

Abstract
Cardiac muscle cells have an intrinsic ability to sense and respond to mechanical load through a process known as mechanotransduction. In the heart, this process involves the conversion of mechanical stimuli into biochemical events that induce changes in myocardial structure and function. Mechanotransduction and its downstream effects function initially as adaptive responses that serve as compensatory mechanisms during adaptation to the initial load. However, under prolonged and abnormal loading conditions, the remodeling processes can become maladaptive, leading to altered physiological function and the development of pathological cardiac hypertrophy and heart failure. Although the mechanisms underlying mechanotransduction are far from being fully elucidated, human and mouse genetic studies have highlighted various cytoskeletal and sarcolemmal structures in cardiac myocytes as the likely candidates for load transducers, based on their link to signaling molecules and architectural components important in disease pathogenesis. In this review, we summarize recent developments that have uncovered specific protein complexes linked to mechanotransduction and mechanotransmission within (1) the sarcomere, (2) the intercalated disc, and (3) at the sarcolemma. The protein structures acting as mechanotransducers are the first step in the process that drives physiological and pathological cardiac hypertrophy and remodeling, as well as the transition to heart failure, and may provide better insights into mechanisms driving mechanotransduction-based diseases.
Keywords: heart, cardiac muscle, mechanotransduction, sarcomere, cytoskeleton, cardiac hypertrophy/remodeling, heart failure, genetically altered mice, sarcolemma, cell-cell junction
INTRODUCTION
In cardiac muscle, a number of proteins have been proposed as key mechanosensors and mechanotransducers that directly sense and respond to mechanical loads (stress and/or strain) triggering structural, signaling and functional alterations associated with various cellular processes. These processes can include regulation of electrophysiology via stretch-sensitive channels, myocardial material properties and fibrosis, contractile function and calcium regulation, and the important downstream effect of mechanical signaling: cardiac muscle growth via hypertrophy and atrophy. In addition to transducing a mechanical signal, proteins and complexes can act as structural transmission conduits for mechanical stress and strain (mechanotransmission). For example, transferring stress generated at the actin-myosin complex, through the sarcomere, Z-disc and cytoskeleton, to the sarcolemma. These structural proteins can thus be critical components of a mechanotransduction pathway. This review will summarize evidence for mechanotransduction at three key sites within the cardiac myocyte/cytoskeleton: the sarcomere, the intercalated disc and the sarcolemma.
An abundance of evidence points to the sarcomere as a key site for mechanotransduction. The sarcomere is the basic contractile unit of cardiac muscle, and is made up of a complex assembly of myofilament proteins (1). The interaction between myosin heads (thick filament) and actin thin filament proteins is the key process that drives force-generation in cardiac muscle (2). In addition to their role in force generation, the direct connection of the cytoskeleton to the sarcomeric Z-disc and myosin's stabilization by titin and other structural proteins, suggest that forces can be transmitted in and out of the sarcomere, reinforcing the idea that sarcomeric proteins play a role in mechanotransduction (3, 4). Growing evidence points to specific complexes within the sarcomeric Z-disc as well as within the I band (linked to titin) in sensing and responding to mechanical loads driving downstream events including those that regulate gene expression, protein synthesis and degradation within cardiac muscle cells (1). Mechanical forces generated by the sarcomere are transmitted both longitudinally and laterally to the sarcolemma of the cardiac muscle cell (5), as there is evidence for directional-dependence of myocyte stress sensing (6, 7), hence there may be directional dependence of mechanotransduction within a myocyte. Such directional dependence in load sensing may be related to different modes of hypertrophic growth (8).
In addition to the complex protein structures within the sarcomere and intracellular cytoskeleton, the intercalated disc and sarcolemma at the outer boundaries of the cardiac myocyte are possible key sites for transduction of mechanical forces. Mechanical force or stress can be transmitted both outside-in and inside-out though the membrane components of a cell. Since the internal cytoskeleton is coupled via transmembrane proteins to the extracellular matrix (ECM) (9) and hence the rest of the myocardium, membrane-associated protein complexes have been widely investigated as sites regulating mechanotransduction. In terms of lateral connections, the sarcomere connects to the sarcolemma by cytoskeletal components that link the Z-disc with the membrane-spanning integrin and dystroglycan complexes, which then bind to components of the ECM (9). The intermediate filament desmin also connects the Z-disc to the nucleus (10). On the other hand, longitudinal connections also take place between cytoskeletal actin and intermediate filaments with fascia adherens and desmosomal junctions, respectively, both of which are components of the specialized cardiac muscle cell-cell junction, the intercalated disc (11).
Although the mechanisms underlying mechanotransduction in cardiac muscle are not known, putative proteins and regional complexes have been suggested to be involved in this highly sensitive and critical regulatory pathway within the cardiac myocyte. These structural connections provide a conduit in which to further explore and uncover protein complexes and signals that impact the cellular response to mechanical cues involved in cardiac hypertrophy, as well as calcium/contractile function (12), regulation of fibroblasts and material properties (13), and electrophysiology (14). Dysregulation in these functional processes can lead to an altered cellular response to external forces and possibly the transition to heart disease.
(1) SARCOMERE-MEDIATED MECHANOTRANSDUCTION
The sarcomere is the molecular motor of the cardiac myocyte, and force generated by actin-myosin interactions are transmitted in a complex fashion through the sarcomere itself, and then to the rest of the cytoskeleton, sarcolemma and eventually through the ECM to the surrounding myocardium. As forces from outside of the cell are transmitted back through the structural proteins to the sarcomere, several sarcomere-level molecules could be involved with load sensing and transduction of both internal and externally generated forces (Figure 1). Titin and muscle LIM protein (MLP, also known as cysteine rich protein 3 (CSRP3)) are likely two critical players in force transmission and sensing within the sarcomere. Both proteins have a vast network of interacting proteins, and form complex signalosomes that have been implicated in mechanotransduction pathways and hypertrophy-related signaling (15, 16).
Figure 1.
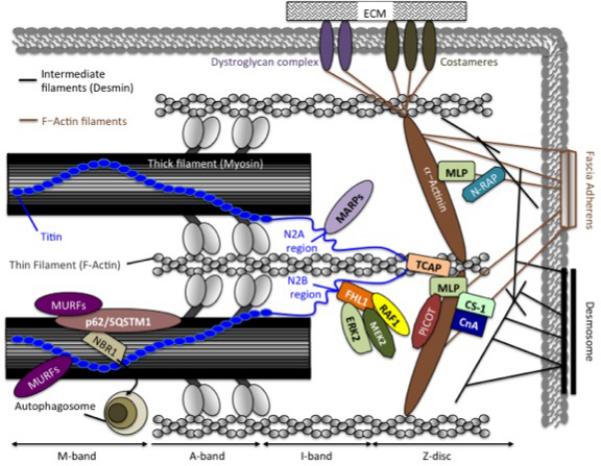
A schematic representation of the specific protein complexes linked to sarcomere-mediated mechanotransduction and mechanotransmission in cardiac muscle. NBR1: Neighbor to BRCA1, p62/SQSTM1: p62/sequestosome-1, MURFs: muscle-specific RING-finger proteins, FHL1: Four-and-a-half LIM domain protein 1, ERK2: Extracellular signal-regulated kinase-2, MEK2: Mitogen-activated protein kinase kinase-2, RAF1: Rapid accelerated fibrosarcoma-1, MARPs: Muscle-specific ankyrin repeat proteins, CS-1: Calsarcin-1, CnA: Calcineurin A, TCAP: Titin Cap, MLP: Muscle-specific LIM domain protein, PICOT: Protein kinase C-interacting cousin of thioredoxin, N-RAP: Nebulin-related anchoring protein. (Illustration Credit: Ben Smith)
Titin-Directed Mechanotransduction
Titin is a giant sarcomeric protein that spans from the Z-disc to the M-line (17). Multiple roles for titin have been established, which include functioning as a molecular ruler for sarcomere assembly, generating passive stiffness in the sarcomere during stretch as well as serving as signal transducer in response to mechanical overload (18-21). Unique regions within titin have been shown to serve as anchoring sites for a variety of cytoskeletal proteins that orchestrate and possibly transmit biomechanical stress responses during cardiac hypertrophy and failure. These proteins include MLP, titin-Cap (TCAP), calsarcin-1 (CS-1), four-and-half-LIM domain protein-1 (FHL1) and muscle-specific ankyrin repeat protein (MARP) family members that have all been implicated to play important roles in titin directed mechanotransduction (22).
MLP/TCAP/Titin Complex
It has been proposed that the Z-disc MLP/TCAP/titin complex functions as a stretch sensor in cardiac muscle cells, and that defects in the complex can lead to cardiomyopathy and heart failure development (16). MLP is composed of two LIM domains and is localized to the Z-disc through direct binding to α-actinin (23, 24). Cardiac muscles cells deficient in MLP respond to treatment with the hypertrophic agent endothelin-1 (ET-1) with a characteristic upregulation of the fetal gene markers, including atrial natriuretic factor (ANF) and brain natriuretic peptide (BNP) (16). The absence of this fetal gene response in MLP-deficient cells exposed to mechanical stress (16) suggests that MLP may play a specific role in mechanosensing and signaling.
MLP knockout mice display a severe dilated cardiomyopathy phenotype resulting in heart failure and premature death (25). A common dilated cardiomyopathy-associated polymorphism found in human MLP (W4R-MLP), resides within the TCAP binding region of MLP and has been shown to abolish binding of TCAP to MLP (16). It has been proposed that loss of this MLP/TCAP interaction induces abnormal intrinsic elastic properties of titin resulting in the inability of cardiac muscle cells to properly sense mechanical stress (16). In support of this concept, a MLPW4R/W4R knock-in mouse model generated by Knöll and colleagues displayed hypertrophic cardiomyopathy and developed a heart failure phenotype (26). The observation of ventricular hypertrophy in these mice in the absence of pressure overload suggests that the W4R-MLP polymorphism impairs mechanotransduction signaling in response to basal levels of load. Mutant hearts displayed reduced MLP mRNA and protein levels, as well as increased nuclear localization of W4R-MLP. Furthermore, in vitro studies demonstrated reduced binding of TCAP to W4R-MLP, which could suggest a plausible mechanism for the translocation of W4R-MLP from the Z-disc into the nucleus. However, the role of MLP within the nucleus of cardiac muscle cells has yet to be clearly defined. MLP does not have the ability to bind DNA directly, but following the discovery of cysteine rich protein 2 (CSRP2), a protein closely related to MLP (MLP is also known as CSRP3), which could enhance gene expression in smooth muscle cells through binding to GATA and serum response factor (SRF) transcription factors (27), there reigns the possibility that MLP may have the ability to bind and modulate important cardiac transcription factors such as GATA-4 and SRF in cardiac muscle cells in response to mechanical stimulation. The discovery that the potassium channel β-subunit minK interacts with TCAP at the sarcomere, suggests that TCAP may also serve to link myofibrillar components to the sarcolemma. Although not fully explored, it has been proposed that this interaction could potentially connect titin deformation with potassium influx in cardiac muscle (28). Equally as important, with the emergence of evidence suggesting that MLP has a predominantly cytosolic localization (29), and studies demonstrating that the titin-TCAP complex is a rigid superstable complex at the Z-disc that is optimized to resist applied loads (30), is the need for further research to asses whether MLP-TCAP is in fact a direct mechanosensor complex that has the required capacity to undergo conformational changes under physiological forces.
MARP/Titin Complex
MARP family members, which include three members; cardiac ankyrin repeat protein (CARP), ankyrin repeat domain protein 2 (ANKRD2) and diabetes related ankyrin repeat protein (DARP) have all been shown to interact at the N2A region of titin and proposed to serve as a stress response signalosome (31). Miller and colleagues demonstrated that rat cardiac muscle cells respond to passive stretch by initiating both an increase in and redistribution of MARP proteins, which included translocation of CARP and DARP into the nucleus (31). Given the fact that CARP has been shown to act as a negative regulator of cardiac gene expression (32), the authors proposed that this redistribution of MARPs into the nucleus in response to myofibrillar stress/strain is an example of stretch-based sensing and signaling (31). In support of the concept of increased MARPs in response to stress, it was found that CARP and ANKRD2 gene expression is increased in response to a single bout of eccentric contractions in the mouse (33). CARP has been shown to be upregulated in heart failure (34, 35), hypertrophy (36), and mutations in the ANKRD1 gene encoding CARP have been shown to be causative for human dilated and hypertrophic cardiomyopathy (37-39). The latter of these studies showed in rat embryonic myocardial cells in vitro that human ANKRD1 mutations can lead to differential stretch-induced gene expression when compared with controls (39), providing further evidence for a role for CARP in stretch sensing. However, recently generated MARP triple knockout mice, have cast doubt on the importance of MARP proteins for cardiac function (40). Under basal conditions and in response to biomechanical stress induced by mechanical pressure overload for 14 days, the mice were found to be viable with normal cardiac function (40), suggesting that compensatory mechanisms may counteract the global loss of MARPs. Therefore, it would be interesting to examine the effects of cardiac specific loss of MARPs at postnatal stages in response to biomechanical stress. Other potential reasons for these contrasting results could arise from both species differences (rat vs. mouse) and/or the mode of stimulation used (cyclic stretch vs. TAC). Hence it would be interesting to examine whether MARP triple KO mice display a cardiac phenotype after extended periods of TAC (greater than 14 days), or in response to other cardiac stress and injuries (e.g., myocardial infarction, etc.). Recent studies have also highlighted a role for MARP proteins in signaling pathways relating to PKC and PKA, thus looking at mouse models deficient in these signaling proteins under mechanical stress may be a useful research avenue to explore (41).
FHL1/Titin Complex
The titin N2B region resides within the extensible I band region of titin, which contributes to the myofibrillar passive/diastolic tension generated upon stretch (42). FHL1 has been shown to bind to titin at this region (43), and has been shown to be upregulated in mouse models in response to both pressure overload induced hypertrophy and treatment with hypertrophic agents (44, 45). This upregulation has also been shown to occur in the hearts of human patients exhibiting hypertrophic cardiomyopathy (46-48), together suggesting that FHL1 may play an important role in biomechanical stress responses in cardiac hypertrophy and heart failure. In support of, and extending upon this idea, we showed FHL1 to be part of a biochemical stress sensing signalosome that scaffolds components of the mitogen activated protein kinase signaling pathway (RAF1 (rapid accelerated fibrosarcoma-1)/ MEK2 (mitogen-activated protein kinase kinase-2)/ ERK2 (extracellular regulated kinase-2)), specifically at the N2B stretch sensor domain of titin (43). These studies showed that mouse hearts devoid of FHL1 did not develop a basal cardiac phenotype but demonstrated a blunted hypertrophic response and preserved LV function in response to pressure overload, which was associated with loss of ERK2 phosphorylation (43). Interestingly, FHL1 deficient cardiac muscles also exhibited an increase in cardiac muscle compliance, consistent with loss of function of the titin N2B stretch sensor domain (43). Given the importance of Gαq (Gq) pathways in pressure overload-induced hypertrophy (49), we further sought to cross FHL1 deficient mice with the transgenic mouse model harboring constitutively active Gq overexpression (43). The results suggest a direct link between FHL1-mediated mechanotransduction pathways and Gq signaling pathways as FHL1 deficiency was shown to prevent the cardiomyopathy and pathological ERK2 phosphorylation, caused by constitutively active Gq overexpression (43). Recent studies showed that FHL1 deficient mice crossed with a hypertrophic cardiomyopathy mouse model (MHC403/+) resulted in increased cardiac hypertrophy (50), suggesting that there may be distinct stress-induced hypertrophic signaling cascades mediated by FHL1. Further mechanistic studies by revealed that FHL1 is a negative regulator of titin N2B phosphorylation (51). We recently showed that titin N2B is a novel substrate of ERK2 and FHL1, and directly interferes with ERK2-mediated titin-N2B phosphorylation (51). Together our results suggest the working hypothesis that FHL1 restricts titin based cardiac muscle compliance by masking/interfering with kinases (ERK2) that bind to titin N2B in order to limit titin N2B phosphorylation and compliance to “physiological ranges” with stretch (51). It may also act as a scaffold for MAPK-mediated hypertrophic signaling (51). However, loss of FHL1 may render titin N2B sites “open” to kinases that bind titin N2B resulting in increased titin N2B phosphoryation and compliance beyond “physiological ranges”, which then destabilize the MAPK scaffold to inactivate hypertrophic signaling (51).
Titin kinase
The M-line-associated region of titin contains a protein kinase domain (52) that has been proposed to play a role in mechanosensation (53). Using single molecule analytical techniques, Puchner and colleagues demonstrated that physiological levels of mechanical stress can initiate the catalytic activity of titin kinase by releasing its active site to allow binding of its co-factor, adenosine triphosphate (54). Mechanically induced titin kinase interacts with a protein complex composed of the autophagosomal receptors, NBR1 (neighbor of BRCA1) and p62, also called sequestosome-1 (SQSTM1), and the E3-ubiquitin ligase, muscle-specific RING finger-2 (MURF2) (55). In vitro studies in cardiac muscle cells demonstrated dissociation of this signalosome in response to mechanical inactivity induced by hyperkalemic depolarization, leading to translocation of MURF2 to the nucleus (55). Within the nucleus, MURF2 induces both downregulation and nuclear export of SRF, suggesting that MURF2 acts as a repressor of SRF-dependent gene expression in response to a loss of mechanical loading (55). Conversely, a more recent study has emerged that suggests the kinase domain of titin is in fact a pseudokinase with undetectable levels of catalysis (56). The authors propose that the titin pseudokinase functions as a scaffold that supports the recruitment of proteins to the sarcomeric M-line. This finding, coupled with the fact that MURF2 knock out mice do not display a detectable basal cardiac phenotype (57), highlights the need for additional studies to establish whether the mechanoactivation of titin kinase and subsequent phosphorylation of downstream targets coupled with the NBR1/SQSTM1/MURF pathway regulates cardiac mechanosensing, or whether any potential signaling function of titin via NBR1/SQSTM1/MURF is solely due to a scaffolding role performed by the kinase domain.
An increasing body of literature suggests that SQSTM1 and NBR1 also serve to provide a link between protein ubiquitination and the process of selective autophagy, which is responsible for the regulated removal of proteins and organelles (58). SQSTM1 and NBR1 mediate direct interactions between polyubiquitinated proteins and microtubule-associated protein light chain 3 (LC3)-II, which is required for autophagosome recruitment (59). Recent evidence suggests an emerging role for autophagy in the clearance of signaling proteins (59, 60), which raises the possibility that changes in SQSTM1/NBR1 localization in response to mechanical stimuli could directly influence autophagic protein turnover and cardiac muscle cell signal transduction pathways. Support for the importance of titin kinase in the regulation of protein turnover via SQSTM1/NBR1 was provided in a human patient with a point mutation in the αR1 helix, R279W (Arg34091Trp) of titin kinase, which abrogates binding to NBR1 (55). This mutation was originally associated with heredity myopathy with early respiratory failure (HMERF), that is characterized by focal myofibrillar breakdown (61), as well as aberrant localization and aggregation of SQSTM1 (55). More recently, titin domain A150/Fn3 119 has emerged as a hotspot for mutations causing HMERF (62). Indeed, patients harboring the originally identified R279W mutation in the kinase domain were also found to contain a missense mutation (Pro30091Leu) in A150/Fn3 119 in cis (63). The controversy whether either one or both mutations are causative of the hereditary myopathy (63-65), and the underlying molecular mechanisms contributing to disease pathology, remain to be further analyzed. However, a recently identified titin kinase-W260R (p.Trp34072Arg) mutation that causes early-onset cardiomyopathy may support the importance of titin kinase for proper muscle function. Indeed, this mutation was also found to abrogate NBR1 binding, thereby further implicating defects in titin kinase to aberrant protein turnover and the development of myopathies (66).
MLP-Calcineurin-NFAT
MLP has also been shown to be essential for anchoring the Ca2+ activated phosphatase, calcineurin (CnA) at the Z-disc (24). In response to activation, CnA dephosphorylates the NFAT (nuclear factor of activated T-cells) family of transcription factors, leading to their translocation into the nucleus and the activation of a pro-hypertrophic gene program (67, 68). Studies using MLP heterozygous deficient mice have shown that mislocalization of CnA in response to reduced MLP levels is associated with reduced NFAT signaling, pronounced LV dilatation and cardiac dysfunction after myocardial infarction (24). These findings suggest that MLP has the ability to transduce biochemical stress to the nucleus via CnA-NFAT activation, and that reduced MLP-CnA signaling can lead to adverse remodeling in response to myocardial infarction.
MLP-PICOT
Protein kinase C-interacting cousin of thioredoxin (PICOT) is a protein found at the Z-disc that can inhibit CnA-NFAT hypertrophic signaling in response to hypertrophy induction by phenylephrine (69). PICOT localizes to the Z-disc through an interaction with MLP, which in turn abrogates the binding of CnA to MLP, leading to its displacement from the Z-disc. The displacement of CnA from the Z-disc has been shown to prevent CnA-NFAT activation and thus inhibit cardiac hypertrophy (69). This mechanism has also been postulated to protect against pressure overload-induced cardiac hypertrophy, with NFAT gene upregulation shown to be significantly diminished in the hearts of PICOT-overexpressing transgenic mice exposed to pressure overload (69). However, it has yet to be established whether PICOT binding to MLP at the Z-disc prevents any nuclear translocation events in response to mechanical stimulation.
Calsarcin-1
Calsarcin-1 (CS-1) is an additional Z-disc localized protein that interacts with and negatively regulates calcineurin activity. CS-1 knockout mice display enhanced calcineurin activation, and develop both increased cardiac hypertrophy and an exacerbated cardiomyopathy in response to pressure overload (70). Mutations in CS-1 have been linked in humans to a form of hypertrophic cardiomyopathy characterized by early onset and cardiac arrhythmias (71).
(2) INTERCALATED DISC-MEDIATED MECHANOTRANSDUCTION
Force developed within the sarcomere is transmitted longitudinally to interdigitated distal ends of the cardiac muscle at specialized junctions known as the intercalated discs, which play a key role in maintaining mechanical and electrical coupling between cardiomyocytes (11). A study performed in rabbit hearts undergoing cyclical volume overload and unload revealed that the intercalated disc undergoes dynamic ultrastructural changes associated with sarcomere assembly/disassembly in response to volume overload, supporting a role for the intercalated disc as a site of mechanotransduction, which is associated with cardiac muscle cell growth and hypertrophic responses (72). The two most prominent structures within the intercalated disc that are thought to sense and process mechanical stress are the fascia adherens and desmosomal junctions, based on their integral links and mechanotransmission to cytoskeletal actin and intermediate filaments, respectively (22) (Figure 2).
Figure 2.

A schematic representation of the specific protein complexes linked to cell-cell junction and sarcolemma-mediated mechanotransduction and mechanotransmission in cardiac muscle. Dotted arrows highlight cross-talk between integrin and caveolin as well as integrin and the dystroglycan complex. DSC2: Desmocollin-2, DSG2: Desmoglein-2, JUP: Plakoglobin, PKP2: Plakophillin-2, DSP: Desmoplakin, VIN: Vinculin, β-CAT: β-catenin, α-CAT: α-catenin, N-CAD: N-Cadherin, CAR: Cocksackievirus-associated receptor, CAV3: Caveolin-3, ILK: Integrin-linked kinase, FAK: Focal adhesion kinase, PAX: Paxilin, CAS: p130 CRK-associated substrate, ECM: Extracellullar matrix. (Illustration Credit: Ben Smith).
Fascia adherens junctions
Fascia adherens junctions are anchoring junctions between cells that connect the membrane bound cadherins that span the extracellular space at the junction to cytoskeletal actin filaments to provide strong adhesion between neighboring cells (73). In the heart, the proteins bound to the fascia adherens junctional complex include (i) transmembrane proteins that are mainly composed of N-cadherin (NCAD), which are calcium-dependent, but also include coxsackievirus and adenovirus receptor (CAR) and lysosomal integral protein 2 (LIMP2)), which are then intracellularly linked to (ii) catenins (α (α-CAT) , β (β-CAT) ανδγ (πλακογλοβιν, JUP)), which regulate cadherin-based activity as well as (ii) catenin binding proteins such as muscle specific mouse Xin alpha (mXinα), vinculin(VIN)/metavinculin(MV) and α-actinin, which modulate catenin activity or act to link the fascia adherens junction to cytoskeletal actin (73, 74). Thus, by scaffolding multi-molecular complexes, that include components with known signaling roles (eg., β-CAT) and anchoring the actin cytoskeleton, it is thought that several aspects of mechanotransduction converge at the fascia adherens junction(74, 75).
A mechanotransduction role has been postulated for N-CAD within the fascia adherens junction complex. N-CAD is upregulated in response to applied mechanical stretch(76), and N-CAD–catenin complexes have been shown to transmit mechanical forces by forming attachment sites between adjacent cardiac myofibrils(74). Elegant in vitro studies performed by Chopra and colleagues revealed a direct role for N-CAD in cardiac muscle mechanotransduction (76). Specifically, by exploiting cardiomyocytes on a N-CAD-substrate, their studies revealed that N-CAD mediated adhesions were capable of eliciting a cytoskeletal-mediated mechanical remodeling response, which included changes in cardiomyocyte shape, myofibrillar organization and function (as measured by traction forces and cortical stiffness), suggesting the importance of adhesion-contractile balance in cardiac myocyte growth (76). These findings were reinforced when inhibitors of myosin contractility were shown to lower N-CAD-mediated effects on cardiomyocyte stiffness, highlighting an adaptive response of the cardiomyocyte cytoskeleton to changes in mechanical stimuli (76). Interestingly, the spreading and stiffness adaptations of cardiac muscle cells were more enhanced when N-CAD was engaged as opposed to integrin-based substrates, further suggesting that different adhesion systems may mediate differential cytoskeletal adaptive responses based on the perceived forces (76). These concepts may also have implications in cardiac disease settings where differential cardiomyocyte cell growth responses are observed (e.g, dilated cardiomyopathy-axial cell lengthening (eccentric) versus hypertrophic cardiomyopathy-transverse (concentric) cell growth) (76). Potential evidence to support this comes from studies focused on the specific contribution of N-CAD in the adult heart, which exploited a cardiac-specific and inducible N-CAD knockout mouse model (77). Loss of N-CAD in adult mouse cardiomyocytes resulted in the complete absence of identifiable intercalated disc structures (loss of fascia adherens and desmosomal junctions as well as reduction in levels of the gap junction protein, connexin43), culminating in cardiac morphological and functional defects, associated with a modest form of dilated cardiomyopathy (77). However, unlike typical dilated cardiomyopathy, which reveals an enlargement of left ventricular chambers in the short axis, the enlargement was more pronounced in the long axis(77), highlighting an in vivo switch in cardiomyocyte growth response that may be consistent with loss of a mechanotransductive role for N-CAD. Interestingly, adult N-CAD knockout mice also displayed ventricular arrhythmias leading to sudden death (77). N-CAD knockout hearts also displayed decreased sarcomere length and wider but less dense Z-discs, consistent with loss of muscle function due to the absence of N-CAD and anchoring of myofibrils to the plasma membrane(77), which highlight that alterations in cardiomyocyte cell-cell mechanosensing can directly impact sarcomere alignment and protein assembly. Interestingly, increased β-1 integrin levels were also observed in adult N-CAD knockout hearts (77), further highlighting the engagement of a differential cytoskeletal adaptive response associated with integrin-based function. Although a role for N-CAD in human cardiac disease remains to be clarified, these studies suggest a contributing role for N-CAD in multiple mechanotransductive pathways in cardiac muscle.
Alpha-catenins (α-CAT) are key molecules that link the cytoplasmic domain of cadherin to the actin cytoskeleton (78). Recent studies have also implicated a role for α-CAT as a force transducer that is important for mechanotransduction at cadherin-based junctions (79). Specifically, in vitro studies by Yonemura and colleagues showed that α-CAT recruits VIN, another main actin-binding protein of the fascia adherens junction, through force-dependent changes in α-CAT conformation that unmask VIN binding sites to promote adherens junction development (79). Interestingly, our group has also demonstrated that α-E-catenin is required for VIN localization to the fascia adherens junction in cardiomyocytes in vivo (80). Specifically we showed that adult cardiomyocytes from cardiac-specific α-E-catenin knockout mice exhibited specific loss of VIN at the intercalated disc but not the costamere, highlighting a requirement for the cadherin/catenin/VIN complex at the fascia adherens junction (80). In vivo studies focused on the cardiac-specific α-E-catenin knockout mice further revealed that loss of α-E-catenin and resulting VIN loss lead to defects in cardiac intercalated disc structure, morphology and function, associated with a progressive form of dilated cardiomyopathy that encompassed right ventricular wall thinning (80). We also showed that cardiac-specific α-E-catenin knockout mice exhibited an increased propensity to ventricular wall rupture and decreased survival following myocardial infarction at stages prior to disease manifestation (80), highlighting a loss in the cardiomyocyte's ability to adapt to mechanical load with α-E-catenin deficiency. Similar observations related to increased vulnerability to infarct rupture were observed in transgenic mice harboring a C-terminal truncated α-E-catenin (81). Interestingly, defective expression and localization of α-E-catenin at the intercalated disc is a feature of human patients prone to ventricular rupture following myocardial infarction (81), highlighting the relevance of these mechanotransduction-associated pathways in human cardiac disease. Given the molecular crosstalk between α-E-catenin and VIN at the fascia adherens junction, it is also important to note that cardiac-specific and heterozygous conventional VIN knockout mice also exhibit cardiac intercalated disc and functional abnormalities either at baseline, which were associated with dilated cardiomyopathy or after hemodynamic stress associated with increased susceptibility to pressure (mechanical) overload, respectively (82, 83). Human dilated cardiomyopathy patients with MV mutations also exhibited similar intercalated disc defects to the cardiac-specific α-E-catenin knockout mice (80, 84), highlighting that the α-E-catenin/VIN protein complex harbors signals important in maintaining the structural and functional integrity of the intercalated disc and heart, respectively, that also translate to human disease settings. These studies focused on uncovering a mechanistic link between the cellular alterations and functional deficits associated with VIN deficiency in mice, and uncovered a novel mechanism linked to the role of VIN at the costamere (5), which will be discussed in more detail in the Sarcolemma section.
Biochemistry-based studies have also associated a role for the striated muscle-specific protein, nebulin-related anchoring protein (N-RAP) in cardiac muscle mechanotransduction at sites that intersect between the fascia adherens junction and sarcomere (85, 86). Part of this evidence comes from studies that have identified N-RAP to contain a C-terminal actin-binding domain and N-terminal LIM domain, which highlight mechanosignaling domains within N-RAP (86). A physical association between N-RAP and fascia adherens junctions was revealed when N-RAP was shown to co-purify with actin-based intercalated disc components and the fascia adherens junction fragments positively staining for N-RAP and VIN (86). Interestingly, detergent-stripped intercalated disc fractions, which rendered the fraction devoid of actin and VIN, still showed an association between cellular N-RAP within the intercalated disc fraction (containing, N-CAD, α-actinin, desmin, connexin 43), highlighting that N-RAP's association with the fascia adherens junction is mediated by cadherins (86). Gel overlay assays also revealed an association between N-RAP and α-actinin (86). Although a role for N-RAP has not been determined in cardiac muscle in vivo, N-RAP expression was found to be highly upregulated and abnormally distributed in MLP knockout hearts(85), raising the possibility that N-RAP functions may also intersect with the sarcomere via MLP-related mechanotransduction pathways associated with heart failure.
Desmosomes
Desmosomes are anchoring junctions that serve to mechanically couple cells in tissues that undergo constant mechanical stress, such as the heart (11). They tether the cytoskeletal intermediate filament network between cardiomyocytes (11). In cardiac muscle, these organized, disc-shaped, electron-dense structures are composed of extracellular transmembrane-based cadherins, desmocollin-2 (DSC2) and desmoglein-2 (DSG2), which provide a platform via their cytoplasmic tails to members of the armadillo family, JUP and plakophilin-2 (PKP2), that are then in turn bound to the plakin family member, desmoplakin (DSP), which is the central cytoplasmic link to the load-bearing intermediate filament network composed of desmin (11, 73). A role for desmosomes in cardiac mechanotransduction and long range force transmission across cells stems from its association with the desmin intermediate filaments, which were originally termed as ‘mechanical integrators of cellular space’ because of connections to various parts of the cell, including the nucleus, mitochondria, desmosome and sarcomeric Z-disc (10). Desmosomes could also be downstream mechanical transducers of fascia adherens junctions as desmosomal assembly and function were shown to be dependent on N-CAD in the adult heart (77). Studies have also identified hybrid junctions known as the area composita that are composed of proteins from both the fascia adherens and desmosomal junctions, highlighting a convergence of pathways (87). However, desmosomal proteins appear to have a distinct and robust role in intermediate filament-based mechanotransduction, based on growing evidence from genetic data that point to a distinct cardiomyocyte remodeling response associated with desmosomal deficiencies and mutations (separate from the fascia adherens junction), that link to the human cardiac disease, arrhythmogenic right ventricular cardiomyopathy (11).
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a genetic-based cardiac disease tightly associated with desmosomal abnormalities(88). Recent studies have also highlighted that mutations in the spring element of titin (IG10) cause an ARVC-like disease (89). A connection between titin and the desmosome was recently highlighted as titin filaments have been shown to connect to a new subcellular domain of the intercalated disc, termed the transitional junction (90), highlighting a potential for titin-based sarcomere pathways to intersect with the desmosome and ARVC. Further studies, possibly through the generation of knock-in mouse models, will be required to determine the direct role for these mutations in ARVC and whether these pathways directly intersect with the desmosome. Hallmarks of ARVC include life-threatening arrhythmias, cardiac dilation and dysplasia of one or both ventricles that are worsened by strenuous exercise, as well as cardiomyocyte death and replacement of myocardial tissue with fibro-fatty infiltration. Ventricle contractility is compromised in ARVC hearts with late-stage ARVC culminating into heart failure(88). Mutations in desmosomal components can account for up to 58% of ARVC cases(91). A number of studies have exploited genetic mouse models to reveal an important role for desmosomes in transducing mechanical cues into a wide variety of cellular responses important for cardiac structure, function and disease features reminiscent of human ARVC. Mutations in the desmosomal cadherins, DSC2 and DSG2 are associated with human ARVC (11, 92). Studies carried out in transgenic mice expressing the human ARVC-associated DSG2 mutation (N266S) revealed that within two weeks after birth, transgenic mice developed spontaneous ventricular arrhythmias, conduction slowing, ventricular dilation and aneurysms, fibrosis and calcification, leading to sudden death (93), which was reminiscent of a biventricular form of ARVC. Supporting this study, targeted deletion in mice of the extracellular domain of DSG2, which is a domain likely capable of sensing mechanical loads and known to be important in desmosomal adhesive activity as well as signaling, also leads to a biventricular form of ARVC encompassing upregulation of heart failure markers, fibrosis, biventricular dilatation and dysfunction, and spontaneous death(94). At the ultrastructural level, mouse hearts displayed an enlargement of the intercellular gap at the intercalated disc associated with the loss of desmosomal structure which seemed to coincide with visible heart lesions at the macroscopical level(95). These studies together highlight a key role for desmosomal cadherins in sensing and responding to mechanical stresses associated with cardiac muscle contraction.
An important cytoplasmic intermediary that interconnects the desmosomal junction to the cytoskeletal-based intermediate filament system is DSP (11). We and others have revealed through the generation of various genetic mouse models harboring mutations and/or loss of DSP (96-98), that DSP is critical for maintaining cardiac desmosomal cell-cell adhesion (intercalated disc) integrity and function. Cardiac-specific loss of DSP in mice in vivo (using a ventricular myosin light chain-2 Cre recombinase (MLC2v-Cre) mouse line) resulted in early ultrastructural defects that were associated with loss of desmosomal but not N-CAD based junctional proteins (98). Furthermore, these mice recapitulated the postnatal onset of human ARVC at the histological (cardiomyocyte death, fibrosis, fatty infiltration), physiological (biventricular dysfunction and heart failure) and electrophysiological (arrhythmias and premature death) levels that was also observed in an ARVC patient harboring a recessive DSP mutation (98, 99). DSP knockout mice (using MLC2v-Cre) also exhibited sarcomeric defects including loss and widened Z-discs, consistent with loss of muscle function (98), revealing that alterations in desmosomal cell-cell mechanosensing (load) and adhesion can directly impact sarcomere structure. Interestingly, cardiac-specific transgenic mouse models harboring a human DSP mutation that impacted its binding to intermediate filaments also resulted in a cardiac disease reminiscent of ARVC (97), further highlighting that intermediate filament-based mechanotransduction associated with the desmosome may be linked to the cardiac disease, ARVC.
Several studies have also probed for signaling targets at the desmosome that contribute to the various hallmarks of ARVC. We and others have highlighted that developmental signals that elicit transdifferentiation of cardiac muscle to adipocytes play a contributory role towards the fibrofatty infiltration associated with ARVC (98, 100). Although the mechanisms have not been fully elucidated, loss of JUP from the desmosomal cell-cell junction and resulting loss in Wnt/β-CAT signaling (due to presence of nuclear JUP or pathogenic Hippo signaling) has been postulated as a hallmark of ARVC (101) and thought to play a role in promoting adipogenic/fibrogenic gene expression in ARVC, respectively (102, 103). A recent study has highlighted a potential direct link between JUP and PKP2 in cardiac muscle shear stress responses (104). Specifically, this study revealed that shear stress can trigger cardiomyocyte junctional remodeling and that neonatal rat ventricular cardiomyocytes overexpressing JUP and PKP2 mutations associated with ARVC displayed an abnormal response to shear stress (104). Expression of the JUP mutant 2057del2 also displayed an increased propensity for cardiomyocyte apoptosis in response to shear stress (104), which was also observed with cyclical stretch (105). Interestingly, stress responses could be reversed by treatment with the glycogen synthase kinase-3-β inhibitor, SB216763 (104, 105), highlighting convergence between mechanical and signaling responses at the desmosome. Studies in genetic mouse models have also revealed a role for JUP in stabilizing mechanical properties of cardiac muscle in vivo, which include a distinct desmosomal cellular remodeling response associated with features associated with ARVC. Aged global heterozygous JUP knockout mice exhibited right ventricular enlargement and slowed conduction associated with spontaneous arrhythmias that were exacerbated with exercise (106). Interestingly, the exercise training-induced ARVC exhibited by these mice could be rescued with load-reducing therapy (loop diuretic furosemide and nitrates) that reduced cardiac ventricular pressure and volume overload (107). Cardiac-specific JUP knockout mediated by alpha myosin heavy chain Cre (αMHC-Cre) largely recapitulated the features of a biventricular form of ARVC, which included progressive biventricular dilation and dysfunction associated with fibrotic replacement of the myocardium, cardiomyocyte death and spontaneous arrhythmias resulting in sudden death in mice as early as two months of age (108). Although sarcomeric structure, fascia adherens junctions and gap junctions were preserved, desmosomal structures were absent from the intercalated disc of JUP knockout hearts (108). Independent studies generating a cardiac-specific plakoglobin knockout model (using tamoxifen inducible αMHC-Mer-Cre-Mer inducible mouse), highlighted similar findings except that no cardiac arrhythmias were observed despite gap junction remodeling and that mice survived longer (109). Interestingly, discrepancies in links to β-CAT signaling were also reported between both cardiac-specific JUP deficient mouse models (108, 109), which have been similarly observed between cardiac-specific DSP deficient mouse models (96, 98), requiring further evaluation. Nonetheless, these studies altogether highlight that an absence of JUP and desmosomes renders cardiomyocytes unable to properly respond to high mechanical stress resulting in myocyte dissociation and a cellular remodeling responses uniquely associated to ARVC. Since increased β-CAT was observed and proposed to be a compensatory response to the loss of JUP in cardiac-specific JUP-deficient mice (108), double cardiac-specific JUP (αMHC-Cre) and β-CAT (using αMHC-Mer-CreMer inducible mouse) knockout mice were generated (110). JUP and β-CAT double knockout mice caused a loss of fascia adherens and desmosomal junctional proteins resulting in mice exhibiting cardiomyopathy, fibrous replacement of the myocardium and strong arrhythmogenic phenotypes, with 100% of them succumbing to sudden death within 3 to 5 months after tamoxifen injection (110). Interestingly, transgenic mouse models overexpressing JUP (wild type or mutant associated with ARVC), also exhibited fibrofatty replacement of the ventricle and higher prevalence of sudden death(102). Thus, it has been suggested that even small levels of JUP overexpression are sufficient to disrupt the mechanical and signaling functions of JUP in cardiac muscle (111).
Loss of the intermediate filament protein desmin in mice in vivo also leads to skeletal and cardiac muscle abnormalities (112, 113). Specific to the heart, desmin null mice exhibited cardiomyocyte ultrastructural defects (intercalated disc and sarcomere) and cardiomyoycte death resulting in fibrosis and calcification preferentially in the interventricular septum and right ventricle, which lead to cardiomyocyte hypertrophy, ventricular dilatation and systolic dysfunction (113, 114), reminiscent of ARVC. Human mutations in desmin are also associated with familial forms of skeletal and cardiac myopathies (115), including ARVC (116). Recent studies have revealed that some of these severe disease altering mutations may directly impact the force bearing properties of desmin filaments (eg., overstretching at low forces or premature stiffening at low deformations) resulting in defects in the mechanosensing and transduction abilities of desmin (117).
(3) SARCOLEMMA-MEDIATED MECHANOTRANSDUCTION
Besides the intercalated discs at the ends of cardiac myocytes, sarcolemma-associated proteins and complexes along the lateral surfaces of elongated myocytes have been described as foci of force transmission and mechanotransduction(118). Equivalent to non-muscle focal adhesions, costameres are crucial in the lateral transmission of force from the Z-disc of the sarcomere to the sarcolemma and ECM (118, 119). By forming a connection between the ECM and the contractile apparatus, costameric structures facilitate the maintenance of mechanical integrity of the sarcolemma (119, 120). A complex network of cytoskeletal and signaling proteins converge at these specialized cell junctions, and therefore costameres are likely key mediators of mechanical related signaling, and possibly involved with transducing mechanical signals bi-directionally between the extracellular space and intracellular signaling networks (119) (Figure 1).
Integrins
Communication between the ECM and the intracellular portion of cardiac costameres is facilitated by transmembrane integrin receptors attached in the extracellular space to ECM ligands, and intracellularly to the cytoskeleton by proteins such as talin, VIN, and α-actinin(121, 122). Integrin receptors are dimers of α and β subunits, and different combinations of these subunits confer the receptor specificity for ECM ligands(123). The patterns of expression of specific integrin subunits in cardiac muscle cells can be altered and resemble fetal patterns under specific pathologic conditions (124). Specifically, α5 and α7 subunits can be significantly upregulated in ischemia or following myocardial infarction (125), while α1, α5, α7, and β1D subunits are increased in response to pressure overload (126), suggesting that the myocardium may respond to mechanical stress through the modulation of integrin related signaling molecules. In support of this, dilated cardiomoyopathy with extensive myocardial fibrosis and reduced tolerance to loading was observed in a mouse model with excision of β1 integrin using the MLC2v-Cre mouse line (127). Furthermore, reduction of β1 integrin in adult mouse cardiomyocytes resulted in impaired hypertrophic response to pressure overload as a result of modified adrenergic-mediated signaling downstream of the hypertrophic stress (128). Integrins are also fundamental nodal points of key signaling pathways related to mechanotransduction. Integrin-linked kinase (ILK) is an important transducer of integrin signaling. Mutations in ILK have been reported in human dilated cardiomyopathy (129) and tetralogy of Fallot (130). Mice bearing a cardiac knockout of ILK develop dilated cardiomyopathy and spontaneous heart failure(131). Studies performed in human induced pluripotent stem cell-derived cardiomyocytes indicated that ILK mediated cardiomyocyte force transduction via regulation of the sarcoplasmic/endoplasmic reticulum Ca(2+)ATPase isoform 2a (SERCA-2a) and phosphorylation of phospholamban (PLN) in the human heart(132). Thus, ILK links mechanoreception to the dynamic modulation of cardiac contractility by interacting with the functional SERCA-2a/PLN axis.
Vinculin-Talin-Integrin Complex
VIN is a cytoskeletal actin-binding protein that links the actin cytoskeleton to the sarcolemma. VIN is specifically tethered at the costamere, where it binds to integrins via talin. Mutations in the muscle-specific isoform of VIN, MV were found in patients with both dilated and hypertrophic cardiomyopathy, highlighting its importance for normal cardiac function (84). The presence of VIN at the intercalated disc, which is another important site of mechanical coupling, has made it difficult to precisely dissect specific VIN functions between these two cellular regions (124, 133). However, studies in mouse models have demonstrated that VIN is essential for mediating the response to mechanical stress in the heart. Homozygous global loss of VIN in mice leads to embryonic lethality (at embryonic day 10) (134). However, mice with heterozygous loss of VIN display normal basal cardiac function but are more susceptible to cardiac dysfunction and display increased mortality in response to mechanically mediated dysfunction induced by pressure overload of the left ventricle (82).
Talin is a cytoskeletal protein that links integrins to the actin cytoskeleton through the cytoplasmic domain of the β-integrin subunit, and modulates integrin activation and ligand binding (124). Talin mediates the recruitment of key signal transduction partners such as focal adhesion kinase (FAK) and phosphatidylinositol-4-phosphate-5 kinase type Iγ to the costameres (135). Studies performed in mice have revealed that talin1 protein expression and targeting to the costameres is increased in response to cardiac hypertrophy induced by pressure overload (119). Furthermore, it was demonstrated that cardiomyocyte-specific knock-out of talin 1 in mice leads to a blunted hypertrophy response and preserved cardiac function in response to pressure overload when compared to littermate controls (119). The results suggested that talin 1 mediates the hypertrophy response to mechanical stress in the heart, and that a reduction of talin1 expression in cardiac muscle cells can lead to improved cardiac remodeling following pressure overload.
In addition to a role at the costameres, FAK and its C-terminal binding partners p130Cas (p130 CRK-associated substrate) and paxillin (PAX) also redistribute to the Z-disc with hypertrophic stress. FAK has been shown to mediate integrin signaling leading to hypertrophy, while also being activated by α-1 adrenergic stimulation (136). Furthermore, in neonatal cardiomyocytes, tyrosine phosphorylation of FAK and p130Cas has been shown to increase upon ET-1 stimulation (a hypertrophic agonist), which also promotes redistribution of FAK, Cas and PAX to sarcomeric Z-discs (137). Cas alone or in cooperation with Src non-receptor tyrosine kinase modulated basal and ET-1-stimulated ANF gene expression. Interaction of Cas with FAK, as well as their localization to Z-discs, appears critical for sarcomeric assembly in cardiac myocytes. Therefore, the assembly of signaling complexes that include the focal adhesion proteins, Cas, FAK and PAX at Z-discs in the cardiac myocytes may regulate, either directly or indirectly, both cytoskeletal organization and gene expression associated with cardiac myocyte hypertrophy (137). Further work indicated that expression of different Cas mutants leads to severe sarcomeric disarray in cardiomyocytes(138). Expression of the C-terminal focal adhesion-targeting domain of FAK disrupted sarcomeric organization and mislocalization of endogenous Cas to Z-discs, suggesting that the association of FAK and Cas as well as preservation of multiple protein-interaction motifs of Cas was required for proper sarcomeric assembly in cardiac myocytes(138). Interestingly, in skeletal muscle, changes in the expression of FAK levels and signaling were also associated with sarcomeric reorganization in response to overload (139).
Caveolin 3
Integrin signaling in response to mechanical load can also be regulated by caveolin 3 (CAV3) (140). CAV3 is highly expressed in cardiac muscle cells, where it can be found both at the focal adhesion complex(141), or as a component of lipid raft microdomains, termed caveolae (142), that can regulate signal transduction by compartmentalizing receptors and their ligands (143). Loss of CAV3 in cardiac muscle cells has been shown to lead to reduced β1 integrin expression leading to perturbed basal and stretch mediated signaling responses (140).
Dystroglycan
The dystroglycan complex also serves as an ECM receptor in cardiac muscle cells. β-dystroglycan interacts with α-dystroglycan on the cell surface, which facilitates interactions, with the ECM protein laminin (144). β-dystroglycan also tethers the actin binding protein dystrophin within the cytosol (145). Mutations in dystrophin have been shown to lead to Duchene's muscular dystrophy, which is characterized by muscle (skeletal and cardiac) degeneration (146, 147), highlighting the importance of dystrophin as linker between the ECM and the cytoskeleton. Combinatorial loss of dystrophin with β1-Integrin has been shown to exacerbate the cardiomyopathic changes observed with loss of dystrophin and integrins alone in response to isoproterenol treatment, suggesting cross talk between these two complexes in response to cardiac stress (148).
CONCLUSIONS/FUTURE DIRECTIONS
Force transmission and sensing within the cardiac myocyte is a complex process, and a multitude of proteins and protein complexes have been implicated in the mechanisms of both stress transmission and stress transduction. The putative mechanotransduction-related proteins exist at many different locations within the cell, from the outer sarcolemmal membrane down to the force-generating sarcomere and associated structures. It appears likely that multiple structural pathways may be involved with the overall processes of load-mediated remodeling of cardiac myocytes, with redundant or overlapping function.
It is intriguing to consider modulation of a mechanosensing pathway in order to alter the downstream effects, for example, adverse remodeling in myocardium could be controlled at the force-sensing level, altering the downstream molecular signals and improving long-term outcomes in cardiac disease. Mediating the molecular signals associated with a specific mechanotransduction pathway could also be a way of modifying downstream remodeling outcomes, without directly changing the structural properties of the force transmission pathways. For example, as described above, several of the hypertrophy-inducing pathways in cardiomyocytes are, at least in part, calcium-dependent: among others, calcium-calmodulin kinase II (CaMKII) and CnA are activated by increased local calcium concentrations. These calcium-mediated signaling pathways have been investigated as mediators of sarcomeric force generation, and are clearly described. Thus small molecule modulations of these well-described pathways are underway and represent a promising direction for therapeutic applications for cardiac hypertrophy and failure (149, 150). Inhibitors of CaMKII have proven excellent research tools and some have been investigated in early clinical trials (151). Amongst those, the CaMKIIN peptide has shown minimal off-target effects, but has yet to be tested in patients (151). CnA inhibitiors cyclosporin A (CsA) and FK506 were shown to prevent the phenotypic manifestations of hypertrophic cardiomyopathy in mice overexpressing tropomodulin, or fetal β-tropomyosin (152, 153). Partial or complete inhibition of cardiac hypertrophy has been achieved with CsA and FK506 following pressure overload in rats and mice (154-156), although it has also been reported that under specific settings a significant rescue could not be achieved (154-157). Studies in rats indicated that CsA prevented exercise-induced cardiac hypertrophy, attenuated hypertrophy and histopathology in double transgenic transgenic mice overexpressing human renin and angiotensinogen (158) as well as attenuated myocardial infarction–induced cardiac hypertrophy(152, 159). CsA also reduced cardiac hypertrophy in constitutively activated Gq transgenic mice(160). Nevertheless the known side effects of CsA and FK506 in humans have prevented further exploration of these small molecules(152). A similar approach could be used for other signaling kinases implicated in mechanotransduction pathways such as ILK, which mediates force transduction in cardiomyocytes by modulating SERCA-2a/PLN function (132). The R211A mutation in ILK has been shown to promote enhanced cardiomyocyte contractility compared to its wild-type counterpart, including increased releasable SR calcium content. Such positive effects were proposed to stem from higher expression levels of ILK and SERCA-2a compared with that resulting from ILK WT overexpression in vitro and in transgenic mice in vivo. Thus, gene therapy with ILK R211A has been proposed as a possible future management strategy for dilated cardiomyopathy (132).
Acknowledgments
SOURCES OF FUNDING
R.C.L is supported by a NIH T32 training grant. F.Z. is a recipient of an American Heart Association Postdoctoral Fellowship. F.S. is supported by NIH/NHLBI (HL09780) and American Heart Association (GRNT22940045) grants. J.H.O. is supported by NIH grants HL103566 and HL105242.
Nonstandard Abbreviations and Acronyms
- α-CAT
α-catenin
- αMHC-Cre
α-myosin heavy chain Cre recombinase
- αMHC-Mer-Cre-Mer
Tamoxifen inducible α-myosin heavy chain Cre recombinase
- β-CAT
β-catenin
- ANF
Atrial natriuretic factor
- ANKRD1
Ankyrin repeat domain protein 1
- ANKRD2
Ankyrin repeat domain protein 2
- ARVC
Arrhythmogenic right ventricular cardiomyopathy
- BNP
Brain natriuretic peptide
- CAR
Coxsackievirus and adenovirus receptor
- CARP
Cardiac Ankyrin Repeat Protein
- CAMKII
Calcium-calmodulin kinase II
- CAV3
Caveolin-3
- CnA
Calcineurin
- CsA
Cyclosporine A
- CS-1
Calsarcin-1
- CSRP2
Cysteine rich protein 2
- CSRP3
Cysteine rich protein 3
- DARP
Diabetes related ankyrin repeat protein
- DSC2
Desmocollin-2
- DSG2
Desmoglein-2
- DSP
Desmoplakin
- ECM
Extracellular Matrix
- ET-1
Endothelin-1
- ERK2
Extracellular regulated kinase-2
- FAK
Focal adhesion kinase
- FHL1
Four and a half LIM domain protein-1
- Gq
Gαq signaling pathway
- HMERF
Heredity myopathy with early respiratory failure
- ILK
Integrin linked kinase
- JUP
γ-catenin (plakoglobin)
- LC3-II
Microtubule-associated protein light chain 3-II
- LIMP2
Lysosomal integral protein 2
- MARP
Muscle-specific Ankyrin Repeat protein
- MEK2
Mitogen-activated protein kinase kinase-2
- MLC2v-Cre
Ventricular myosin light chain-2 Cre recombinase
- MLP
Muscle LIM protein
- MURF2
Muscle-specific RING finger-2
- MV
Metavinculin
- NBR1
Neighbor to BRCA1
- N-CAD
N-cadherin
- NFAT
Nuclear factor of activated T-cells
- NRAP
Nebulin-related anchoring protein
- p62/SQSTM1
p62/sequestosome-1
- p130Cas
p130 CRK-associated substrate
- PAX
Paxillin
- PICOT
Protein kinase C-interacting cousin of thioredoxin
- PKP2
Plakophilin-2
- PLN
Phospholamban
- RAF1
Rapid accelerated fibrosarcoma-1
- SERCA-2a
Sarcoplasmic/endoplasmic reticulum Ca(2+)ATPase isoform 2a
- SRF
Serum Response Factor
- TCAP
Titin-Cap
- VIN
Vinculin
- W4R-MLP
Dilated cardiomyopathy-associated polymorphism found in human MLP
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Gautel M. The sarcomeric cytoskeleton: who picks up the strain? Current opinion in cell biology. 2011;23:39–46. doi: 10.1016/j.ceb.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Spudich JA. The myosin swinging cross-bridge model. Nature reviews. Molecular cell biology. 2001;2:387–392. doi: 10.1038/35073086. [DOI] [PubMed] [Google Scholar]
- 3.Buyandelger B, Mansfield C, Knoll R. Mechano-signaling in heart failure. Pflugers Archiv : European journal of physiology. 2014;466:1093–1099. doi: 10.1007/s00424-014-1468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frank D, Frey N. Cardiac Z-disc signaling network. The Journal of biological chemistry. 2011;286:9897–9904. doi: 10.1074/jbc.R110.174268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tangney JR, Chuang JS, Janssen MS, Krishnamurthy A, Liao P, Hoshijima M, Wu X, Meininger GA, Muthuchamy M, Zemljic-Harpf A, Ross RS, Frank LR, McCulloch AD, Omens JH. Novel role for vinculin in ventricular myocyte mechanics and dysfunction. Biophysical journal. 2013;104:1623–1633. doi: 10.1016/j.bpj.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simpson DG, Majeski M, Borg TK, Terracio L. Regulation of cardiac myocyte protein turnover and myofibrillar structure in vitro by specific directions of stretch. Circulation research. 1999;85:e59–69. doi: 10.1161/01.res.85.10.e59. [DOI] [PubMed] [Google Scholar]
- 7.Gopalan SM, Flaim C, Bhatia SN, Hoshijima M, Knoell R, Chien KR, Omens JH, McCulloch AD. Anisotropic stretch-induced hypertrophy in neonatal ventricular myocytes micropatterned on deformable elastomers. Biotechnology and bioengineering. 2003;81:578–587. doi: 10.1002/bit.10506. [DOI] [PubMed] [Google Scholar]
- 8.Kerckhoffs RC, Omens J, McCulloch AD. A single strain-based growth law predicts concentric and eccentric cardiac growth during pressure and volume overload. Mechanics research communications. 2012;42:40–50. doi: 10.1016/j.mechrescom.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peter AK, Cheng H, Ross RS, Knowlton KU, Chen J. The costamere bridges sarcomeres to the sarcolemma in striated muscle. Progress in pediatric cardiology. 2011;31:83–88. doi: 10.1016/j.ppedcard.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lazarides E. Intermediate filaments as mechanical integrators of cellular space. Nature. 1980;283:249–256. doi: 10.1038/283249a0. [DOI] [PubMed] [Google Scholar]
- 11.Sheikh F, Ross RS, Chen J. Cell-cell connection to cardiac disease. Trends in cardiovascular medicine. 2009;19:182–190. doi: 10.1016/j.tcm.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prosser BL, Ward CW. Mechano-chemo transduction tunes the heartstrings. Science signaling. 2014;7:e7. doi: 10.1126/scisignal.2005214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abramochkin DV, Lozinsky IT, Kamkin A. Influence of mechanical stress on fibroblast-myocyte interactions in mammalian heart. Journal of molecular and cellular cardiology. 2014;70:27–36. doi: 10.1016/j.yjmcc.2013.12.020. [DOI] [PubMed] [Google Scholar]
- 14.Reed A, Kohl P, Peyronnet R. Molecular candidates for cardiac stretch-activated ion channels. Global cardiology science & practice. 2014;2014:9–25. doi: 10.5339/gcsp.2014.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson BR, Granzier HL. Titin-based tension in the cardiac sarcomere: molecular origin and physiological adaptations. Progress in biophysics and molecular biology. 2012;110:204–217. doi: 10.1016/j.pbiomolbio.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, McKenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP, Chien KR. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–955. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- 17.Wang K, McClure J, Tu A. Titin: major myofibrillar components of striated muscle. Proceedings of the National Academy of Sciences of the United States of America. 1979;76:3698–3702. doi: 10.1073/pnas.76.8.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linke WA, Kruger M. The giant protein titin as an integrator of myocyte signaling pathways. Physiology (Bethesda) 2010;25:186–198. doi: 10.1152/physiol.00005.2010. [DOI] [PubMed] [Google Scholar]
- 19.Granzier HL, Labeit S. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circulation research. 2004;94:284–295. doi: 10.1161/01.RES.0000117769.88862.F8. [DOI] [PubMed] [Google Scholar]
- 20.Kruger M, Linke WA. Titin-based mechanical signalling in normal and failing myocardium. Journal of molecular and cellular cardiology. 2009;46:490–498. doi: 10.1016/j.yjmcc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Tskhovrebova L, Trinick J. Titin: properties and family relationships. Nature reviews. Molecular cell biology. 2003;4:679–689. doi: 10.1038/nrm1198. [DOI] [PubMed] [Google Scholar]
- 22.Hoshijima M. Mechanical stress-strain sensors embedded in cardiac cytoskeleton: Z disk, titin, and associated structures. American journal of physiology. Heart and circulatory physiology. 2006;290:H1313–1325. doi: 10.1152/ajpheart.00816.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arber S, Halder G, Caroni P. Muscle LIM protein, a novel essential regulator of myogenesis, promotes myogenic differentiation. Cell. 1994;79:221–231. doi: 10.1016/0092-8674(94)90192-9. [DOI] [PubMed] [Google Scholar]
- 24.Heineke J, Ruetten H, Willenbockel C, Gross SC, Naguib M, Schaefer A, Kempf T, Hilfiker-Kleiner D, Caroni P, Kraft T, Kaiser RA, Molkentin JD, Drexler H, Wollert KC. Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1655–1660. doi: 10.1073/pnas.0405488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arber S, Hunter JJ, Ross J, Jr., Hongo M, Sansig G, Borg J, Perriard JC, Chien KR, Caroni P. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell. 1997;88:393–403. doi: 10.1016/s0092-8674(00)81878-4. [DOI] [PubMed] [Google Scholar]
- 26.Knoll R, Kostin S, Klede S, Savvatis K, Klinge L, Stehle I, Gunkel S, Kotter S, Babicz K, Sohns M, Miocic S, Didie M, Knoll G, Zimmermann WH, Thelen P, Bickeboller H, Maier LS, Schaper W, Schaper J, Kraft T, Tschope C, Linke WA, Chien KR. A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circulation research. 2010;106:695–704. doi: 10.1161/CIRCRESAHA.109.206243. [DOI] [PubMed] [Google Scholar]
- 27.Chang DF, Belaguli NS, Iyer D, Roberts WB, Wu SP, Dong XR, Marx JG, Moore MS, Beckerle MC, Majesky MW, Schwartz RJ. Cysteine-rich LIM-only proteins CRP1 and CRP2 are potent smooth muscle differentiation cofactors. Developmental cell. 2003;4:107–118. doi: 10.1016/s1534-5807(02)00396-9. [DOI] [PubMed] [Google Scholar]
- 28.Furukawa T, Ono Y, Tsuchiya H, Katayama Y, Bang ML, Labeit D, Labeit S, Inagaki N, Gregorio CC. Specific interaction of the potassium channel beta-subunit minK with the sarcomeric protein T-cap suggests a T-tubule-myofibril linking system. Journal of molecular biology. 2001;313:775–784. doi: 10.1006/jmbi.2001.5053. [DOI] [PubMed] [Google Scholar]
- 29.Geier C, Gehmlich K, Ehler E, Hassfeld S, Perrot A, Hayess K, Cardim N, Wenzel K, Erdmann B, Krackhardt F, Posch MG, Osterziel KJ, Bublak A, Nagele H, Scheffold T, Dietz R, Chien KR, Spuler S, Furst DO, Nurnberg P, Ozcelik C. Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Human molecular genetics. 2008;17:2753–2765. doi: 10.1093/hmg/ddn160. [DOI] [PubMed] [Google Scholar]
- 30.Bertz M, Wilmanns M, Rief M. The titin-telethonin complex is a directed, superstable molecular bond in the muscle Z-disk. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13307–133310. doi: 10.1073/pnas.0902312106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller MK, Bang ML, Witt CC, Labeit D, Trombitas C, Watanabe K, Granzier H, McElhinny AS, Gregorio CC, Labeit S. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. Journal of molecular biology. 2003;333:951–964. doi: 10.1016/j.jmb.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 32.Jeyaseelan R, Poizat C, Baker RK, Abdishoo S, Isterabadi LB, Lyons GE, Kedes L. A novel cardiac-restricted target for doxorubicin. CARP, a nuclear modulator of gene expression in cardiac progenitor cells and cardiomyocytes. The Journal of biological chemistry. 1997;272:22800–22808. doi: 10.1074/jbc.272.36.22800. [DOI] [PubMed] [Google Scholar]
- 33.Barash IA, Mathew L, Ryan AF, Chen J, Lieber RL. Rapid muscle-specific gene expression changes after a single bout of eccentric contractions in the mouse. American journal of physiology. Cell physiology. 2004;286:C355–364. doi: 10.1152/ajpcell.00211.2003. [DOI] [PubMed] [Google Scholar]
- 34.Torrado M, Lopez E, Centeno A, Castro-Beiras A, Mikhailov AT. Left-right asymmetric ventricular expression of CARP in the piglet heart: regional response to experimental heart failure. European journal of heart failure. 2004;6:161–172. doi: 10.1016/j.ejheart.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 35.Zolk O, Frohme M, Maurer A, Kluxen FW, Hentsch B, Zubakov D, Hoheisel JD, Zucker IH, Pepe S, Eschenhagen T. Cardiac ankyrin repeat protein, a negative regulator of cardiac gene expression, is augmented in human heart failure. Biochemical and biophysical research communications. 2002;293:1377–1382. doi: 10.1016/S0006-291X(02)00387-X. [DOI] [PubMed] [Google Scholar]
- 36.Aihara Y, Kurabayashi M, Saito Y, Ohyama Y, Tanaka T, Takeda S, Tomaru K, Sekiguchi K, Arai M, Nakamura T, Nagai R. Cardiac ankyrin repeat protein is a novel marker of cardiac hypertrophy: role of M-CAT element within the promoter. Hypertension. 2000;36:48–53. doi: 10.1161/01.hyp.36.1.48. [DOI] [PubMed] [Google Scholar]
- 37.Arimura T, Bos JM, Sato A, Kubo T, Okamoto H, Nishi H, Harada H, Koga Y, Moulik M, Doi YL, Towbin JA, Ackerman MJ, Kimura A. Cardiac ankyrin repeat protein gene (ANKRD1) mutations in hypertrophic cardiomyopathy. Journal of the American College of Cardiology. 2009;54:334–342. doi: 10.1016/j.jacc.2008.12.082. [DOI] [PubMed] [Google Scholar]
- 38.Duboscq-Bidot L, Charron P, Ruppert V, Fauchier L, Richter A, Tavazzi L, Arbustini E, Wichter T, Maisch B, Komajda M, Isnard R, Villard E. Mutations in the ANKRD1 gene encoding CARP are responsible for human dilated cardiomyopathy. European heart journal. 2009;30:2128–2136. doi: 10.1093/eurheartj/ehp225. [DOI] [PubMed] [Google Scholar]
- 39.Moulik M, Vatta M, Witt SH, Arola AM, Murphy RT, McKenna WJ, Boriek AM, Oka K, Labeit S, Bowles NE, Arimura T, Kimura A, Towbin JA. ANKRD1, the gene encoding cardiac ankyrin repeat protein, is a novel dilated cardiomyopathy gene. Journal of the American College of Cardiology. 2009;54:325–333. doi: 10.1016/j.jacc.2009.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bang ML, Gu Y, Dalton ND, Peterson KL, Chien KR, Chen J. The muscle ankyrin repeat proteins CARP, Ankrd2, and DARP are not essential for normal cardiac development and function at basal conditions and in response to pressure overload. PloS one. 2014;9:e93638. doi: 10.1371/journal.pone.0093638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lun AS, Chen J, Lange S. Probing muscle ankyrin-repeat protein (MARP) structure and function. Anat Rec (Hoboken) 2014;297:1615–1629. doi: 10.1002/ar.22968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Linke WA, Rudy DE, Centner T, Gautel M, Witt C, Labeit S, Gregorio CC. I-band titin in cardiac muscle is a three-element molecular spring and is critical for maintaining thin filament structure. The Journal of cell biology. 1999;146:631–644. doi: 10.1083/jcb.146.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheikh F, Raskin A, Chu PH, Lange S, Domenighetti AA, Zheng M, Liang X, Zhang T, Yajima T, Gu Y, Dalton ND, Mahata SK, Dorn GW, 2nd, Brown JH, Peterson KL, Omens JH, McCulloch AD, Chen J. An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. The Journal of clinical investigation. 2008;118:3870–3880. doi: 10.1172/JCI34472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chu PH, Ruiz-Lozano P, Zhou Q, Cai C, Chen J. Expression patterns of FHL/SLIM family members suggest important functional roles in skeletal muscle and cardiovascular system. Mechanisms of development. 2000;95:259–265. doi: 10.1016/s0925-4773(00)00341-5. [DOI] [PubMed] [Google Scholar]
- 45.Gaussin V, Tomlinson JE, Depre C, Engelhardt S, Antos CL, Takagi G, Hein L, Topper JN, Liggett SB, Olson EN, Lohse MJ, Vatner SF, Vatner DE. Common genomic response in different mouse models of beta-adrenergic-induced cardiomyopathy. Circulation. 2003;108:2926–2933. doi: 10.1161/01.CIR.0000101922.18151.7B. [DOI] [PubMed] [Google Scholar]
- 46.Hwang DM, Dempsey AA, Wang RX, Rezvani M, Barrans JD, Dai KS, Wang HY, Ma H, Cukerman E, Liu YQ, Gu JR, Zhang JH, Tsui SK, Waye MM, Fung KP, Lee CY, Liew CC. A genome-based resource for molecular cardiovascular medicine: toward a compendium of cardiovascular genes. Circulation. 1997;96:4146–4203. doi: 10.1161/01.cir.96.12.4146. [DOI] [PubMed] [Google Scholar]
- 47.Hwang DM, Dempsey AA, Lee CY, Liew CC. Identification of differentially expressed genes in cardiac hypertrophy by analysis of expressed sequence tags. Genomics. 2000;66:1–14. doi: 10.1006/geno.2000.6171. [DOI] [PubMed] [Google Scholar]
- 48.Lim DS, Roberts R, Marian AJ. Expression profiling of cardiac genes in human hypertrophic cardiomyopathy: insight into the pathogenesis of phenotypes. Journal of the American College of Cardiology. 2001;38:1175–1180. doi: 10.1016/s0735-1097(01)01509-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wettschureck N, Rutten H, Zywietz A, Gehring D, Wilkie TM, Chen J, Chien KR, Offermanns S. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nature medicine. 2001;7:1236–1240. doi: 10.1038/nm1101-1236. [DOI] [PubMed] [Google Scholar]
- 50.Christodoulou DC, Wakimoto H, Onoue K, Eminaga S, Gorham JM, DePalma SR, Herman DS, Teekakirikul P, Conner DA, McKean DM, Domenighetti AA, Aboukhalil A, Chang S, Srivastava G, McDonough B, De Jager PL, Chen J, Bulyk ML, Muehlschlegel JD, Seidman CE, Seidman JG. 5'RNA-Seq identifies Fhl1 as a genetic modifier in cardiomyopathy. The Journal of clinical investigation. 2014;124:1364–1370. doi: 10.1172/JCI70108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Raskin A, Lange S, Banares K, Lyon RC, Zieseniss A, Lee LK, Yamazaki KG, Granzier HL, Gregorio CC, McCulloch AD, Omens JH, Sheikh F. A novel mechanism involving four-and-a-half LIM domain protein-1 and extracellular signal-regulated kinase-2 regulates titin phosphorylation and mechanics. The Journal of biological chemistry. 2012;287:29273–29284. doi: 10.1074/jbc.M112.372839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Obermann WM, Gautel M, Steiner F, van der Ven PF, Weber K, Furst DO. The structure of the sarcomeric M band: localization of defined domains of myomesin, M-protein, and the 250-kD carboxy-terminal region of titin by immunoelectron microscopy. The Journal of cell biology. 1996;134:1441–1453. doi: 10.1083/jcb.134.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gautel M. Cytoskeletal protein kinases: titin and its relations in mechanosensing. Pflugers Archiv : European journal of physiology. 2011;462:119–134. doi: 10.1007/s00424-011-0946-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Puchner EM, Alexandrovich A, Kho AL, Hensen U, Schafer LV, Brandmeier B, Grater F, Grubmuller H, Gaub HE, Gautel M. Mechanoenzymatics of titin kinase. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13385–13390. doi: 10.1073/pnas.0805034105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, Kristensen J, Brandmeier B, Franzen G, Hedberg B, Gunnarsson LG, Hughes SM, Marchand S, Sejersen T, Richard I, Edstrom L, Ehler E, Udd B, Gautel M. The kinase domain of titin controls muscle gene expression and protein turnover. Science. 2005;308:1599–1603. doi: 10.1126/science.1110463. [DOI] [PubMed] [Google Scholar]
- 56.Bogomolovas J, Gasch A, Simkovic F, Rigden DJ, Labeit S, Mayans O. Titin kinase is an inactive pseudokinase scaffold that supports MuRF1 recruitment to the sarcomeric M-line. Open biology. 2014;4:140041. doi: 10.1098/rsob.140041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Witt CC, Witt SH, Lerche S, Labeit D, Back W, Labeit S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. The EMBO journal. 2008;27:350–360. doi: 10.1038/sj.emboj.7601952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lyon RC, Lange S, Sheikh F. Breaking down protein degradation mechanisms in cardiac muscle. Trends in molecular medicine. 2013;19:239–249. doi: 10.1016/j.molmed.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Backues SK, Klionsky DJ. Autophagy gets in on the regulatory act. Journal of molecular cell biology. 2011;3:76–77. doi: 10.1093/jmcb/mjq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Edstrom L, Thornell LE, Albo J, Landin S, Samuelsson M. Myopathy with respiratory failure and typical myofibrillar lesions. Journal of the neurological sciences. 1990;96:211–228. doi: 10.1016/0022-510x(90)90134-9. [DOI] [PubMed] [Google Scholar]
- 62.Hedberg C, Melberg A, Dahlbom K, Oldfors A. Hereditary myopathy with early respiratory failure is caused by mutations in the titin FN3 119 domain. Brain : a journal of neurology. 2014;137:e270. doi: 10.1093/brain/awt305. [DOI] [PubMed] [Google Scholar]
- 63.Lange S, Edstrom L, Udd B, Gautel M. Reply: Hereditary myopathy with early respiratory failure is caused by mutations in the titin FN3 119 domain. Brain : a journal of neurology. 2014;137:e279. doi: 10.1093/brain/awu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pfeffer G, Griffin H, Pyle A, Horvath R, Chinnery PF. Reply: Hereditary myopathy with early respiratory failure is caused by mutations in the titin FN3 119 domain. Brain : a journal of neurology. 2014;137:e271. doi: 10.1093/brain/awu034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pfeffer G, Chinnery PF. Reply: Hereditary myopathy with early respiratory failure is caused by mutations in the titin FN3 119 domain. Brain : a journal of neurology. 2014;137:e280. doi: 10.1093/brain/awt306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chauveau C, Bonnemann CG, Julien C, Kho AL, Marks H, Talim B, Maury P, Arne-Bes MC, Uro-Coste E, Alexandrovich A, Vihola A, Schafer S, Kaufmann B, Medne L, Hubner N, Foley AR, Santi M, Udd B, Topaloglu H, Moore SA, Gotthardt M, Samuels ME, Gautel M, Ferreiro A. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Human molecular genetics. 2014;23:980–991. doi: 10.1093/hmg/ddt494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 68.Frank D, Kuhn C, Katus HA, Frey N. The sarcomeric Z-disc: a nodal point in signalling and disease. J Mol Med (Berl) 2006;84:446–468. doi: 10.1007/s00109-005-0033-1. [DOI] [PubMed] [Google Scholar]
- 69.Jeong D, Kim JM, Cha H, Oh JG, Park J, Yun SH, Ju ES, Jeon ES, Hajjar RJ, Park WJ. PICOT attenuates cardiac hypertrophy by disrupting calcineurin-NFAT signaling. Circulation research. 2008;102:711–719. doi: 10.1161/CIRCRESAHA.107.165985. [DOI] [PubMed] [Google Scholar]
- 70.Frey N, Barrientos T, Shelton JM, Frank D, Rutten H, Gehring D, Kuhn C, Lutz M, Rothermel B, Bassel-Duby R, Richardson JA, Katus HA, Hill JA, Olson EN. Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nature medicine. 2004;10:1336–1343. doi: 10.1038/nm1132. [DOI] [PubMed] [Google Scholar]
- 71.Osio A, Tan L, Chen SN, Lombardi R, Nagueh SF, Shete S, Roberts R, Willerson JT, Marian AJ. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circulation research. 2007;100:766–768. doi: 10.1161/01.RES.0000263008.66799.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoshida M, Sho E, Nanjo H, Takahashi M, Kobayashi M, Kawamura K, Honma M, Komatsu M, Sugita A, Yamauchi M, Hosoi T, Ito Y, Masuda H. Weaving hypothesis of cardiomyocyte sarcomeres: discovery of periodic broadening and narrowing of intercalated disk during volume-load change. The American journal of pathology. 2010;176:660–678. doi: 10.2353/ajpath.2010.090348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mezzano V, Sheikh F. Cell-cell junction remodeling in the heart: possible role in cardiac conduction system function and arrhythmias? Life sciences. 2012;90:313–321. doi: 10.1016/j.lfs.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Michaelson JE, Huang H. Cell-cell junctional proteins in cardiovascular mechanotransduction. Annals of biomedical engineering. 2012;40:568–577. doi: 10.1007/s10439-011-0439-6. [DOI] [PubMed] [Google Scholar]
- 75.Nagafuchi A. Molecular architecture of adherens junctions. Current opinion in cell biology. 2001;13:600–603. doi: 10.1016/s0955-0674(00)00257-x. [DOI] [PubMed] [Google Scholar]
- 76.Chopra A, Tabdanov E, Patel H, Janmey PA, Kresh JY. Cardiac myocyte remodeling mediated by N-cadherin-dependent mechanosensing. American journal of physiology. Heart and circulatory physiology. 2011;300:H1252–1266. doi: 10.1152/ajpheart.00515.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kostetskii I, Li J, Xiong Y, Zhou R, Ferrari VA, Patel VV, Molkentin JD, Radice GL. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circulation research. 2005;96:346–354. doi: 10.1161/01.RES.0000156274.72390.2c. [DOI] [PubMed] [Google Scholar]
- 78.Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:8813–8817. doi: 10.1073/pnas.92.19.8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yonemura S, Wada Y, Watanabe T, Nagafuchi A, Shibata M. alpha-Catenin as a tension transducer that induces adherens junction development. Nature cell biology. 2010;12:533–542. doi: 10.1038/ncb2055. [DOI] [PubMed] [Google Scholar]
- 80.Sheikh F, Chen Y, Liang X, Hirschy A, Stenbit AE, Gu Y, Dalton ND, Yajima T, Lu Y, Knowlton KU, Peterson KL, Perriard JC, Chen J. alpha-E-catenin inactivation disrupts the cardiomyocyte adherens junction, resulting in cardiomyopathy and susceptibility to wall rupture. Circulation. 2006;114:1046–1055. doi: 10.1161/CIRCULATIONAHA.106.634469. [DOI] [PubMed] [Google Scholar]
- 81.van den Borne SW, Narula J, Voncken JW, Lijnen PM, Vervoort-Peters HT, Dahlmans VE, Smits JF, Daemen MJ, Blankesteijn WM. Defective intercellular adhesion complex in myocardium predisposes to infarct rupture in humans. Journal of the American College of Cardiology. 2008;51:2184–2192. doi: 10.1016/j.jacc.2008.02.056. [DOI] [PubMed] [Google Scholar]
- 82.Zemljic-Harpf AE, Ponrartana S, Avalos RT, Jordan MC, Roos KP, Dalton ND, Phan VQ, Adamson ED, Ross RS. Heterozygous inactivation of the vinculin gene predisposes to stress-induced cardiomyopathy. The American journal of pathology. 2004;165:1033–1044. doi: 10.1016/S0002-9440(10)63364-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zemljic-Harpf AE, Miller JC, Henderson SA, Wright AT, Manso AM, Elsherif L, Dalton ND, Thor AK, Perkins GA, McCulloch AD, Ross RS. Cardiac-myocyte-specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Molecular and cellular biology. 2007;27:7522–7537. doi: 10.1128/MCB.00728-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Olson TM, Illenberger S, Kishimoto NY, Huttelmaier S, Keating MT, Jockusch BM. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation. 2002;105:431–437. doi: 10.1161/hc0402.102930. [DOI] [PubMed] [Google Scholar]
- 85.Ehler E, Horowits R, Zuppinger C, Price RL, Perriard E, Leu M, Caroni P, Sussman M, Eppenberger HM, Perriard JC. Alterations at the intercalated disk associated with the absence of muscle LIM protein. The Journal of cell biology. 2001;153:763–772. doi: 10.1083/jcb.153.4.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang JQ, Elzey B, Williams G, Lu S, Law DJ, Horowits R. Ultrastructural and biochemical localization of N-RAP at the interface between myofibrils and intercalated disks in the mouse heart. Biochemistry. 2001;40:14898–14906. doi: 10.1021/bi0107445. [DOI] [PubMed] [Google Scholar]
- 87.Pieperhoff S, Franke WW. The area composita of adhering junctions connecting heart muscle cells of vertebrates - IV: coalescence and amalgamation of desmosomal and adhaerens junction components - late processes in mammalian heart development. European journal of cell biology. 2007;86:377–391. doi: 10.1016/j.ejcb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 88.James CA, Calkins H. Update on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C) Current treatment options in cardiovascular medicine. 2013;15:476–487. doi: 10.1007/s11936-013-0251-8. [DOI] [PubMed] [Google Scholar]
- 89.Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, Pinamonti B, Salcedo EE, Sauer W, Pyxaras S, Anderson B, Simon B, Bogomolovas J, Labeit S, Granzier H, Mestroni L. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation. 2011;124:876–885. doi: 10.1161/CIRCULATIONAHA.110.005405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bennett PM, Maggs AM, Baines AJ, Pinder JC. The transitional junction: a new functional subcellular domain at the intercalated disc. Molecular biology of the cell. 2006;17:2091–2100. doi: 10.1091/mbc.E05-12-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marcus FI, Edson S, Towbin JA. Genetics of arrhythmogenic right ventricular cardiomyopathy: a practical guide for physicians. Journal of the American College of Cardiology. 2013;61:1945–1948. doi: 10.1016/j.jacc.2013.01.073. [DOI] [PubMed] [Google Scholar]
- 92.Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli GA, Rampazzo A. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–1179. doi: 10.1161/CIRCULATIONAHA.105.583674. [DOI] [PubMed] [Google Scholar]
- 93.Pilichou K, Remme CA, Basso C, Campian ME, Rizzo S, Barnett P, Scicluna BP, Bauce B, van den Hoff MJ, de Bakker JM, Tan HL, Valente M, Nava A, Wilde AA, Moorman AF, Thiene G, Bezzina CR. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. The Journal of experimental medicine. 2009;206:1787–1802. doi: 10.1084/jem.20090641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Krusche CA, Holthofer B, Hofe V, van de Sandt AM, Eshkind L, Bockamp E, Merx MW, Kant S, Windoffer R, Leube RE. Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic research in cardiology. 2011;106:617–633. doi: 10.1007/s00395-011-0175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kant S, Krull P, Eisner S, Leube RE, Krusche CA. Histological and ultrastructural abnormalities in murine desmoglein 2-mutant hearts. Cell and tissue research. 2012;348:249–259. doi: 10.1007/s00441-011-1322-3. [DOI] [PubMed] [Google Scholar]
- 96.Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. The Journal of clinical investigation. 2006;116:2012–2021. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, Ge S, Nadvoretskiy VV, DeFreitas G, Carabello B, Brandon LI, Godsel LM, Green KJ, Saffitz JE, Li H, Danieli GA, Calkins H, Marcus F, Towbin JA. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation research. 2006;99:646–655. doi: 10.1161/01.RES.0000241482.19382.c6. [DOI] [PubMed] [Google Scholar]
- 98.Lyon RC, Mezzano V, Wright AT, Pfeiffer E, Chuang J, Banares K, Castaneda A, Ouyang K, Cui L, Contu R, Gu Y, Evans SM, Omens JH, Peterson KL, McCulloch AD, Sheikh F. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Human molecular genetics. 2014;23:1134–1150. doi: 10.1093/hmg/ddt508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Uzumcu A, Norgett EE, Dindar A, Uyguner O, Nisli K, Kayserili H, Sahin SE, Dupont E, Severs NJ, Leigh IM, Yuksel-Apak M, Kelsell DP, Wollnik B. Loss of desmoplakin isoform I causes early onset cardiomyopathy and heart failure in a Naxos-like syndrome. Journal of medical genetics. 2006;43:e5. doi: 10.1136/jmg.2005.032904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lombardi R, Marian AJ. Arrhythmogenic right ventricular cardiomyopathy is a disease of cardiac stem cells. Current opinion in cardiology. 2010;25:222–228. doi: 10.1097/HCO.0b013e3283376daf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, McKenna WJ, Calkins H, Saffitz JE. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. The New England journal of medicine. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 102.Lombardi R, da Graca Cabreira-Hansen M, Bell A, Fromm RR, Willerson JT, Marian AJ. Nuclear plakoglobin is essential for differentiation of cardiac progenitor cells to adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circulation research. 2011;109:1342–1353. doi: 10.1161/CIRCRESAHA.111.255075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen SN, Gurha P, Lombardi R, Ruggiero A, Willerson JT, Marian AJ. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circulation research. 2014;114:454–468. doi: 10.1161/CIRCRESAHA.114.302810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hariharan V, Asimaki A, Michaelson JE, Plovie E, MacRae CA, Saffitz JE, Huang H. Arrhythmogenic right ventricular cardiomyopathy mutations alter shear response without changes in cell-cell adhesion. Cardiovascular research. 2014;104:280–289. doi: 10.1093/cvr/cvu212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Asimaki A, Kapoor S, Plovie E, Karin Arndt A, Adams E, Liu Z, James CA, Judge DP, Calkins H, Churko J, Wu JC, MacRae CA, Kleber AG, Saffitz JE. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Science translational medicine. 2014;6:240ra274. doi: 10.1126/scitranslmed.3008008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kirchhof P, Fabritz L, Zwiener M, Witt H, Schafers M, Zellerhoff S, Paul M, Athai T, Hiller KH, Baba HA, Breithardt G, Ruiz P, Wichter T, Levkau B. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation. 2006;114:1799–1806. doi: 10.1161/CIRCULATIONAHA.106.624502. [DOI] [PubMed] [Google Scholar]
- 107.Fabritz L, Hoogendijk MG, Scicluna BP, van Amersfoorth SC, Fortmueller L, Wolf S, Laakmann S, Kreienkamp N, Piccini I, Breithardt G, Noppinger PR, Witt H, Ebnet K, Wichter T, Levkau B, Franke WW, Pieperhoff S, de Bakker JM, Coronel R, Kirchhof P. Load-reducing therapy prevents development of arrhythmogenic right ventricular cardiomyopathy in plakoglobin-deficient mice. Journal of the American College of Cardiology. 2011;57:740–750. doi: 10.1016/j.jacc.2010.09.046. [DOI] [PubMed] [Google Scholar]
- 108.Li D, Liu Y, Maruyama M, Zhu W, Chen H, Zhang W, Reuter S, Lin SF, Haneline LS, Field LJ, Chen PS, Shou W. Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Human molecular genetics. 2011;20:4582–4596. doi: 10.1093/hmg/ddr392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li J, Swope D, Raess N, Cheng L, Muller EJ, Radice GL. Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of {beta}-catenin signaling. Molecular and cellular biology. 2011;31:1134–1144. doi: 10.1128/MCB.01025-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Swope D, Cheng L, Gao E, Li J, Radice GL. Loss of cadherin-binding proteins beta-catenin and plakoglobin in the heart leads to gap junction remodeling and arrhythmogenesis. Molecular and cellular biology. 2012;32:1056–1067. doi: 10.1128/MCB.06188-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lodder EM, Rizzo S. Mouse models in arrhythmogenic right ventricular cardiomyopathy. Frontiers in physiology. 2012;3:221. doi: 10.3389/fphys.2012.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li Z, Colucci-Guyon E, Pincon-Raymond M, Mericskay M, Pournin S, Paulin D, Babinet C. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Developmental biology. 1996;175:362–366. doi: 10.1006/dbio.1996.0122. [DOI] [PubMed] [Google Scholar]
- 113.Thornell L, Carlsson L, Li Z, Mericskay M, Paulin D. Null mutation in the desmin gene gives rise to a cardiomyopathy. Journal of molecular and cellular cardiology. 1997;29:2107–2124. doi: 10.1006/jmcc.1997.0446. [DOI] [PubMed] [Google Scholar]
- 114.Milner DJ, Taffet GE, Wang X, Pham T, Tamura T, Hartley C, Gerdes AM, Capetanaki Y. The absence of desmin leads to cardiomyocyte hypertrophy and cardiac dilation with compromised systolic function. Journal of molecular and cellular cardiology. 1999;31:2063–2076. doi: 10.1006/jmcc.1999.1037. [DOI] [PubMed] [Google Scholar]
- 115.Goldfarb LG, Park KY, Cervenakova L, Gorokhova S, Lee HS, Vasconcelos O, Nagle JW, Semino-Mora C, Sivakumar K, Dalakas MC. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nature genetics. 1998;19:402–403. doi: 10.1038/1300. [DOI] [PubMed] [Google Scholar]
- 116.Klauke B, Kossmann S, Gaertner A, Brand K, Stork I, Brodehl A, Dieding M, Walhorn V, Anselmetti D, Gerdes D, Bohms B, Schulz U, Zu Knyphausen E, Vorgerd M, Gummert J, Milting H. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Human molecular genetics. 2010;19:4595–4607. doi: 10.1093/hmg/ddq387. [DOI] [PubMed] [Google Scholar]
- 117.Kreplak L, Bar H. Severe myopathy mutations modify the nanomechanics of desmin intermediate filaments. Journal of molecular biology. 2009;385:1043–1051. doi: 10.1016/j.jmb.2008.10.095. [DOI] [PubMed] [Google Scholar]
- 118.Danowski BA, Imanaka-Yoshida K, Sanger JM, Sanger JW. Costameres are sites of force transmission to the substratum in adult rat cardiomyocytes. The Journal of cell biology. 1992;118:1411–1420. doi: 10.1083/jcb.118.6.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Manso AM, Li R, Monkley SJ, Cruz NM, Ong S, Lao DH, Koshman YE, Gu Y, Peterson KL, Chen J, Abel ED, Samarel AM, Critchley DR, Ross RS. Talin1 has unique expression versus talin 2 in the heart and modifies the hypertrophic response to pressure overload. The Journal of biological chemistry. 2013;288:4252–4264. doi: 10.1074/jbc.M112.427484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sharp WW, Simpson DG, Borg TK, Samarel AM, Terracio L. Mechanical forces regulate focal adhesion and costamere assembly in cardiac myocytes. The American journal of physiology. 1997;273:H546–556. doi: 10.1152/ajpheart.1997.273.2.H546. [DOI] [PubMed] [Google Scholar]
- 121.Kostin S, Scholz D, Shimada T, Maeno Y, Mollnau H, Hein S, Schaper J. The internal and external protein scaffold of the T-tubular system in cardiomyocytes. Cell and tissue research. 1998;294:449–460. doi: 10.1007/s004410051196. [DOI] [PubMed] [Google Scholar]
- 122.Imanaka-Yoshida K, Enomoto-Iwamoto M, Yoshida T, Sakakura T. Vinculin, Talin, Integrin alpha6beta1 and laminin can serve as components of attachment complex mediating contraction force transmission from cardiomyocytes to extracellular matrix. Cell motility and the cytoskeleton. 1999;42:1–11. doi: 10.1002/(SICI)1097-0169(1999)42:1<1::AID-CM1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 123.Carter WG, Wayner EA, Bouchard TS, Kaur P. The role of integrins alpha 2 beta 1 and alpha 3 beta 1 in cell-cell and cell-substrate adhesion of human epidermal cells. The Journal of cell biology. 1990;110:1387–1404. doi: 10.1083/jcb.110.4.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Israeli-Rosenberg S, Manso AM, Okada H, Ross RS. Integrins and integrin-associated proteins in the cardiac myocyte. Circulation research. 2014;114:572–586. doi: 10.1161/CIRCRESAHA.114.301275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nawata J, Ohno I, Isoyama S, Suzuki J, Miura S, Ikeda J, Shirato K. Differential expression of alpha 1, alpha 3 and alpha 5 integrin subunits in acute and chronic stages of myocardial infarction in rats. Cardiovascular research. 1999;43:371–381. doi: 10.1016/s0008-6363(99)00117-0. [DOI] [PubMed] [Google Scholar]
- 126.Babbitt CJ, Shai SY, Harpf AE, Pham CG, Ross RS. Modulation of integrins and integrin signaling molecules in the pressure-loaded murine ventricle. Histochemistry and cell biology. 2002;118:431–439. doi: 10.1007/s00418-002-0476-1. [DOI] [PubMed] [Google Scholar]
- 127.Shai SY, Harpf AE, Babbitt CJ, Jordan MC, Fishbein MC, Chen J, Omura M, Leil TA, Becker KD, Jiang M, Smith DJ, Cherry SR, Loftus JC, Ross RS. Cardiac myocyte-specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circulation research. 2002;90:458–464. doi: 10.1161/hh0402.105790. [DOI] [PubMed] [Google Scholar]
- 128.Li R, Wu Y, Manso AM, Gu Y, Liao P, Israeli S, Yajima T, Nguyen U, Huang MS, Dalton ND, Peterson KL, Ross RS. beta1 integrin gene excision in the adult murine cardiac myocyte causes defective mechanical and signaling responses. The American journal of pathology. 2012;180:952–962. doi: 10.1016/j.ajpath.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Knoll R, Postel R, Wang J, Kratzner R, Hennecke G, Vacaru AM, Vakeel P, Schubert C, Murthy K, Rana BK, Kube D, Knoll G, Schafer K, Hayashi T, Holm T, Kimura A, Schork N, Toliat MR, Nurnberg P, Schultheiss HP, Schaper W, Schaper J, Bos E, Den Hertog J, van Eeden FJ, Peters PJ, Hasenfuss G, Chien KR, Bakkers J. Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation. 2007;116:515–525. doi: 10.1161/CIRCULATIONAHA.107.689984. [DOI] [PubMed] [Google Scholar]
- 130.Lu H, Fedak PW, Dai X, Du C, Zhou YQ, Henkelman M, Mongroo PS, Lau A, Yamabi H, Hinek A, Husain M, Hannigan G, Coles JG. Integrin-linked kinase expression is elevated in human cardiac hypertrophy and induces hypertrophy in transgenic mice. Circulation. 2006;114:2271–2279. doi: 10.1161/CIRCULATIONAHA.106.642330. [DOI] [PubMed] [Google Scholar]
- 131.White DE, Coutu P, Shi YF, Tardif JC, Nattel S, St Arnaud R, Dedhar S, Muller WJ. Targeted ablation of ILK from the murine heart results in dilated cardiomyopathy and spontaneous heart failure. Genes & development. 2006;20:2355–2360. doi: 10.1101/gad.1458906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Traister A, Li M, Aafaqi S, Lu M, Arab S, Radisic M, Gross G, Guido F, Sherret J, Verma S, Slorach C, Mertens L, Hui W, Roy A, Delgado-Olguin P, Hannigan G, Maynes JT, Coles JG. Integrin-linked kinase mediates force transduction in cardiomyocytes by modulating SERCA2a/PLN function. Nature communications. 2014;5:4533. doi: 10.1038/ncomms5533. [DOI] [PubMed] [Google Scholar]
- 133.Zemljic-Harpf A, Manso AM, Ross RS. Vinculin and talin: focus on the myocardium. Journal of investigative medicine : the official publication of the American Federation for Clinical Research. 2009;57:849–855. doi: 10.231/JIM.0b013e3181c5e074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xu W, Baribault H, Adamson ED. Vinculin knockout results in heart and brain defects during embryonic development. Development. 1998;125:327–337. doi: 10.1242/dev.125.2.327. [DOI] [PubMed] [Google Scholar]
- 135.Critchley DR. Biochemical and structural properties of the integrin-associated cytoskeletal protein talin. Annual review of biophysics. 2009;38:235–254. doi: 10.1146/annurev.biophys.050708.133744. [DOI] [PubMed] [Google Scholar]
- 136.Pham CG, Harpf AE, Keller RS, Vu HT, Shai SY, Loftus JC, Ross RS. Striated muscle-specific beta(1D)-integrin and FAK are involved in cardiac myocyte hypertrophic response pathway. American journal of physiology. Heart and circulatory physiology. 2000;279:H2916–2926. doi: 10.1152/ajpheart.2000.279.6.H2916. [DOI] [PubMed] [Google Scholar]
- 137.Kovacic-Milivojevic B, Roediger F, Almeida EA, Damsky CH, Gardner DG, Ilic D. Focal adhesion kinase and p130Cas mediate both sarcomeric organization and activation of genes associated with cardiac myocyte hypertrophy. Molecular biology of the cell. 2001;12:2290–2307. doi: 10.1091/mbc.12.8.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kovacic-Milivojevic B, Damsky CC, Gardner DG, Ilic D. Requirements for the localization of p130 Cas to Z-lines in cardiac myocytes. Cellular & molecular biology letters. 2002;7:323–329. [PubMed] [Google Scholar]
- 139.Durieux AC, D'Antona G, Desplanches D, Freyssenet D, Klossner S, Bottinelli R, Fluck M. Focal adhesion kinase is a load-dependent governor of the slow contractile and oxidative muscle phenotype. The Journal of physiology. 2009;587:3703–3717. doi: 10.1113/jphysiol.2009.171355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Israeli-Rosenberg S, Chen C, Li R, Deussen DN, Niesman IR, Okada H, Patel HH, Roth DM, Ross RS. Caveolin modulates integrin function and mechanical activation in the cardiomyocyte. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2014 doi: 10.1096/fj.13-243139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Head BP, Patel HH, Insel PA. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochimica et biophysica acta. 2014;1838:532–545. doi: 10.1016/j.bbamem.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chidlow JH, Jr., Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovascular research. 2010;86:219–225. doi: 10.1093/cvr/cvq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Insel PA, Head BP, Patel HH, Roth DM, Bundey RA, Swaney JS. Compartmentation of G-protein-coupled receptors and their signalling components in lipid rafts and caveolae. Biochemical Society transactions. 2005;33:1131–1134. doi: 10.1042/BST20051131. [DOI] [PubMed] [Google Scholar]
- 144.Ervasti JM, Campbell KP. Dystrophin-associated glycoproteins: their possible roles in the pathogenesis of Duchenne muscular dystrophy. Molecular and cell biology of human diseases series. 1993;3:139–166. doi: 10.1007/978-94-011-1528-5_6. [DOI] [PubMed] [Google Scholar]
- 145.Jung D, Yang B, Meyer J, Chamberlain JS, Campbell KP. Identification and characterization of the dystrophin anchoring site on beta-dystroglycan. The Journal of biological chemistry. 1995;270:27305–27310. doi: 10.1074/jbc.270.45.27305. [DOI] [PubMed] [Google Scholar]
- 146.Towbin JA, Hejtmancik JF, Brink P, Gelb B, Zhu XM, Chamberlain JS, McCabe ER, Swift M. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation. 1993;87:1854–1865. doi: 10.1161/01.cir.87.6.1854. [DOI] [PubMed] [Google Scholar]
- 147.Hoffman EP, Brown RH, Jr., Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 148.Elsherif L, Huang MS, Shai SY, Yang Y, Li RY, Chun J, Mekany MA, Chu AL, Kaufman SJ, Ross RS. Combined deficiency of dystrophin and beta1 integrin in the cardiac myocyte causes myocardial dysfunction, fibrosis and calcification. Circulation research. 2008;102:1109–1117. doi: 10.1161/CIRCRESAHA.108.173153. [DOI] [PubMed] [Google Scholar]
- 149.Kirchhof P, Fortmuller L, Waldeyer C, Breithardt G, Fabritz L. Drugs that interact with cardiac electro-mechanics: old and new targets for treatment. Progress in biophysics and molecular biology. 2008;97:497–512. doi: 10.1016/j.pbiomolbio.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 150.Yu H, van Berkel TJ, Biessen EA. Therapeutic potential of VIVIT, a selective peptide inhibitor of nuclear factor of activated T cells, in cardiovascular disorders. Cardiovascular drug reviews. 2007;25:175–187. doi: 10.1111/j.1527-3466.2007.00011.x. [DOI] [PubMed] [Google Scholar]
- 151.Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Frontiers in pharmacology. 2014;5:21. doi: 10.3389/fphar.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Molkentin JD. Calcineurin and beyond: cardiac hypertrophic signaling. Circulation research. 2000;87:731–738. doi: 10.1161/01.res.87.9.731. [DOI] [PubMed] [Google Scholar]
- 153.Sussman MA, Lim HW, Gude N, Taigen T, Olson EN, Robbins J, Colbert MC, Gualberto A, Wieczorek DF, Molkentin JD. Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science. 1998;281:1690–1693. doi: 10.1126/science.281.5383.1690. [DOI] [PubMed] [Google Scholar]
- 154.Ding B, Price RL, Borg TK, Weinberg EO, Halloran PF, Lorell BH. Pressure overload induces severe hypertrophy in mice treated with cyclosporine, an inhibitor of calcineurin. Circulation research. 1999;84:729–734. doi: 10.1161/01.res.84.6.729. [DOI] [PubMed] [Google Scholar]
- 155.Zhang W, Kowal RC, Rusnak F, Sikkink RA, Olson EN, Victor RG. Failure of calcineurin inhibitors to prevent pressure-overload left ventricular hypertrophy in rats. Circulation research. 1999;84:722–728. doi: 10.1161/01.res.84.6.722. [DOI] [PubMed] [Google Scholar]
- 156.Luo Z, Shyu KG, Gualberto A, Walsh K. Calcineurin inhibitors and cardiac hypertrophy. Nature medicine. 1998;4:1092–1093. doi: 10.1038/2578. [DOI] [PubMed] [Google Scholar]
- 157.Wang X, Culotta VC, Klee CB. Superoxide dismutase protects calcineurin from inactivation. Nature. 1996;383:434–437. doi: 10.1038/383434a0. [DOI] [PubMed] [Google Scholar]
- 158.Mervaala E, Muller DN, Park JK, Dechend R, Schmidt F, Fiebeler A, Bieringer M, Breu V, Ganten D, Haller H, Luft FC. Cyclosporin A protects against angiotensin II-induced end-organ damage in double transgenic rats harboring human renin and angiotensinogen genes. Hypertension. 2000;35:360–366. doi: 10.1161/01.hyp.35.1.360. [DOI] [PubMed] [Google Scholar]
- 159.Oie E, Bjornerheim R, Clausen OP, Attramadal H. Cyclosporin A inhibits cardiac hypertrophy and enhances cardiac dysfunction during postinfarction failure in rats. American journal of physiology. Heart and circulatory physiology. 2000;278:H2115–2123. doi: 10.1152/ajpheart.2000.278.6.H2115. [DOI] [PubMed] [Google Scholar]
- 160.Mende U, Kagen A, Cohen A, Aramburu J, Schoen FJ, Neer EJ. Transient cardiac expression of constitutively active Galphaq leads to hypertrophy and dilated cardiomyopathy by calcineurin-dependent and independent pathways. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:13893–13898. doi: 10.1073/pnas.95.23.13893. [DOI] [PMC free article] [PubMed] [Google Scholar]