

Abstract
Adverse experiences in early life have the ability to “get under the skin” and affect future health. This study examined the relative influence of adversities during childhood and adulthood in accounting for individual differences in pro-inflammatory gene expression in late life. Using a pilot-sample from the Health and Retirement Study (N=114) aged from 51-95, OLS regression models were run to determine the association between a composite score from three proinflammatory gene expression levels (PTGS2, ILIB, and IL8) and 1) childhood trauma, 2) childhood SES, 3) childhood health, 4) adult traumas, and 5) low SES in adulthood. Our results showed that only childhood trauma was found to be associated with increased inflammatory transcription in late life. Furthermore, examination of interaction effects showed that childhood trauma exacerbated the influence of low SES in adulthood on elevated levels of inflammatory gene expression—signifying that having low SES in adulthood was most damaging for persons who had experienced traumatic events during their childhood. Overall our study suggests that traumas experienced during childhood may alter the stress response, leading to more sensitive reactivity throughout the lifespan. As a result, individuals who experienced greater adversity in early life may be at higher risk of late life health outcomes, particularly if adulthood adversity related to SES persists.
Keywords: Social Genomics, Inflammation, Stress
Introduction
Adverse life experiences have been linked to poorer health and increased risk of cardiovascular disease, diabetes, cancer, neurodegeneration, arthritis, and frailty (Kiecolt-Glaser et al., 2002; Glass et al, 2010; Finch, 2007). Through eliciting neural and endocrine responses, stress has the ability to “get under the skin”, altering physiological functioning and disrupting homeostasis (McEwen, 2012). As environmental challenges arise, the body initiates a cascade of physiological responses in order to adapt to the changing environment. However, under chronic activation, this response—termed allostatic load—can lead to dysregulation of a number of systems, including the neuroendocrine and immune systems.
Stress-related alterations in pro-inflammatory pathways have been hypothesized to be one of the major biological links through which the environment influences health (Miller et al., 2009b). In both human and animal models, exposure to psycho-social stress has been shown to elicit increases in circulating levels of pro-inflammatory cytokines, and a number of studies have provided evidence to support the links between inflammation and adversity. Neuroendocrine responses to stress can up-regulate inflammation by enhancing the transcription of pro-inflammatory genes such as interleukin-1 beta (IL1B), interleukin-6 (IL6), interleukin-8 (IL8), cyclooxygenase 2 (COX2/PTGS2) and tumor necrosis factor alpha (TNF) (Cole et al., 2013; Irwin & Cole, 2011). Through this mechanism, persistent stress may contribute to a chronic inflammatory environment, leading to downstream health consequences (Miller et al., 2009; Cole 2013).
In addition to the direct connections between chronic psychosocial stress and physiological functioning, there is also evidence that adversity during early life may influence health as individuals age. Recent studies have reported that persons with traumatic events, low SES, or poor health during childhood are at increased risk of morbidity and mortality as adults (Barnes et al., 2012; Montez et al., 2013; Warner & Hayward, 2006; Hayward & Gorman, 2004; Banks et al., 2011; Blackwell et al., 2001). Some plausible non-stress pathways which might also contribute to differential gene expression include increased drinking and smoking as well as higher rates of obesity—by individuals who face significant or chronic adversity (Anda et al., 2010). Additionally, there is also evidence that stress during critical periods of development may have lasting molecular effects on transcription levels throughout late life—implying that stress that has the potential to “get under the skin” may persist and possibly exacerbate responses to future events (Cole et al., 2012; Gluckman et al., 2008; Miller et al., 2009a; Levine, 2005; Zhang et al., 2006). It has been suggested that physiological adaptations in response to adversity during development may contribute to “environmental programming” of the stress response via alterations in DNA methylation, histone acetylation, and transcription factor binding (Meaney & Szyf, 2005). Such effects have been well documented in animal models which demonstrate that elevated hypothalamic-pituitary-adrenal axis (HPA) activation in adulthood can be traced back to alterations in chromatin structure that resulted from a stressful early-life environment (Meaney et al., 2007; Weaver et al., 2004; Fish et al., 2004).
The idea that early life events prompt biological changes with the potential to alter physiological responses to adversity is in line with the diathesis-stress model, which assumes individuals vary in their degree of vulnerability to everyday stressors (Meaney & Szyf, 2005). For instance, individuals who experienced stressful life events in childhood may be more reactive to changes in their environment later on, thus instigating an increased inflammatory response. There is evidence from both animal and human studies documenting the links between stressful conditions in early life and increased inflammatory response. Rhesus macaques reared under adverse conditions have been shown to have higher gene expression related to T-lymphocyte activation, inflammation, and cytokine signaling (Cole et al., 2012). Among humans, ages 25-40, low socioeconomic status (SES) during childhood was found to be associated with increased production of interleukin-6, as well as higher expression of genes bearing response elements for the pro-inflammatory transcription factor NF-kappaB (Miller, 2009a).
Nevertheless, in examining inflammatory gene expression in late life, it is often difficult to disentangle the influences of adversity in early life and adulthood, especially in light of evidence of cumulative disadvantage, indicating that individuals who experience disadvantages in early life are more likely to experience adversity in late life (O'Rand, 1996, 2003). Therefore, it is important to adopt a life course approach to studying the influence of stress on increased transcription of pro-inflammatory genes—by comparing the differential and cumulative effects of stress experienced at different points in the life course.
Using a pilot-sample from the Health and Retirement Study (HRS), we examined the associations between various stressors in childhood and adulthood and the quantitative expression level of three cardinal pro-inflammatory genes: cyclo-oxygenase 2 (COX2, also known as prostaglandin-endoperoxide synthase 2, PTGS2), ILIB, and IL8. We hypothesize that childhood exposure to adversity will be associated with more pronounced increase in inflammatory gene expression than later life exposures, and that early life adversity will also sensitize inflammatory gene expression to the effects of adversity encountered later in life.
Methods
Sample Description
Participants were a randomly-selected subsample of individuals from the HRS which is a longitudinal study of the U.S. population 50 years of age and older, conducted by the University of Michigan, under the sponsorship of the National Institute on Aging. The pilot sample consisted of about 200 respondents to the 2010 interview randomly selected from a group of 1000 respondents living in 13 areas who had completed a face-to-face interview in either 2006 or 2008. Selected respondents were asked to provide a blood sample in the near future. At the time of the interview, 15.9% declined to participate; an additional 14.8% did not complete the blood draw; the final completion rate was 69.3%, or 122 respondents who provided a blood sample. Of these 122, our final analytic sample was N=113. Excluded participants were those with missing data on childhood or adulthood adversity.
RNA Sample collection and measurement
This analysis uses blood samples collected into PAXgene RNA tubes and shipped overnight to a central lab for storage at -80C. RNA was extracted in parallel for all samples using an automated nucleic acid processing system (Qiagen QIAcube), and all samples were tested for suitable mass and purity (by spectrophotemetery on a Nanodrop ND1000 instrument) and for suitable RNA integrity (using an Agilent Bioanalzyer). 121 of the total 122 PAXgene samples yielded suitable quantities of high-quality (intact) RNA. Six mRNA species were assayed by quantitative real-time reverse transcriptase polymerase chain reaction (RT-PCR). In duplicate RT-PCR assays of all 121 samples, results showed excellent reliability, with test-retest correlations averaging .87 (range .73-.96). The present analyses focus on the three assayed transcripts that assess inflammation (IL1B, IL8, PTGS2) and a housekeeping / normalization marker—ACTB. PTGS2 over-expression is often induced by proinflammatory stimuli and is one of the main targets of NSAIDs (Sakoda et al., 2006), while ILIB and IL8 are both cytokines that are released by macrophages during an inflammatory response (Barksby et al., 2007). The two other genes assayed yet not included in the present study are indicators of distinct non-inflammatory biological processes: SOD2 as an indicator of oxidative stress, and IFI27 as an indicator of Type I interferon response.
Transcript abundance within each sample was quantified by standard threshold cycle analysis (Miller et al., 2009a), with values corresponding to approximately a log-2 transformed value of the relative mRNA quantity. Replicate assays were run for each biological sample, yielding two measures per gene for each participant, and values were then averaged across the two runs. Normalized difference scores were calculated by subtracting participants' values for ACTB from their values for IL1B, IL8, and PTGS2. A composite score of pro-inflammatory gene expression was created by averaging across the normalized difference scores for PTGS2, IL1B, and IL8. Composite scores were generated 1) to reduce issues with multiple testing and 2) based on findings from previous studies showing that gene expression differences may be better observed when combining related genes rather than observing them individually, particularly given that individual gene expression is often vulnerable to noise and thus using combined measures of a similar process might yield a more accurate picture (Mootha et al., 2003; Segal et al., 2004; Nam et al., 2006). Finally, since threshold cycle numbers are inversely proportional to the quantity of RNA, composite values were centered and multiplied by -1.
Childhood and Later-Life Adversity
Three variables were used to represent adversity in early life—experience of traumatic events before age 18, low childhood SES, and poor childhood health—while two variables were used to represent adversity in adulthood—low SES, and experience of traumatic events after age 18 (Table 1). Information on childhood traumas and SES, as well as adult traumas were taken from the HRS Psychosocial Leave-Behind Participant Lifestyle Questionnaire. Half of the HRS sample were randomly assigned to receive the questionnaire in 2006 and the other half received it in 2008. Information on traumas used in this analysis was self-reported using 9 items. Two questions were asked regarding particular traumas in childhood—with participants being prompted to answer yes or no for each. These included: having a parent who drank or used drugs so often that it caused problems in the family, and being physically abused by a parent. A simple unweighted sum of the two traumas was calculated and used as a proxy for the level of childhood trauma. Previous reports provide evidence for good reliability for retrospective recall of childhood trauma and abuse (Dill et al., 1991; Turner et al., 1995) and Bernstein et al. (1994) conclude that retrospective reports of trauma tend to remain fairly stable over time and are often in agreement with informant reporting.
Table 1. Self-reported items used to measure adversity.
| Childhood SES (Possible Range 0-5) |
| Before age 16, would you say your family during that time was pretty well off financially, about average, or poor? (poor=1) |
| Before age 16, was there a time when you or your family received help from relatives because of financial difficulties? |
| While you were growing up, before age 16, did financial difficulties ever cause you or your family to move to a different place? |
| Mother had less than 9 years of education |
| Father had less than 9 years of education |
|
|
| Childhood Traumas (Possible Range 0-2) |
| Before you were 18 years old, did either of your parents drink or use drugs so often that it caused problems in the family? |
| Before you were 18 years old, were you ever physically abused by either of your parents? |
|
|
| Childhood Health (Possible Range 0-8) |
| Before you were 16 years old, did you have asthma? |
| Before you were 16 years old, did you have diabetes? |
| Before you were 16 years old, did you have heart problems? |
| Before you were 16 years old, did you have ear problems? |
| Before you were 16 years old, did you have severe headaches/migraines? |
| Before you were 16 years old, did you have epilepsy/seizures? |
| Before you were 16 years old, did you have respiratory problems? |
| Before you were 16 years old, did you have hypertension? |
|
|
| Adulthood Traumas (Possible Range 0-7) |
| Has a child of yours ever died? |
| Have you ever been in a major fire, flood, earthquake, or other natural disaster? |
| Have you ever fired a weapon in combat or been fired upon in combat? |
| Has your spouse, partner, or child ever been addicted to drugs or alcohol? |
| Were you the victim of a serious physical attack or assault in your life? |
| Did you ever have a life-threatening illness or accident? |
| Did your spouse or a child of yours ever have a life-threatening illness or accident? |
|
|
| Adulthood Low SES (0, 1) |
| Participant had less than 9 years of education |
Later-life traumas were measured using seven yes-or-no questions asking whether given events occurred “at any point in [participant's] life”. For each event, participants were also asked when the event occurred, and for our study, only events occurring after age 18 were considered. The events included: having a child who died; being in a major fire, flood, earthquake, or other natural disaster; firing a weapon on being fired upon during combat; having a spouse, partner or child who was addicted to drugs or alcohol; being a victim of a serious physical attack; having a life-threatening illness or accident; and having a spouse or child with a life-threatening illness or accident. Similar to the childhood traumas, a simple unweighted sum was used for the overall measure.
Childhood SES was assessed using a summary score of five dichotomous variables—self-reported poverty, mother had less than 9 years of education, father had less than nine years of education, childhood family received financial help, and childhood family was forced to move for financial reasons. In our sample, 16 participants did not report parental education. In order to retain them, values for these participants were imputed using the nearest neighbor technique. Regression is used to predict a value for each participant, then the entire sample is sorted by predicted value, and cases with missing data are assigned the observed value for the case nearest to them in predicted value. Mother's education was imputed first based on a regression using respondent's year of birth, race/ethnicity and completed education. Father's education was then imputed based on the same variables in addition mother's education. Overall, there is evidence that retrospective assessments of childhood SES tend to be valid measures, and that even in older age, respondents tend to recall conditions with fairly good accuracy (Havari & Mazzonna, 2011; Krieger et al., 1998).
Childhood health was scored as a sum of the number of self-reported conditions, out of eight: asthma, diabetes, heart problems, ear problems, severe headaches/migraines, epilepsy/seizures, respiratory problems, and hypertension. Retrospective measures of childhood health in the HRS survey have previously been found to be reliable and not biased by current health status (Haas, 2007; Smith, 2009). Finally, a single dichotomous variable—having less than 9 years of education—was used to signify low SES in adulthood.
Sociodemographic Covariates
Age, sex, race, smoking, obesity, and drinking were included as covariates in analyses. Age was measured at the 2010 interview for all participants. Sex was based on self-report and coded as 1 for females and 0 for males. A dummy variable for white was coded based on self-reports of race. Three categories were created for smoking—participants reporting that they had not smoked at least 100 cigarettes during their lifetime were classified as never-smokers and used as the reference group; participants reporting that they had smoked at least 100 cigarettes during their lifetime, but no longer smoke were classified as former smokers; and participants reporting that they still smoke were classified as current smokers. A dichotomous variable was created for obesity, with those having a measured body mass index (BMI) of less than 30 coded as 0, and those with a BMI of 30 or greater coded as 1. Finally, a continuous variable was used for drinking based on participants' self-reported average weekly consumption of alcoholic beverages.
Statistical analysis
Ordinary least-squares regression models were used to determine the association between proinflammatory gene expression levels and 1) childhood traumas 2) childhood SES, 3) childhood health, 4) adult traumas, and 5) adult SES, to determine their relative association with expression levels when other stressors and sociodemographic and behavioral variables were controlled. To examine whether increased trauma in childhood moderated the association between later life stress and proinflammatory gene expression levels, we tested for an interaction between early traumas and our two adult conditions—traumatic events and low SES.
Results
Sample Characteristics
As shown in Table 2, the final pilot sample (N=114) was 55.3% female; with a mean age of 73.2 (s.d. 9.5), but with a wide spread (20.2% less than 65 and 29.0% over 80). Most respondents self-identified their race as white (92.1%). Approximately 30% of participants were classified as obese, and on average participants consumed 2.12 alcoholic beverages per week. About half of the participants were former smokers (54.4%) and 10.5% were current smokers.
Table 2. Sample Characteristics (N=114).
| Characteristics | Values |
|---|---|
| Age (years), mean (sd) | 73.2 (9.5) |
| Sex (Female=1), n (%) | 63, 55.3% |
| Race (White=1), n (%) | 105, 92.1% |
| Former Smoker, n (%) | 62, 54.4% |
| Current Smoker, n (%) | 12, 10.5% |
| Obesity, (%) | 34, 29.8% |
| Drinks per Day, mean (sd) | 2.1 (4.5) |
| Childhood Trauma, mean (sd) | 0.2 (0.4) |
| Childhood SES, mean (sd) | 1.5 (1.3) |
| Childhood Health Conditions, mean (sd) | 0.6 (1.0) |
| Adulthood Traumas (count), mean (sd) | 1.2 (1.1) |
| Low Adult SES, n (%) | 8, 7.0% |
| Centered RNA Composite Score, mean (sd) | 0 (0.7) |
The number of adult traumas ranged from 0-5, with a mean of 1.2 (s.d. 1.1), and the number of childhood traumas ranged from 0-2, with a mean of 0.2 (s.d. 0.4). The score for childhood SES ranged from 0-5, with a mean of 1.5 (s.d. 1.3). About 7% of participants had low adult SES, defined as having less than nine years of education. Participants had an average of 0.6 (s.d. 1.0) health conditions in childhood, which ranged from 0-3. The centered mean for our composite score of proinflammatory gene expression was 0, with a range from -5.0 to 5.8 and a standard deviation of 0.7. Finally, Table 3 shows the correlations between our childhood and adulthood adversity variables. Correlation coefficients ranged from 0.04 (for childhood health and adult trauma), to 0.40 (for childhood SES and adult SES).
Table 3. Pearson Correlations Between Childhood and Adulthood Adversity.
| Adulthood Traumas | Low Adult SES | Childhood Trauma | Childhood SES | |
|---|---|---|---|---|
| Adulthood Traumas | ||||
| Low Adult SES | -0.06 | |||
| Childhood Trauma | 0.14 | 0.04 | ||
| Childhood SES | 0.09 | 0.40 | 0.12 | |
| Childhood Health Conditions | 0.04 | -0.09 | 0.16 | 0.04 |
Association between Trauma and Inflammatory Gene Expression
An unadjusted OLS model, which included all five adversity measures simultaneously, as well as a model adjusting for age, sex, race, smoking, obesity, and drinking was used to examine the association between expression levels among inflammatory genes and adversity in both childhood and adulthood (Table 4). Out of the five adversity variables, only childhood trauma was found to be significantly associated with the pro-inflammatory gene expression composite scores in both models. We found that an increase of one early life trauma was associated with a 0.32-0.34 increase in the RNA composite score (Unadjusted: β=0.34, P=.020; Adjusted: β=0.32, P=.045). Furthermore, the R-squared was R2=0.083 for the unadjusted model and R2=0.154 for the fully adjusted model. Childhood health (Unadjusted: β=-0.02, P=.770; Adjusted: β=-0.04, P=.575) and childhood SES were not associated with increased inflammatory gene expression in adulthood Unadjusted: β=0.07, P=.200; Adjusted: β=0.09, P=.146). Additionally, adult trauma (Unadjusted: β=0.04, P=.520; Adjusted: β=0.04, P=.495) and SES (Unadjusted: β=-0.05, P=.835; Adjusted: β=0.10, P=.748) were also not significantly associated with higher RNA composite scores. Subsequently, models containing only one adversity variable at a time were also run, which produced nearly identical results—expression levels were only found to be associated with childhood adversity (results not shown).
Table 4. Coefficients from Ordinary Least-Squares Regression of the Association Between Childhood and Adulthood Adversity and Inflammatory Gene Expression.
| Unadjusted Model | Adjusted Model | |||
|---|---|---|---|---|
|
| ||||
| β Coefficient | P-value | β Coefficient | P-value | |
| Adversity Measure | ||||
| Adulthood Traumas | 0.04 | .520 | 0.04 | .495 |
| Low Adult SES | -0.05 | .835 | 0.10 | .748 |
| Childhood Trauma | 0.34 | .020 | 0.32 | .045 |
| Childhood SES | 0.07 | .200 | 0.09 | .146 |
| Childhood Health Conditions | -0.02 | .770 | -0.04 | .575 |
| Sex (Female=1) | 0.26 | .050 | ||
| Age | 0.01 | .902 | ||
| Race (White=1, Non-White=0) | 0.37 | .201 | ||
| Drinks | -0.01 | .432 | ||
| Obese | 0.04 | .791 | ||
| Smoking | ||||
| Former | 0.21 | .147 | ||
| Current | 0.29 | .253 | ||
|
| ||||
| R-Squared=0.083 | R-Squared=0.154 | |||
Both the unadjusted and adjusted models contain all adversity variables simultaneously. Individual models containing only one adversity variable at a time were also run and produced similar results.
To test our stress-diathesis hypothesis, we examined how late life inflammatory gene expression was associated with the interaction between 1) childhood traumas and adult traumas, and 2) childhood traumas and adult SES (Table 5). In Model 1, which included the interaction between childhood and adult trauma, no significant associations were found for either childhood trauma (β=0.10, P=.66), adult trauma (β=-0.01, P=.87), or the interaction between the two (β=0.18, P=.18). However, for model 2, which included the interaction between childhood trauma and low SES in adulthood, while the main-effects for both variables were no longer significant (Childhood trauma: β=0.21, P=.17; Low SES: β=0.08, P=.79), the interaction between them was positively associated with expression (β=0.81, P=.034). From this model, we estimated predicted means for the RNA composite score based on childhood trauma, adult SES, and their interaction (Figure 1). Results showed that there was no increase in expression levels associated with having low SES in adulthood among participants who reported having no early life traumas. However, for those reporting one early life trauma, proinflammatory gene expression were increased by an average of 1.85-fold for low SES individuals. Finally, for those reporting two early life traumas the increase in gene expression with low SES was 3.24-fold, suggesting that this group may be the most affected by having low SES in adulthood.
Table 5. Association Between Inflammatory Gene Expression and the Interaction Between Childhood Traumas and Adulthood Traumas and SES.
| Main Effects | |||
|---|---|---|---|
|
| |||
| β Coefficient (SE) | P-value | ||
| Model 1 | |||
| Childhood Traumas (Count) | 0.10 | .657 | |
| Adult Traumas (Count) | -0.01 | .872 | |
| Childhood × Adult Traumas | 0.18 | .183 | |
|
| |||
| R-squared | 0.133 | ||
|
| |||
| Model 2 | |||
| Childhood Traumas (Count) | 0.21 | .172 | |
| Low Adult SES | 0.08 | .786 | |
| Childhood × Low Adult SES | 0.81 | .034 | |
|
| |||
| R-squared | .158 | ||
All models run adjusted for age, sex, race, smoking, drinking, and obesity
Figure 1. The Association Between Predicted Means of Proinflammatory Gene Expression and the Interaction of Childhood Traumas and Low SES in Adulthood.
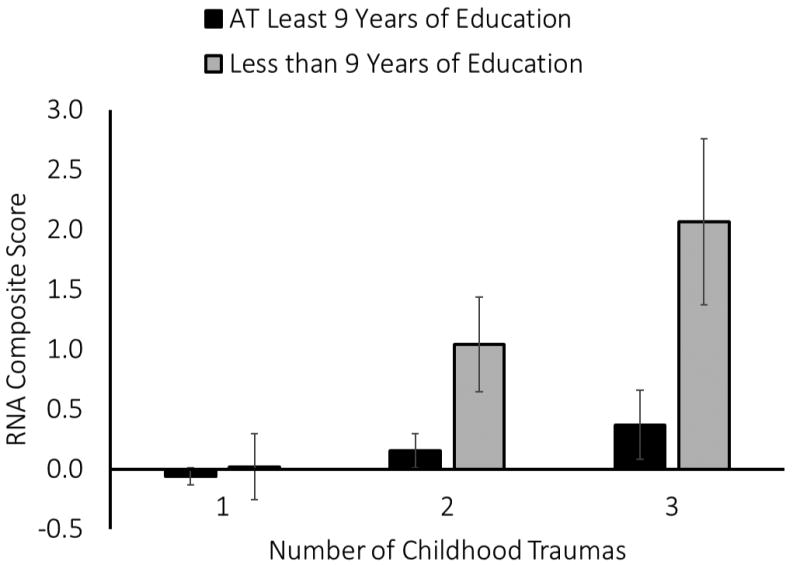
The interaction between childhood traumas and low SES in adulthood was significantly associated with expression (β=0.81, P=.034). Among participants who reported experiencing two early life traumas, low SES in adulthood was positively associated with RNA composite scores of inflammatory gene expression. This association was less strong for participants who only reported experiencing one of the early life traumas. Finally, among those with no reported early life traumas, we found no significant difference in levels of gene expression between individuals with low SES and those with moderate or high SES. Error bars correspond to standard errors of predicted means.
Discussion
Our results suggest that childhood exposure to traumatic events is associated with elevated expression of pro-inflammatory genes in late adulthood, and that this may exacerbate the association between gene expression and low SES in adulthood.. The present results extend earlier research which suggests that negative experiences in early life are associated with increased inflammatory gene expression in adulthood by examining both childhood and adult stresses and their associations with later life gene expression (Miller et al., 2009; Cole et al., 2012; Cole et al., 2013; Irwin & Cole, 2011). This is the first study that has demonstrated evidence that the effects of early life experiences persist even into old age. These chronic inflammatory factors may underlie the link between early life adversity and late life morbidity that has been reported previously in the literature. It is well recognized that continuous low-grade inflammation is associated with increased risks for neurodegeneration, atherosclerosis, cancer, diabetes, chronic-obstructive pulmonary disorder, and arthritis. Chronically elevated inflammatory mechanisms influence diseases of aging via their damaging effects on tissue. Prolonged inflammation has also been shown to alter glucose metabolism, increase insulin resistance, and disrupt homeostasis. As a result, the chronic systemic inflammation associated with early life conditions may accelerate the accumulation of comorbid conditions throughout an individual's life.
The inflammatory response relies on a complex regulatory network, influenced by epigenetic mechanisms, and transcription factors, whose activity in later life may be determined through biological reprogramming in response to early life environment (Bayarsaihan, 2011). For instance, among primates, adversity early in life has been associated with over-expression of genes associated with inflammation, cytokine signaling, and T-lymphocyte activation, which was initiated by increased activation of NF-kB and CREB transcription factors (Cole et al., 2012). Taken together this suggests that individuals who were subject to adverse environments in early life may experience heightened nervous system activity, and increased inflammatory expression, in response to stressors during adulthood, due to differences in persistent gene regulation dynamics established earlier in development.
This is consistent with our results which suggest that traumas experienced in childhood may prime individuals to have more reactive responses to adversity in the future, and as a result, stressors experienced across the lifetime may be more hazardous for this group relative to individuals with advantageous childhood environments. We found that among individuals with early life traumas, having low SES in adulthood was positively associated with levels of inflammatory expression in late life. However, there was no association found between low adult SES and gene expression for individuals who reported having no traumatic events in childhood. This also suggests that having higher SES in adulthood may attenuate the risks of an adverse childhood environment. This may be because the propensity towards a heightened response may only be harmful if events occur that initiate that response. Together, these findings are in line with the stress-diathesis model, which suggests individuals vary in their susceptibility to the harmful effects of stress.
There are a number of explanations for how early life adversity may contribute to differential susceptibility in adulthood. For instance there is evidence that chronic-stress mediated activation has the potential to lead to lasting alteration in the stress response. A study using mice showed that adverse early environments—defined in the study as low maternal investment measured by licking and grooming—caused demethylation at specific target sites, which contributed to increased stress response (Meaney & Szyf, 2005). Furthermore, they showed that these changes were sustained in adulthood. Epigenetic or other alterations in response to stress during “critical periods” of growth and development may modify brain function, and thus sensitize neural responsiveness to traumatic situations later in life. For instance, under stressful conditions, the persistently sensitized brain may elicit a heightened sympathetic nervous system response—the response associated with fight-or-flight—which has been shown to up-regulate sub-populations of immature monocytes, prompting increased leukocyte expression of proinflammatory genes (Powell et al., 2013).
Finally, another potential explanation is that individuals with early traumas may lose the ability to down-regulate inflammatory response, as a result of glucocorticoid resistance. Continued stress exposure is linked to chronic glucocorticoid secretion. Over time, the immune system may lose sensitivity in the receptors that bind to these hormones, and thus may not appropriately respond to secretion from the HPA axis which is meant to “turn-off” inflammatory activation (Miller et al., 2002).
While our study found a main effect for childhood trauma, we did not find significant associations between increased inflammatory expression and any of our other adversity measures. One potential explanation is that our sample size may have limited our ability to detect weaker associations. This study is just a first step in trying to understand how mind-body connections operate over the lifecourse. It is important to acknowledge that the study relied on pilot data and thus it will be critical to replicate these findings using larger and more robust samples. While we did identify a significant association, the pilot study is still subject to the risks and biases associated with relying on a small sample. Nevertheless, with replication, such studies may eventually serve as a proof of concept that environmental influences may be observable on a molecular level even over extended periods of time.
Another potential explanation for the significant finding for traumatic events but not childhood low SES is that while low SES in childhood may be stressful, the childhood traumas we investigated in this study may represent more extreme circumstances—having a physically abusive or substance addicted parent—thus eliciting a stronger persistent inflammatory response. Distinctions between the gravity of these two variables also has the potential to influence differences in the risk of measurement error. Given that both measures rely on retrospective self-reports, measurement error may arise if events are not well remembered or understood. Because of the significance of the traumatic events, this variable may have more reliable recall than other retrospective measures. Additionally, there is a chance that participants may not have been aware of some of the events that were asked about in constructing the childhood SES variable—such as whether their family moved for financial reasons, or received financial help. That being said, we cannot rule out the potential that low SES in childhood influences gene expression in adulthood, and these questions should be continue to be explored.
Our study also did not identify an association between worse childhood health and higher inflammatory gene expression. While it is reasonable to assume that compromised health in childhood may carry an inflammatory load that persists into adulthood, this relationship may be disease and severity dependent. Prior studies have shown a link between childhood asthma and inflammation (Michelson et al., 2009); however, in our sample the majority of the negative health conditions that were reported were ear problems or respiratory problems. Furthermore, of those reporting respiratory problems, none reported concurrent asthma. Thus, one explanation for the lack of association between childhood health and adult inflammatory expression is that the childhood health conditions reported in our sample may have been in reference to acute events—such as ear infections, bronchitis, or pneumonia—which may not exhibit the lasting inflammatory effects seen in other conditions, like childhood asthma.
Likewise, there are additional limitations to this study which we wish to acknowledge. First, as previously noted, our sample size may have limited our statistical power to detect some associations and did not allow for significant multivariate analysis. Nevertheless, studies examining similar relationships at earlier ages have had as few as 31 cases (Chen et al., 2009), suggesting that for this type of analysis, our sample size was acceptable. Second, the use of only three markers of gene expression may not fully capture what is occurring in the association between stress and inflammation. In moving forward, genome-wide RNA profiling would expand our understanding of how environment influences inflammation and health through its interactions with gene regulatory mechanisms. Finally, although we incorporated phenotypic data that accounted for adversity at various points in the life course, with the inclusion of larger sample sizes, analyses would likely benefit from the incorporation of information on event timing, and objective and subjective assessment of severity.
Overall, our study showed that childhood traumas were significantly associated with elevated expression of proinflammatory genes during late life. Furthermore, we showed that experiencing these traumas during childhood may alter the association between later-life inflammatory gene expression and low SES, suggesting that early life adversity may increase later life reactivity to stress.
References
- Anda RF, Butchart A, Felitti VJ, Brown DW. Building a framework for global surveillance of the public health implications of adverse childhood experiences. AM J Prev Med. 2010 Jul;39(1):93–8. doi: 10.1016/j.amepre.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Banks J, Oldfield Z, Smith JP. Childhood Health and Differences in Late-Life Health Outcomes between England and the United States. 2011 (Working Paper). Retrieved from RAND Labor and Population working paper series. http://www.rand.org/content/dam/rand/pubs/working_papers/2011/RAND_WR860.pdf.
- Barksby HE, Lea SR, Preshaw PM, Taylor JJ. The expanding family of interleukin-1 cytokines and their role in destructive inflammatory disorders. Clin Exp Immunol. 2007;149(2):217–25. doi: 10.1111/j.1365-2249.2007.03441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes LL, Wilson RS, Everson-Rose SA, et al. Effects of early-life adversity on cognitive decline in older African Americans and Whites. Neurology. 2012;79(24):2321–7. doi: 10.1212/WNL.0b013e318278b607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein DP, Fink L, Handelsman L, et al. Initial reliability and validity of a new retrospective measure of child abuse and neglect. American Journal of Psychiatry. 1994;151:1132–6. doi: 10.1176/ajp.151.8.1132. [DOI] [PubMed] [Google Scholar]
- Blackwell DL, Hayward MD, Crimmins EM. Does Childhood health affect chronic morbidity in later life? Soc Sci Med. 2001;52(8):1269–84. doi: 10.1016/s0277-9536(00)00230-6. [DOI] [PubMed] [Google Scholar]
- Chen E, Miller GE, Walker HA, Arevalo JM, Sung CY, Cole SW. Genome-wide transcriptional profiling linked to social class in asthma. Thorax. 2009;64:38–43. doi: 10.1136/thx.2007.095091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SW, et al. Social regulation of gene expression in human leukocytes. Genome Biol. 2007;8(9):R189. doi: 10.1186/gb-2007-8-9-r189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SW. Social regulation of human gene expression. Curr Dir Psychol Sci. 2009;18(3):132–137. doi: 10.1111/j.1467-8721.2009.01623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SW, Conti G, Arevalo JM, et al. Transcriptional modulation of the developing immune system by early life social adversity. PNAS. 2012;109(50):20578–83. doi: 10.1073/pnas.1218253109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SW. Social regulation of human gene expression: Mechanisms and implications for public health. American Journal of Public Health. 2013;103:S84–92. doi: 10.2105/AJPH.2012.301183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill DL, Chu JA, Grob MC, et al. The reliability of abuse history reports: a comparison of two inquiry formats. Compr Psychiatry. 1991;32(2):166–9. doi: 10.1016/0010-440x(91)90009-2. [DOI] [PubMed] [Google Scholar]
- Finch C. The Biology of Human Longevity: Inflammation, Nutrition, and Aging in the Evolution of Human Lifespans. Elsevier, Inc; 2007. [Google Scholar]
- Fish EW, Shahrokh D, Bagot R, et al. Epigenetic programming of stress responses through variations in maternal care. Ann N Y Acad Sci. 2004;1036:167–80. doi: 10.1196/annals.1330.011. [DOI] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, et al. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–34. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359(1):61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas SA. The long-term effects of poor childhood health: an assessment and application of retrospective reports. Demography. 2007;44(1):113–35. doi: 10.1353/dem.2007.0003. [DOI] [PubMed] [Google Scholar]
- Havari E, Mazzonna F. Can We trust older people's statements on their childhood circumstances? Evidence from SHARELIFE. SHARE Working Paper. 2011 [Google Scholar]
- Hayward MD, Gorman BK. The long are of childhood: the influence of early-life social conditions on men's mortality. Demography. 2004;41(1):87–107. doi: 10.1353/dem.2004.0005. [DOI] [PubMed] [Google Scholar]
- Irwin MR, Cole SW. Reciprocal regulation of the neural and innate immune systems. Nature Reviews Immunology. 2011;11:625–632. doi: 10.1038/nri3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliman P, Alvarez-Lopez MJ, Cosin-Tomas, et al. Rapid changes in histone deacetylases and inflammatory gene expression in expert meditators. Psychoneuroendocrinology. 2014;40:96–107. doi: 10.1016/j.psyneuen.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiecolt-Glaser JK, McGuire L, Robles TR, Glaser R. Emotions, morbidity, and mortality: New perspectives from psychoneuroimmunology. Annu Rev Psychol. 2002;53:83–107. doi: 10.1146/annurev.psych.53.100901.135217. [DOI] [PubMed] [Google Scholar]
- Krieger N, Okamoto A, Selby JV. Adult female twins' recall of childhood social class and father's education: a validation study for public health research. Am J Epidemiol. 1998;147(7):704–8. doi: 10.1093/oxfordjournals.aje.a009512. [DOI] [PubMed] [Google Scholar]
- Levine S. Developmental determinants of sensitivity and resistance to stress. Psychoneuroendocrinology. 2005;30(10):939–946. doi: 10.1016/j.psyneuen.2005.03.013. [DOI] [PubMed] [Google Scholar]
- McEwan BS. Brain on stress: how the social environment gets under the skin. PNAS. 2012;109(2):17180–5. doi: 10.1073/pnas.1121254109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan PO, Szyf M. The epigenetics of social adversity in early life: implications for mental health outcomes. Neurobiol Dis. 2010;39(1):66–72. doi: 10.1016/j.nbd.2009.12.026. [DOI] [PubMed] [Google Scholar]
- Meaney MJ, Szyf M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci. 2005;7(2):103–23. doi: 10.31887/DCNS.2005.7.2/mmeaney. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney MJ, Szyf M, Seckl JR. Epigenetic mechanism of perinatal programming of hypothalamic-pituitary-adrenal function and health. Trends Mol Med. 2007;13(7):269–77. doi: 10.1016/j.molmed.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Michelson PH, Williams LW, Benjamin DK, Barnato AE. Obesity, inflammation, and asthma severity in childhood: data from the National Health and Nutrition Examination Survey 2001-2004. Ann Allergy Ashma Immunol. 2009;103(5):381–5. doi: 10.1016/S1081-1206(10)60356-0. [DOI] [PubMed] [Google Scholar]
- Miller GE, et al. A functional genomic fingerprint of chronic stress in humans: Blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry. 2008;64(4):266–272. doi: 10.1016/j.biopsych.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E, Fok AK, et al. Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. PNAS. 2009a;106(34):14716–21. doi: 10.1073/pnas.0902971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G, Chen E, Cole SW. Health psychology: developing biologically plausible models linking the social world and physical health. Annu Rev Psychol. 2009b;60:501–524. doi: 10.1146/annurev.psych.60.110707.163551. [DOI] [PubMed] [Google Scholar]
- Miller GE, et al. Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proc Natl Acad Sci USA. 2009;106(34):14716–14721. doi: 10.1073/pnas.0902971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montez JK, Hayward MD. Cumulative Childhood Adversity, Educational Attainment, and Active Life Expectancy. Demography. 2013 doi: 10.1007/s13524-013-0261-x. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003 Jul;34:267. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Nam D, Kim SB, Kim SK, et al. ADGO: analysis of differentially expressed gene sets using composite GO annotation. Bioinformatics. 2006;22(18):2249–53. doi: 10.1093/bioinformatics/btl378. [DOI] [PubMed] [Google Scholar]
- O'Rand AM. The precious and the precocious: Understanding cumulative disadvantage and cumulative advantage over the life course. Gerontologist. 1996;36:230–238. doi: 10.1093/geront/36.2.230. [DOI] [PubMed] [Google Scholar]
- O'Rand AM. Cumulative advantage theory in life course research. In: Crystal S, Shea DF, editors. Annual review of gerontology and geriatrics: Focus on economic outcomes in later life. New York: Springer; 2003. pp. 14–30. [Google Scholar]
- Powell ND, Sloan EK, Bailey MT, et al. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via β-adrenergic induction of myelopoiesis. PNAS. 2013;110(41):16574–9. doi: 10.1073/pnas.1310655110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakoda LC, Gao YT, Chen BE, et al. Prostaglandin-endoperoxide synthase 2 (PTGS2) gene polymorphisms and risk of biliary tract cancer and gallstones: a population-based study in Shanghai, China. Carcinogenesis. 2006;27(6):1251–6. doi: 10.1093/carcin/bgi314. [DOI] [PubMed] [Google Scholar]
- Segal E, et al. A module map showing conditional activity of expression modules in cancer. Nat Genet. 2004;36:1090–1098. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci. 2012;1261:55–63. doi: 10.1111/j.1749-6632.2012.06633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JP. Reconstructing Childhood Health Histories Demography. 2009;46(2):387–403. doi: 10.1353/dem.0.0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JR, Lloyd DA. Lifetime traumas and mental health: The significance of cumulative adversity. Journal of Health and Social Behavior. 1995;36(4):360–376. [PubMed] [Google Scholar]
- Tyrka AR, Price LH, Marsit C, et al. Childhood Adversity and Epigenetic Modulation of the Leukocyte Glucocorticoid Receptor: Preliminary Findings in Healthy Adults. PLoS One. 2012;7(1):e30148. doi: 10.1371/journal.pone.0030148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner DF, Hayward MD. Early-life origins of the race gap in men's mortality. J Health Soc Behav. 2006;47(3):209–26. doi: 10.1177/002214650604700302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7(8):847–54. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Zhang TY, et al. Maternal programming of defensive responses through sustained effects on gene expression. Biol Psychol. 2006;73(1):72–89. doi: 10.1016/j.biopsycho.2006.01.009. [DOI] [PubMed] [Google Scholar]