next explored incorporation of the tetrazine chromophore into a variety of peptides (Scheme 2), possessing spacings (i, i+3–i+28) between the cysteine residues, to test compatibility with various coded as well as d-amino acids comprising the peptide. To this end, the red stapled tetrazine macrocycles 2b–i were prepared employing the phase-transfer protocol with isolation and purification by reverse-phase chromatography; in most cases yields were good. Minor amounts of disulfide (<5%) and dimeric products (<10%) were observed in some cases. Oligomeric products became increasingly problematic as the ring size of the cyclic tetrazine peptides became larger (cf. 2i); separation of the desired monomer could nevertheless be achieved in all cases. Interestingly, vis-à-vis the reaction scope, although early SPPS attempts to incorporate the s-tetrazine chromophore into peptides possessing arginine side chains led to decomposition,20 incorporation of the s-tetrazine chromophore into peptides possessing both the reactive arginine and methionine side chains now proved feasible, given the use of an aqueous buffer during the phase-transfer protocol (2g,i). In the case of peptides containing methionine amino acids, however, we did observe minor (<5%) oxidation of the sulfide side chain.
Scheme 2. Stapled Peptides Prepared by Phase-Transfer Insertion of Dichlorotetrazine into Unprotected Peptides.

Turning next to release of the S,S-tetrazine staple, cyclic peptides 2b–d were subjected to steady-state irradiation with UV-A lamps (λmax = 365 nm). In these larger tetrazine macrocycles, initial yields for the dithiocyanate products were 25–35%. Optimization proved feasible by exploring a variety of solvents, wavelengths, and temperatures; methanol was the best solvent, with the most productive wavelengths ranging from 300 to 350 nm. The effect of oxygen was also examined to improve the photochemical outcome; solutions containing peptides 2a–e were sparged with O2 prior to irradiation. Photolysis with UV-B lamps (λmax = 312 nm) in the presence of O2 provided exclusively the unstapled peptides 3a–e (Scheme 3).28 For peptides containing tryptophan and methionine residues (cf. 2f–h), additional reactivity was observed under the O2 conditions; the dithiocyanate counterpart 3h is provided as an example that could be isolated in 38% yield after purification. In the end, the tetrazine macrocycles could be unstapled photochemically in situ to yield the unconstrained linear dithiocyanate peptides 3b–e,h and then, in turn, converted to their native structures 1b–e,h employing cysteine for nitrile removal in yields of 55–87%.
Scheme 3. Photochemical Unstapling of S,S-Tetrazine Macrocycles and Regeneration of the Native Peptides.
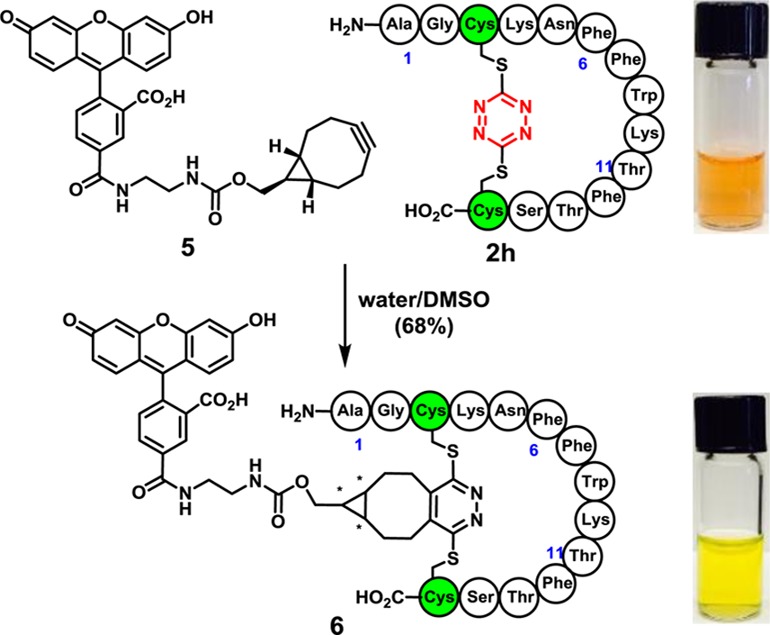
A significant advantage of the S,S-tetrazine staple is reactivity of the incorporated tetrazine to introduce probes exploiting inverse electron demand Diels–Alder reactions.29 To demonstrate this feature, fluorescein dye 5 was prepared and combined with stapled peptide 2h to obtain a diastereomeric mixture of the fluorescein-labeled peptide 6 (Scheme 4). Thus, tetrazine staples hold the promise of dual roles: confining peptide conformation and introducing photophysical and other potential probes.30
Scheme 4. S,S-Tetrazine Staple Introduces a Handle for Inverse Electron Demand Diels–Alder Reactions.
Having established a protocol for the stapling and unstapling of peptides between two cysteine sulfhydryls, we turned to a protein. We selected thioredoxin (Trx), an oxidoreductase that specifically contains a single, solvent-exposed disulfide bond at the active site. To replace the disulfide bond with s-tetrazine, the Trx disulfide bond was reduced with immobilized TCEP, the TCEP removed by filtration, and the reduced Trx stirred with dichlorotetrazine (4 equiv) at pH 5 (acetate buffer/DMSO, 50:1) with an overall protein concentration of 20 μM. After 1 min, tetrazine-Trx resulted, with 90% incorporation of s-tetrazine, as revealed by both C4 LC-MS and MALDI-TOF-MS (m/z = 11757.782) (Scheme 5A).31 The latter could be purified with a desalting column to give an 88% yield of protein, quantified by Bradford assay. To regenerate the protein, the tetrazine-Trx was subjected in situ to photolysis under an ambient atmosphere with UV-B lamps. After 1 h, loss of the very pale pink color occurred; MALDI-TOF-MS revealed not only the loss of N2 but also partial loss of nitrile groups from the thiocyanates. To complete the removal of the nitriles, a solution of cysteine and TCEP was added. After 4 h, the TCEP was removed via centrifugal filtration. Addition of Ellman’s reagent32 then furnished the disulfide bond, again as observed by C4 LC-MS and MALDI-TOF-MS. Fast protein liquid chromatography (FPLC) and C4 LC-MS revealed a 73% yield and 52% purity of the sample after photolysis (see Supporting Information). To interrogate the regenerated protein, and mindful that previous synthetic modifications were all directed at the site of catalysis, bioactivity measurements were performed employing insulin as a substrate. After evaluation, the regenerated protein did not display substantial catalytic turnover for the reduction of insulin. However, the regenerated protein remained reactive toward electrophiles, such as dichlorotetrazine, when resubjected to the above-described stapling conditions (Scheme 5A). To understand the loss of activity, we subjected native Trx to the photochemical conditions identical to those employed to release the tetrazine staple, only to find a similar loss of biological activity. Thus, it is difficult to evaluate whether photolysis of the tetrazine moiety damaged Trx beyond what would be incurred by photolysis alone.
Scheme 5. S,S-Tetrazine Stapling, Unstapling, and Probe Introduction with the Thioredoxin Protein.
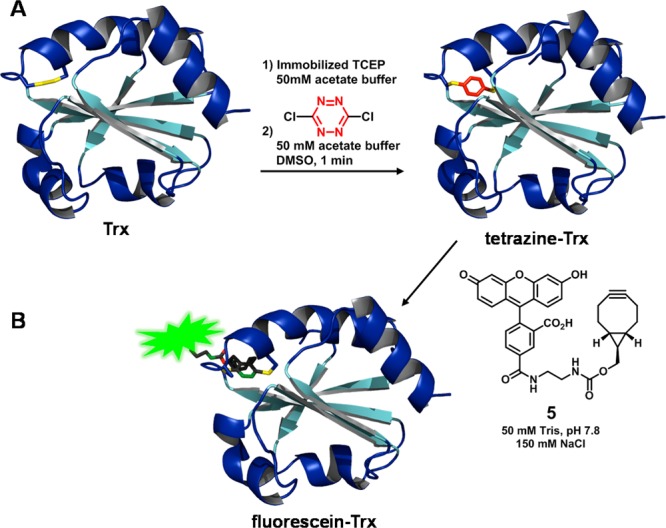
With the tetrazine-Trx adduct in hand, we next evaluated the feasibility of incorporating the fluorescein dye 5 (Scheme 5B). Toward this end, simply permitting 5 to stand in a buffered solution of the tetrazine-Trx provided the fluorescein-Trx, as witnessed both by the retention of green color from fluorescein in the protein upon separation by centrifugal filtration and by MALDI-TOF-MS. Thus, the mildly reactive bicyclononyne33 does indeed add to the tetrazine once incorporated within the secondary structure of thioredoxin.
In summary, we have developed facile protocols for the incorporation of s-tetrazine between proximate cysteine sulfhydryls of unprotected peptides and the protein thioredoxin. The tetrazine-transfer reaction in turn enables the construction of macrocyclic peptides with a wide range of functionality and ring topology, bridging from 1 to 27 amino acid residues adjoining cysteines. Importantly, the stapled conformations can be released photochemically to their thiocyanate counterparts and, in turn, the resulting thiocyanates removed to regenerate the peptide. The s-tetrazine thus comprises a readily removable peptide staple. Finally, the stapling and unstapling protocol has been extended to include thioredoxin as an example of a protein with an incorporated S,S-tetrazine construct that can also serve a useful role in conjugation strategies.
Acknowledgments
Financial support was provided by the NIH GM 12694 and the RLBL facility grant (NIH P41 GM 104605, S.B.). The MALDI-TOF-MS instrumentation was supported by NSF MRI-0820996. We thank Drs. George Furst and Rakesh Kohli for assistance in obtaining NMR and high-resolution MS, respectively; Christopher Lanci and the BCRC facility for automated peptide synthesis; Matthew J. Eibling for assisting with FPLC separations of thioredoxin; and Yanxin Wang for helpful discussions. We also thank Prof. Feng Gai for helpful discussions and encouragement.
Supporting Information Available
Experimental procedures and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bock J. E.; Gavenonis J.; Kritzer J. A. ACS Chem. Biol. 2013, 8, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsault E.; Peterson M. L. J. Med. Chem. 2011, 54, 1961. [DOI] [PubMed] [Google Scholar]
- White C. J.; Yudin A. K. Nat. Chem. 2011, 3, 509. [DOI] [PubMed] [Google Scholar]
- Miriam Góngora-Benítez M.; Tulla-Puche J.; Albericio F. Chem. Rev. 2014, 114, 901. [DOI] [PubMed] [Google Scholar]
- Walensky L. D.; Bird G. H. J. Med. Chem. 2014, 57, 6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q. Z.; Tian Y.; Lao Y. Z.; Li Z. G. Curr. Med. Chem. 2014, 21, 2438. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Holly F. W.; Paleveda W. J.; Nutt R. F.; Bergstrand S. J.; Torchiana M.; Glitzer M. S.; Saperstein R.; Hirschmann R. Proc. Natl. Acad. Sci. U.S.A. 1978, 75, 2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veber D. F.; Holly F. W.; Nutt R. F.; Bergstrand S. J.; Brady S. F.; Hirschmann R.; Glitzer M. S.; Saperstein R. Nature 1979, 280, 512. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Freidinger R. M.; Perlow D. S.; Paleveda W. J.; Holly F. W.; Strachan R. G.; Nutt R. F.; Arison B. H.; Homnick C.; Randall W. C.; Glitzer M. S.; Saperstein R.; Hirschmann R. Nature 1981, 292, 55. [DOI] [PubMed] [Google Scholar]
- Hruby V. J.; Alobeidi F.; Kazmierski W. Biochem. J. 1990, 268, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruby V. J.; Balse P. M. Curr. Med. Chem. 2000, 7, 945. [DOI] [PubMed] [Google Scholar]
- Kessler H. Angew. Chem., Int. Ed. 1982, 21, 512. [Google Scholar]
- Schafmeister C. E.; Po J.; Verdine G. L. J. Am. Chem. Soc. 2000, 122, 5891. [Google Scholar]; Kim Y.-W.; Grossmann T. N.; Verdine G. L. Nat. Protoc. 2011, 6, 761. [DOI] [PubMed] [Google Scholar]; Verdine G. L.; Hilinski G. J. Drug Discovery Today Technol. 2012, 9, e41. [Google Scholar]
- Lau Y. H.; de Andrade P.; Wu Y.; Spring D. R. Chem. Soc. Rev. 2015, 44, 91. [DOI] [PubMed] [Google Scholar]
- a Kawamoto S. A.; Coleska A.; Ran X.; Yi H.; Yang C. Y.; Wang S. M. J. Med. Chem. 2012, 55, 1137. [DOI] [PMC free article] [PubMed] [Google Scholar]; b de Araujo A. D.; Hoang H. N.; Kok W. M.; Diness F.; Gupta P.; Hill T. A.; Driver R. W.; Price D. A.; Liras S.; Fairlie D. P. Angew. Chem., Int. Ed. 2014, 53, 6965. [DOI] [PubMed] [Google Scholar]
- a Haney C. M.; Horne W. S. Chem.—Eur. J. 2013, 19, 11342. [DOI] [PubMed] [Google Scholar]; b Haney C. M.; Horne W. S. J. Pept. Sci. 2014, 20, 108. [DOI] [PubMed] [Google Scholar]
- Spokoyny A. M.; Zou Y.; Ling J. J.; Yu H.; Lin Y. S.; Pentelute B. L. J. Am. Chem. Soc. 2013, 135, 5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Collins J.; Tanaka J.; Wilson P.; Kempe K.; Davis T. P.; McIntosh M. P.; Whittaker M. R.; Haddleton D. M. Bioconjugate Chem. 2015, 10.1021/bc5006202. [DOI] [PubMed] [Google Scholar]; See also:; b Smith M. E. B.; Schumacher F. F.; Ryan C. P.; Tedaldi L. M.; Papaioannou D.; Waksman G.; Caddick S.; Baker J. R. J. Am. Chem. Soc. 2010, 132, 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker M. J.; Courter J. R.; Chen J.; Atasoylu O.; Smith A. B.; Hochstrasser R. M. Angew. Chem., Int. Ed. 2010, 49, 3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdo M.; Brown S. P.; Courter J. R.; Tucker M. J.; Hochstrasser R. M.; Smith A. B. III. Org. Lett. 2012, 14, 3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courter J. R.; Abdo M.; Brown S. B.; Tucker M. J.; Hochstrasser R. M.; Smith A. B. J. Org. Chem. 2014, 79, 759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker M. J.; Abdo M.; Courter J. R.; Chen J. X.; Smith A. B.; Hochstrasser R. M. J. Photochem. Photobiol., A 2012, 234, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker M. J.; Abdo M.; Courter J. R.; Chen J. X.; Brown S. P.; Smith A. B.; Hochstrasser R. M. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 17314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asplund M. C.; Zanni M. T.; Hochstrasser R. M. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catsimpoolas N.; Wood J. L. J. Biol. Chem. 1964, 239, 4132. [PubMed] [Google Scholar]; Catsimpoolas N.; Wood J. L. J. Biol. Chem. 1966, 241, 1790. [PubMed] [Google Scholar]
- Degani Y.; Neumann H.; Patchorn A. J. Am. Chem. Soc. 1970, 92, 6969. [DOI] [PubMed] [Google Scholar]
- Dawson P. E.; Muir T. W.; Clark-Lewis I.; Kent S. B. H. Science 1994, 266, 776. [DOI] [PubMed] [Google Scholar]
- O2 is suspected to quench minor amounts radicals formed after photochemical excitation. The productive photochemical pathway, leading to the generation of the dithiocyanates, is expected to occur without long-lived radical intermediates.22 See also:Dellinger B.; King D. S.; Hochstrasser R. M.; Smith A. B. III. J. Am. Chem. Soc. 1977, 99, 3197. [Google Scholar]
- Carboni R. A.; Lindsey R. V. J. Am. Chem. Soc. 1959, 81, 4342. [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. J. Am. Chem. Soc. 2008, 130, 13518. [DOI] [PMC free article] [PubMed] [Google Scholar]; Selvaraj R.; Fox J. M. Curr. Opin. Chem. Biol. 2013, 17, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wu Z.; Liu S.; Hassink M.; Nair I.; Park R.; Li L.; Todorov I.; Fox J. M.; Li Z.; Shively J. E.; Conti P. S.; Kandeel F. J. Nucl. Med. 2013, 54, 244. [DOI] [PubMed] [Google Scholar]
- Images created using data from PDB ID 1XOB and the following:; Jeng M. F.; Campbell A. P.; Begley T.; Holmgren A.; Case D. A.; Wright P. E.; Dyson H. J. Structure 1994, 2, 853. [DOI] [PubMed] [Google Scholar]; The PyMOL Molecular Graphics System, Version 1.5.0.4; Schrödinger, LLC, 2013.
- Ellman G. L. Arch. Biochem. Biophys. 1959, 82, 70. [DOI] [PubMed] [Google Scholar]
- Lang K.; Davis L.; Wallace S.; Mahesh M.; Cox D. J.; Blackman M. L.; Fox J. M.; Chin J. W. J. Am. Chem. Soc. 2012, 134, 10317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.