

Abstract
The stimuli for neuronal cell death in neurodegenerative disorders are multi-factorial and may include genetic predisposition, environmental factors, cellular stressors such as oxidative stress and free radical production, bioenergy failure, glutamate-induced excitotoxicity, neuroinflammation, disruption of Ca2+-regulating systems, mitochondrial dysfunction and misfolded protein accumulation. Cellular stress disrupts functioning of the endoplasmic reticulum (ER), a critical organelle for protein quality control, leading to induction of the unfolded protein response (UPR). ER stress may contribute to neurodegeneration in a range of neurodegenerative disorders. This review summarizes the molecular events occurring during ER stress and the unfolded protein response and it specifically evaluates the evidence suggesting the ER stress response plays a role in neurodegenerative disorders.
Keywords: neurodegeneration, ER stress, UPR, apoptosis, autophagy, Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, prions disease
Introduction
Chronic neurodegenerative diseases are a group of progressive disorders characterized by gradual loss of neuronal function in distinct areas of the central nervous system, leading to impaired brain functioning [1–4]. They include Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and prion diseases [5]. Emerging evidence suggests that ER stress may play a pivotal role in the development or pathology of many neurodegenerative diseases.
ER stress and the UPR
Physiological or pathological processes that disturb protein folding in the ER cause ER stress. The cells initial and rapid response to ER stress is the activation of a set of pro-survival signalling pathways termed the UPR. Activation of the UPR causes a shutdown of global protein synthesis and activates mechanisms that allow the cell to deal with the accumulation of unfolded proteins. For example, it enhances the protein folding capacity by increasing the expression of ER chaperones and it up-regulates the degradation of misfolded proteins. This co-ordinated biochemical response to ER stress allows cells to deal with ER stress–however, if the stress is prolonged or excessive, apoptosis ensues.
In mammals, the three major ER stress sensors are IRE1 (inositol requiring 1; ERN1, endoplasmic reticulum-to-nucleus signalling 1), PERK [double-stranded RNA-activated protein kinase (PKR)-like ER kinase; PEK, pancreatic eukaryotic initiation factor 2α kinase; EIF2AK3] and ATF6 (activating transcription factor 6) [6]. IRE1 and PERK are type I transmembrane proteins with protein kinase activity, whereas ATF6 is a type II transmembrane protein encoding a transcription factor [7]. The ER-luminal domain of PERK, IRE1 and ATF6 interacts with the ER chaperone glucose-regulated protein 78 (GRP78); however, upon accumulation of unfolded proteins, GRP78 dissociates from these molecules, leading to their activation [7]. Activation of PERK, IRE1 and ATF6 initiates a network of intracellular signalling pathways during the UPR (Fig. 1).
Fig 1.
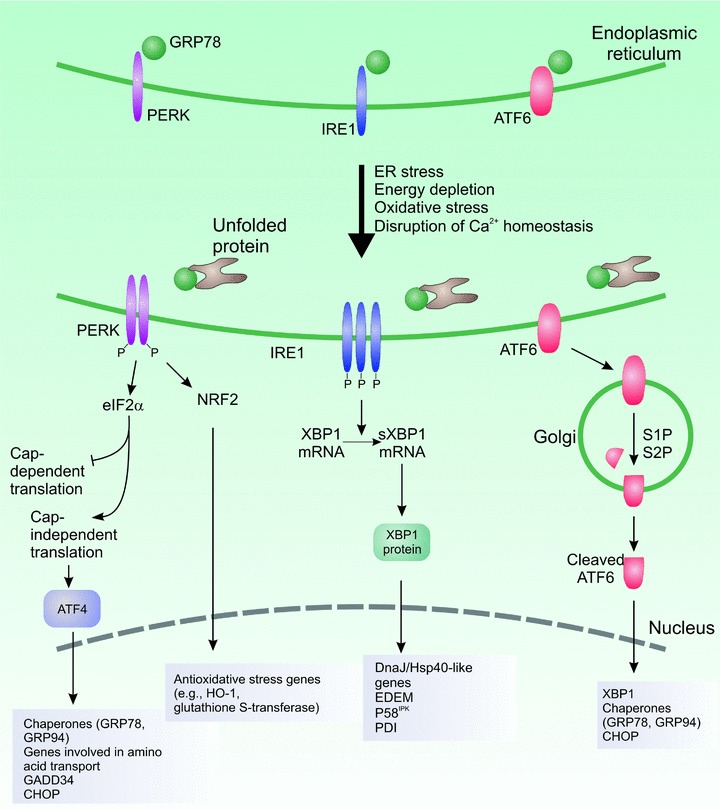
The unfolded protein response. ER stress such as presence of misfolded proteins leads to activation of the UPR sensors, PERK, IRE1 and ATF6. The individual arms have distinct roles but the overall aim is to relieve the stress and restore homeostasis. Activation of PERK leads to inhibition of cap-dependent translation but paradoxical increased translation of the potent transcription factor, AFT4. This leads to increased expression of genes involved in amino acid metabolism and transport and in redox chemistry through cap-independent translation. Activation of IRE1 is associated with non-conventional splicing of XBP1 which translocates to the nucleus to increase expression of components of the ERAD system and molecular chaperones. ATF6 translocates to the Golgi apparatus following activation where it is cleaved by site 1 and site 2 proteases. In the nucleus, ATF6 activates transcription of XBP1 and molecular chaperones such as GRP78 and GRP94.
The IRE1 axis: non-conventional splicing of XBP1 mRNA
IRE1 exists in two highly conserved isoforms: IRE1α and IRE1β. IRE1α is expressed ubiquitously, whereas the expression of IRE1β is limited to gut epithelial cells [8]. The cytoplasmic domain of IRE1 contains a serine/threonine kinase domain and a C-terminal endoribonuclease domain [9]. ER stress leads to dissociation of GRP78 from IRE1, resulting in autophosphorylation of IRE1α and activation of its RNAse activity. The downstream consequence of IRE1-mediated endoribonuclease activity is non-conventional splicing of XBP1 [9]. Activated IRE1 excises a 26-nucleotide sequence from XBP1 mRNA. This post-transcriptional mRNA processing is unique in that it does not use traditional mRNA splicing mechanisms. IRE1-mediated XBP1 mRNA splicing causes a shift in the reading frame, such that spliced XBP1 (XBP1s) mRNA encoding a 376 amino acid protein is produced. XBP1s possesses a potent transcriptional transactivation domain in its C-terminal region [9]. In addition, activated IRE1 can bind to tumour necrosis factor (TNF)-receptor-associated factor 2 (TRAF2), an adaptor protein that promotes activation of JUN N-terminal kinase (JNK) through apoptosis signal-regulating kinase 1 (ASK1) [10]. JNK activation results in enhanced autophagy [11]. This might allow cells to adapt to stress by initiating autophagy.
The IRE1 axis of the UPR is modulated by several interacting proteins (Fig. 2, inset) [12]. The pro-apoptotic B-cell lymphoma 2 (BCL-2) family members BCL-2–associated X protein (BAX) and BCL-2 antagonist/killer (BAK) augment both the kinase and endoribonuclease activities of IRE1. BAX and BAK form a protein complex with the cytosolic domain of IRE1, which requires their conserved BH1 and BH3 domains [12]. Protein tyrosine phosphatase 1B (PTP1B), which is present mostly in the ER, also influences IRE1 activity. The absence of PTP1B caused impaired XBP1 splicing, JNK phosphorylation and attenuated up-regulation of XBP1 target genes such as ER degradation enhancing α-mannosidase–like protein (EDEM) [12]. ASK1-interacting protein 1 (AIP1) was recently shown to specifically regulate and enhance IRE1 signalling. The pleckstrin homology (PH) domain of AIP1 is critical for IRE1 binding. AIP1-deficient cells displayed impaired IRE1 signalling after exposure to ER stress agents [12]. The IRE1 pathway is negatively modulated by the ER-located protein BAX inhibitor-1 (BI-1). BI-1 forms a protein complex with IRE1, and BI-1−/− cells show hyperactivation of IRE1 and a subsequent increase in XBP1 mRNA splicing [12]. We have recently shown that the stress-inducible form of HSP70 (HSP72) can interact with IRE1 and increase XBP1 mRNA splicing, thus modulating the expression of XBP1’s target genes causing attenuated apoptosis under ER stress conditions [13]. It is likely that additional proteins can interact with IRE1 to alter its activity.
Fig 2.

ER stress-induced pro-apoptotic signalling. ER stress leads to activation of JNK and induction of CHOP. JNK and CHOP alter the balance between pro-apoptotic and anti-apoptotic BCL-2 family members. CHOP causes up-regulation of BIM transcription and down-regulation of BCL-2 transcription. JNK phosphorylates and activates BIM. Consequently, BAX and BAK are activated resulting in release of calcium from the ER and opening of the PTP, loss of mitochondrial membrane potential with consequent release of cytochrome c which interacts with Apaf-1, pro-caspase-9 and cytochrome c to form the apoptosome.
The PERK axis: attenuation of translation
PERK is an ER-associated transmembrane serine/threonine protein kinase. Upon accumulation of unfolded proteins in the ER lumen, PERK dimerization and trans-autophosphorylation leads to activation of its kinase domain [14]. PERK-mediated phosphorylation of the α subunit of eukaryotic translation initiation factor 2 α (eIF2α) at Ser51 leads to translational attenuation [14]. Although phosphorylation of eIF2α inhibits general translation initiation, it paradoxically increases translation of activating transcription factor 4 (ATF4) [15] through a cap-independent process (Fig. 1). Recent studies suggest that in addition to eIF2α, the bZiP Cap ‘n’ Collar transcription factor, nuclear respiratory factor 2 (NRF2) is also a substrate of PERK [16]. NRF2 is retained in the cytoplasm through its association with the microtubule-associated protein KEAP1 (Kelch-like Ech-associated protein 1) [16]. Upon ER stress, PERK-mediated phosphorylation of NRF2 promotes its dissociation from KEAP1, leading to the nuclear accumulation of NRF2 [16]. NRF2 binds to the antioxidant response element to activate transcription of genes encoding detoxifying enzymes such as A1 and A2 subunits of glutathione S-transferase, NAD(P)H:quinone oxidoreductase, γ-glutamylcysteine synthetase, heme oxygenase-1 and UDP-glucoronosyl transferase [16].
The ATF6 axis: regulated proteolytic activation
In mammals, there are two alleles of ATF6, ATF6α (90 kD) and ATF6β (110 kD), both are synthesized in all cell types as ER transmembrane proteins. In unstressed cells, ATF6 is localized at the ER membrane and bound to GRP78 [17]. In response to ER stress, GRP78 dissociation permits trafficking of ATF6 to the Golgi complex, where ATF6 is sequentially cleaved by two proteases [17]. The site-1 protease cleaves ATF6 in the luminal domain. The N-terminal portion is subsequently cleaved by the site-2 protease. The processed forms of ATF6α and ATF6β translocate to the nucleus and bind to the ATF/cAMP response element and to the ER stress responsive element to activate target genes (Fig. 1). Studies of ATF6−/− cells have recently shown that ATF6 is responsible for transcriptional induction of a cohort of ER proteins which includes chaperones, folding enzymes and ER-associated degradation (ERAD) components [18]. A number of other bZIP transcription factors that localize to the ER have been identified including OASIS, CREBH, LUMAN/CREB3, CREB4 and BBF2H7 [12]. All of these ATF6-related bZIP factors are processed at the Golgi in a similar manner to ATF6. The function of each factor in the UPR is poorly characterized, although OASIS has been shown to be involved in bone formation [19] and BBF2H7 targets Sec23a, which governs protein transport from the ER to the Golgi. BBF2H7−/− mice exhibit chondroplasia and die due to immature development of the chest cavity [20].
The most salient feature of the UPR is to increase the transactivation function of an array of bZIP transcription factors such as ATF6, ATF4, ATF3, NRF2 and XBP1. Once activated, these transcription factors co-ordinate transcriptional induction of ER chaperones and genes involved in ERAD, to enhance the protein folding capacity of the cell and to decrease the unfolded protein load of the ER, respectively [6]. However, if the damage is too severe and ER homeostasis cannot be restored, apoptosis ensues [21].
ER stress–induced apoptosis
If the aforementioned pro-survival mechanisms fail to rescue the cell then apoptosis can occur. It is not clear at which point the switch between pro-survival and pro-apoptotic signalling occurs, nor are the mechanisms which underlie cell death fully elucidated. Overall, it is thought that the apoptotic signals generated from excessive activation of the UPR converge on the mitochondria resulting in opening of the permeability transmembrane pore (PTP) and loss of mitochondrial membrane potential (ΔΨm) with consequent release of pro-apoptotic factors, including cytochrome c (Fig. 2). In conjunction with apoptotic protease activating factor 1 (Apaf-1), pro-caspase-9 and cytochrome c form the apoptosome [22, 23]. The apoptosome is a complex consisting of adaptor proteins, which mediate the activation of initiator caspases at the onset of apoptosis. Specifically it processes pro-caspase-9 to its active form, which then activates downstream effector caspases including caspase-3, -7 and -6 [24], leading to apoptosis. Caspase-12 is an ER resident caspase; however, its role in ER stress–mediated apoptosis is subject to controversy as the human gene contains several inactivating mutations producing a truncated caspase-12 [25]. In addition, caspase-12 expression has no effect on cell viability in B16/B16 melanoma cells when treated with the ER stress inducer thapsigargin [26]. Caspase-4 has high homology to caspase-12 and its expression and cleavage is increased during ER stress [2]. Activation of caspase-4 has also been reported in response to disturbances in Ca2+ homeostasis as a Ca2+ chelator, EGTA, reduces the cleavage of caspase-4 in a concentration-dependent manner [27]. Caspase-2 is cleaved in response to excessive ER stress. Inhibition of caspase-2 confers resistance to ER stress–induced apoptosis [28].
It is currently understood that the cross-talk between the ER and mitochondria in apoptosis is predominantly mediated by the BCL-2 protein family. Experimental evidence supports a role for the BCL-2 family in ER stress–induced apoptosis. Overexpression of BCL-2 can protect cells from ER stress–induced cell death [29]. Also, many of the BCL-2 family members associate with the ER where they function to regulate Ca2+ homeostasis. BCL-2 family members are classified into anti-apoptotic members (BCL-2, BCL-XL and MCL-1), which have all four BH domains and pro-apoptotic BCL-2–homology domain 3 (BH3)-only proteins family members (BAD, BIM, BIK, BID, PUMA and NOXA) and multi-domain members BAX and BAK [23]. The balance between pro-apoptotic and anti-apoptotic BCL-2 family members is thought to play a critical role in regulating the transition from a protective to an apoptotic UPR response [23, 29]. Pro-apoptotic members BAX and BAK cause mitochondrial outer membrane permeabilization (MOMP) and formation of the PTP, in a process which ultimately leads to release of pro-apoptotic molecules such as second mitochondria-derived activator of caspases and cytochrome c. Pro-apoptotic family members sequester anti-apoptotic members such as BCL-2, thereby tipping the balance towards death [30]. Interaction between the anti-apoptotic BCL-2 proteins and pro-apoptotic proteins neutralizes the action of the pro-apoptotic molecules [23]. ER stress induces expression of the BH3-only proteins BIM, PUMA and NOXA and can also down-regulate expression of BCL-2 and cause cell death. Overexpression of PUMA induced apoptosis, whereas PUMA−/− cells were resistant to ER stress–induced apoptosis [31]. Numerous other studies exist which provide evidence for the involvement of BCL-2 family members in ER stress–induced cell death (reviewed in Ref. [32]). In summary, ER stress–induced cell death is thought to be primarily mediated via the BCL-2 family of proteins. However, the molecular switch signalling cells to change from a survival response to cell death is still not understood.
CHOP, also known as growth arrest and DNA damage-inducible gene 153 (GADD153), is a member of the C/EBP family that heterodimerizes with other members of the C/EBP transcription factor family. This 29 kD factor is expressed at low levels in unstressed cells and is strongly induced in response to ER stress [33]. It can be induced by all three arms of the UPR. It has been shown that mouse embryonic fibroblasts derived from CHOP−/− animals exhibited significantly less cell death when challenged with ER stress–inducing agents compared to wild type [33]. CHOP’s pro-apoptotic effects are linked to down-regulation of BCL-2 and enhanced production of reactive oxygen species (ROS) [34]. Caspase-11 has been reported to act downstream of CHOP to induce cell death by activating death effector caspases-1 and -3 [35]. CHOP can also bind to the promoter region of pro-apoptotic BIM, increasing its expression as well as transcriptionally down-regulating BCL-2 and in this way it induces cell death. Paradoxically, PERK−/− cells, which do not express CHOP are sensitive to ER stress–induced apoptosis, indicating redundancy in the system and CHOP-independent cell death mechanisms [36].
ER stress and autophagy
Autophagy, similar to ER stress has both pro-death and -survival functions. Accumulating evidence indicates that autophagy may confer neuroprotection by enhancing clearance of soluble and aggregated misfolded proteins and conversely, deregulation of autophagy may lead to neurodegeneration [37].
Synthesis of proteins in the ER is monitored by an elaborate quality control mechanism that allows only correctly folded proteins to be transported to their final destination, and misfolded or unassembled proteins are retained in the ER and subsequently degraded by ERAD. In the ERAD pathway, ER-resident chaperones recognize the misfolded proteins and ER reductases remove disulfide bonds in these proteins to facilitate retrograde transport to the cytosol where they are degraded by the proteasome [38]. To remove the aggregates of misfolded proteins that cannot be degraded by the ERAD, the UPR activates autophagy [11]. During ER stress–induced autophagy, portions of the ER and protein aggregates are engulfed in double-membrane structures called autophagosomes and delivered to lysosomes for degradation [39]. The initiation of autophagy requires activation of the ATG1/ULK induction complex [40]. This complex is essential for the formation of a small double membrane structure known as a phagophore, which will eventually mature into a double-membraned vacuole termed an autophagosome [41].
Mammalian target of rapamycin (mTOR) is a key kinase in the regulation of autophagy and activated mTOR inhibits autophagy. mTOR exists in two different complex forms, mTOR complex I (mTORC1) and mTOR complex II [42]. AMP-activated protein kinase (AMPK) negatively regulates mTOR via the tuberous sclerosis complex (TSC). AMPK is activated in response to many stresses such as hypoxia, starvation, heat shock, ischaemia and ER stress [43]. During ER stress, Ca2+ flux from the ER lumen to the cytosol can lead to the activation of Ca2+/calmodulin-dependent protein kinase kinase-β (CaMKK-β) [44]. CaMKK-β activates AMPK, in turn inhibiting mTOR and activating the ATG/ULK induction complex. The inhibition of mTOR during ER stress via AMPK is an important event during ER stress for the induction of autophagy [45].
Different conditions that induce ER stress lead to induction of autophagy [39]. Both the PERK/eIF2α and IRE1 arms of the UPR have been implicated in the regulation of autophagy [46, 47]. Treatment of cells with tunicamycin, thapsigargin or proteasome inhibitors induces autophagy in an IRE1-dependent manner [47]. The pro-autophagic actions of IRE1 seem to rely on the ability of IRE1 to activate JNK. JNK has been shown to regulate autophagy through BCL-2 phosphorylation, which disrupts its interaction with Beclin-1 [48]. Intriguingly, it has been recently shown that XBP1 ablation increases autophagy and protects cells from the toxicity induced by aggregates of the mutant form of enzyme superoxide dismutase 1 (mSOD1) in a model of ALS [49]. These observations suggest that the two distinct signalling pathways emanating from the IRE1 arm of the UPR can regulate autophagy. It has been shown that PERK signalling is also required for autophagy following expression of the Huntington’s disease-associated expanded polyglutamine repeats, which is a result of expansion of a CAG trinucleotide repeat and extension of a polyglutamine tract at the N-terminus of the encoded, ubiquitously expressed protein called huntingtin [50]. PERK-eIF2α–dependent ATG12 up-regulation is required for induction of autophagy in response to polyglutamine protein accumulation [51]. PERK-dependent transcription factors ATF4 and CHOP have been shown to induce transcriptional activation of MAP1LC3B and ATG5 during hypoxia [52]. Further, eIF2α-dependent up-regulation of the transcription factors p8, ATF4, CHOP and TRB3 is required for induction of autophagy [53]. However, the detailed molecular mechanism behind activation of autophagy during ER stress is not yet fully elucidated.
The UPR and neurodegenerative disorders
As outlined earlier, disruption of ER functioning is associated with the accumulation of misfolded proteins. Significantly, the accumulation of misfolded proteins is a characteristic occurrence in many neurodegenerative diseases [3, 54, 55] and neurodegenerative diseases are often described as protein conformational disorders or proteinopathies [1]. Normally, accumulation of misfolded proteins triggers the unfolded protein response, which determines the fate of the cell. In this section of the review we aim to explore the links emerging between factors that trigger the accumulation of misfolded proteins in neurodegenerative diseases, the cellular response to this stress and how this response influences neuronal cell fate. These are summarized in Table 1.
Table 1.
Evidence for disruption of UPR signalling in neurodegenerative disease
| Protein name | Role in UPR | Evidence of disturbed UPR | ||
|---|---|---|---|---|
| Alzheimer’s disease | Parkinson’s disease | ALS | ||
| IRE1 | ER stress sensor erine/threonine kinase: Autophosphorylates itself | PS1 mutants inhibit IRE1 signalling [73] | IRE1/ASK1/JNK pathway activated in PD Paraquat induces phosphorylation of IRE1 [92] | Phosphorylated IRE1 detected in spinal cord of ALS patients [96] |
| Endoribonuclease: splices XBP1 | ALS associated with ASK1-dependent cell death [99] | |||
| Recruits TRAF2-ASK1-JNK complex | Increased IRE1 expression in spinal cord of ALS patients [96] | |||
| XBP1 | Transcription factor increases expression of ERAD genes including EDEM | XBP1 can bind to the promotor the negative regulator of γ-secretase complex and to the promoter of genes involved in APP trafficking [75] | Exogenous expression of the active protein (XBP1s) has protective effects against cell death induced by MPP+ and proteasome inhibitors [120] | XBP1 ablation increases autophagy and protects cells from the toxicity induced by aggregates of mSOD1 in a model of ALS [49] |
| GRP78 | Chaperone protein which controls the activation of the UPR sensors IRE1, ATF6 and PERK | Reduction at mRNA level via inhibition of IRE1 signalling in mice homozygous for PS1 knock-in mutation [121] | ||
| PERK | Inhibits general protein translation | Increased phospho-PERK in AD patients [74] | Increased PERK expression in spinal cord of patients [96] | |
| Increases cap-independent transcripts, for example ATF4 | PS1 mutants inhibit PERK signalling [73] | |||
| Induces antioxidant response via NRF2 | ||||
| ATF4 | Transcription factor: increases expression of genes involved in stress response, redox reactions and CHOP | ATF4 leads to increase in parkin mRNA [87] | ||
| ATF6 | ER stress sensor | PS1 mutation inhibits activation of ATF6 [73] | ALS-associated mutation in VAPB inhibits translocation of ATF6 to Golgi [102] | |
| Transcription factor | ||||
| Increases expression of genes involved in protein folding, protein degradation and protein trafficking | Increased ATF6 expression in spinal cord of sporadic human ALS patients [96] | |||
| Increases XBP1 mRNA | ||||
Protein folding in vivo is an inefficient process and is aided by molecular chaperones, which increase folding efficiency. In addition, degradation systems such as ERAD, the endo-lysosomal pathway, the proteasome and autophagy rapidly remove misfolded proteins. Despite this, accumulation of misfolded proteins can still occur due to spontaneous errors during transcription and translation, genetic mutations, toxic compounds and cellular stresses [38]. In the native conformation, hydrophobic patches are usually buried within the interior of soluble proteins to maintain the lowest energy state [55]. Misfolded proteins have hydrophobic patches exposed allowing them to interact with other proteins leading to aggregation. In most cases, the native monomeric protein is mainly composed of α-helix, whereas the misfolded polymers are rich in β-sheet conformation [55]. Neurons are heavily reliant on the removal of misfolded proteins to maintain homeostasis [56] and accumulation of misfolded proteins is a characteristic feature of many neurodegenerative diseases including AD [57], PD [58], transmissible spongiform encephalopathy [59] and also acute neurodegenerative disorders such as traumatic brain injury [60] and cerebral ischaemia [61]. Because misfolded proteins trigger the UPR, this has prompted several groups to investigate the involvement of ER stress in neurodegenerative disorders. Here the links between misfolded proteins, ER stress and neuronal cell death in some of the major neurodegenerative diseases are reviewed (Table 1). Furthermore, links that have been established between ER stress and autophagy as well as between autophagy and neurodegeneration will also be outlined.
Alzheimer’s disease
AD is characterized by the presence of senile plaques with a core of extracellular β-amyloid protein and intracellular neurofibrillary tangles containing hyperphosphorylated tau [62]. These pathological hallmarks are accompanied by ballooning of neurites and neuronal loss. β-Amyloid is cleaved from its precursor protein, amyloid precursor protein (APP), through the action of β-secretase (BACE) and γ-secretase that results in the production of β-amyloid protein. γ-Secretase can produce β-amyloid of different lengths, most notably Aβ40 and Aβ42 [63]. The Aβ42 form of the protein is the most amyloidogenic and is prevalent in senile plaques. The extracellular deposition of senile plaques may precede the development of neurofibrillary tangles, and has been the subject of much investigative interest [64]. Although the evolution of senile plaques is closely linked to the development of the neurodegeneration and onset and progression of symptoms in AD, there is arguably not a direct causal relationship between β-amyloid deposition and neurodegeneration. The possibility that there may be a lethal intermediate in the process of transition between the soluble normal monomeric protein and the insoluble fibrils has been raised [65].
In particular, soluble oligomeric forms of β-amyloid have been suggested as such lethal intermediates [66–68]. Neurofibrillary tangles contain twisted pairs of helical filaments formed by the aggregation of hyperphosphorylated tau [69]. Hyperphosphorylation of tau impairs its ability to interact with cytoskeletal microfilaments, resulting in disorganization of the cytoskeleton.
The presenilin protein is a component of the γ-secretase complex, which is widely expressed in the ER and Golgi apparatus [70]. Presenilin mutations are linked with the majority of early onset forms of AD [69] with presenilin 1 (PS1) being more highly expressed than PS2, and mutations in the genes coding for a presenilin protein reduce the average age of onset of AD [71]. Evidence also demonstrates that PS1 mutations render cells more susceptible to apoptosis induced by a range of insults [72]. Presenilin is an integral membrane protein that is located primarily in ER and has been shown to influence the activity of two of the key ER stress sensors IRE1 and PERK (Fig. 3A). Presenilin mutations reduced phosphorylation of PERK and eIF2α, resulting in failure to attenuate protein synthesis causing protein accumulation in the ER [73]. However, studies on the brains of AD patients have revealed increased activation of PERK, therefore more work is required to delineate the contribution of PERK signalling in AD pathology [74]. Mutant PS1 is also known to bind, and inhibit IRE1, thereby reducing, or delaying, the transcription of ER chaperones such as GRP78 which has consistently been found to be down-regulated in AD [73]. In fact, the increased sensitivity of neurons to ER stress is attributed to the decreased levels of GRP78 mRNA. We have recently demonstrated that modulation of IRE1 activity and the resultant effect on XBP1 splicing can regulate cell fate [13]. Therefore, it is possible that mutant PS1, acting on IRE1, can reduce or delay splicing of XBP1, thus switching signalling to a pro-death response. Interestingly, genome wide approaches have identified a number of XBP1 target genes that are associated with AD. XBP1 can bind to the promotors of at least one key component of the γ-secretase complex, namely UBQLN1 that is a negative regulator of the γ-secretase complex. It has been suggested that UBQLN1 may control APP trafficking and thus the generation of Aβ. XBP1 can also bind to the promoter of genes involved in APP trafficking and processing as well as genes involved in AD pathogenesis, thereby implicating XBP1 in AD (Fig. 3A) [75]. It is possible therefore that reduced expression of XBP1 in AD influences the generation of Aβ and affects cell fate decisions. Examination of XBP1 splicing in AD models should reveal the role of XBP1 in AD and manipulation of spliced XBP1 levels in these models will indicate if XBP1 is a potential new therapeutic target for AD.
Fig 3.

Schematic diagrams showing links between ER stress, the UPR and neurodegenerative diseases: (A) Alzheimer’s disease, (B) Parkinson’s disease, (C) amyotrophic lateral sclerosis and (D) Prion disease. Refer to text for details.
Ca2+ homeostasis is important for proper functioning of ER chaperones and protein folding. Alterations in Ca2+ homeostasis lead to reduced chaperone activity, protein misfolding and initiation of the UPR. Aβ peptides have been shown to cause depletion of ER Ca2+ stores by triggering release of Ca2+ into the cytoplasm. In addition, PS1 mutations increase Aβ42 levels and have also been shown to impair ER Ca2+ homeostasis. Cells containing human PS1 mutations exhibit increased Ca2+ release from intracellular stores in response to stress in vitro[76]. Therefore, current studies suggest that there is a perturbed UPR response in AD and that presenilins may play a role in influencing this response (Fig. 3A) via a number of different mechanisms.
Autophagosomes and precursor autophagosomes (autophagic vacuoles) are abundant in swollen and dystrophic neurites from human AD brains, suggesting that the later stages of autophagy or the removal of autophagic vacuoles may be deregulated [77]. Autophagic vacuoles contain the proteases and substrates necessary to cleave APP, suggesting that the abnormal accumulation of autophagic vacuoles in affected neurons of the AD brain may act as a reservoir for the production of toxic aggregates and contribute to Aβ42 deposition [77].
Parkinson’s disease
PD is characterized by motor symptoms such as dyskinesia, muscle rigidity, postural instability and resting tremor. In addition, olfactory sensory loss and gastrointestinal disturbance are common in PD sufferers. Degeneration of the dopaminergic neurones of the nigrostriatal pathway and the presence of α-synuclein containing Lewy bodies and Lewy neurites are characteristic of the disease [78]. Within the Lewy plaques, diffuse deposits of misfolded α-synuclein form the core in association with other proteins, notably components of the ubiquitin–proteasome system [3]. PD is mostly sporadic with unknown causes, with monogenic forms representing 5–10%[79]. Genes associated with autosomal dominant PD include α-synuclein, ubiquitin carboxyl-terminal esterase L1 (UCHL1) and leucine-rich repeat kinase 2 (LRRK2); autosomal recessive PD genes include Parkinson protein 2 (PARK2/Parkin); PTEN-induced kinase 1 (PARK6/PINK1); PD (autosomal recessive, early onset) 7 (PARK7/DJ-1) and PD (autosomal recessive) 9; (PARK9/ATP13A2) [80].
α-Synuclein is expressed in synaptic vesicles and on cell membranes in nervous tissue. Post-translational modification of α-synuclein such as phosphorylation and nitrosylation can cause misfolding and subsequent deposition of the protein [78]. With misfolding, the tertiary structure of α-synuclein changes from a predominantly α-helix to β-sheet conformation. There is a suggestion that α-synuclein, although normally contained within cells, may be released upon cell death [78]. Uptake mechanisms may result in a domino-like spread of α-synuclein misfolding to neighbouring cells. Missense mutations in the gene coding for α-synuclein cause dominant familial PD. The A53T mutation is associated with UPR activation as evidenced by increased expression of CHOP and GRP78 and increased phosphorylation of eIF2α, suggesting the UPR is active in these cells (Fig. 3B) [80]. Inhibition of phosphorylation of eIF2α protected the A53T α-synuclein-overexpressing cells from cell death, suggesting that the activated UPR was shifting the balance towards apoptosis [80].
LRRK2 mutations also cause dominant familial PD, and may also account for a number of previously considered sporadic cases of PD [81, 82]. LRRK2 is a large multi-domain protein with kinase and GTPase activity, although its biological function has yet to be elucidated [82]. Mutations in LRRK2 cause impairment of protein degradation pathways with ageing [82]. This can lead to accumulation of α-synuclein and ubiquitinated proteins, impairment of the autophagy-lysosomal pathway, accumulation of oxidised proteins, an inflammatory response and increased apoptosis [82].
UCH-L1 mutations are linked to autosomal-dominant PD, however the mechanism by which it caused the disease is unclear, with conflicting evidence reported for its in vivo functions. Recent studies reveal a role for UCH-L1 in chaperone-mediated autophagy (CMA) and mutant UCH-L1 was shown to inhibit CMA-mediated removal of α-synuclein [83].
Several genetic mutations have been linked to the recessive form of PD including Parkin, an E3 ligase which forms part of the cascade reaction which targets misfolded proteins for degradation by the proteasome [84]. Mutations in Parkin result in loss of ubiquitin-protein ligase activity [85, 86], which can result in the accumulation of misfolded proteins within cells and may underpin the development of PD in people with this genetic mutation [85, 86]. Parkin has been shown to be up-regulated via AFT4, following ER stress and this event is associated with promotion of cell survival. A reciprocal relationship was also shown between JNK and Parkin. In addition, it was found that CHOP could down-regulate Parkin expression [87]. These findings suggest wild-type Parkin plays a protective role following ER stress by preventing stress-induced mitochondrial damage. However, prolonged stress will eventually lead to cell death with mutant Parkin potentially tipping the balance towards cell death [87]. More recently, the role of Parkin in mitophagy (selective degradation of mitochondria via the autophagy pathway) has been implicated in contributing to PD. Mitophagy is required for the removal of damaged mitochondria and to maintain cellular integrity. Parkin is recruited to depolarized mitochondria via PTEN-induced kinase-1 (PINK1) leading to mitophagy. It is proposed that Parkin then causes ubiquitination of voltage-dependent anion channel 1 leading to mitochondrial clearance [88]. Interestingly, disease-associated Parkin mutations disrupted mitophagy at distinct steps highlighting the importance of Parkin-mediated mitophagy in PD [88].
Mutations in PINK1 have also been implicated in the recessive form of PD. Apart from its role in recruiting Parkin to mitochondria for subsequent mitophagy, PINK1 has also been implicated in protein stability and the wild-type protein may protect cells from oxidative stress, mitochondrial dysfunction and apoptosis [89]. Mutations in DJ-1 are also linked to PD. DJ-1 has been suggested to act as an antioxidant or redox sensor protein and defective DJ-1 may predispose to oxidative stress and activation of the ER stress cascade [90]. Paraquat, an agricultural herbicide which is linked to sporadic PD, can induce expression of ER stress markers such as GRP78 and CHOP. Further investigation has revealed that paraquat activates IRE1/ASK1/JNK leading to apoptosis [91].
Therefore, significant evidence indicates that protein products of genes mutated in PD have a role in regulating protein stability such as α-synuclein (proteasome), Parkin (E3 ligase), DJ-1 (redox sensor) and PINK1 (protein stability) (Fig. 3B). In addition, drugs such as 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenylpyridinium (MPP+) which are used to develop animal models of PD, induce ER stress [92] (Fig. 3B). These studies therefore implicate protein quality control and the UPR as a key function that is disrupted in familial and sporadic PD leading to neuronal cell death. In addition, recent evidence points to the involvement of mitophagy influenced by the UPR playing a role in the development of PD.
Amyotrophic lateral sclerosis
ALS is a progressive fatal neurodegenerative disease that principally affects motor neurones. Most cases of ALS are sporadic, but 20% of sufferers have a familial form. Pathological mechanisms such as excitotoxicity, oxidative damage, mitochondrial dysfunction and defective axonal transport have all been implicated as causative factors in the apoptotic death of the motor neurones [93, 94]. Abnormal protein aggregation has also been reported in ALS. Bunina bodies, neurofilament cytoskeletal aggregation and deposition of aggregates of proteins such as ubiquitin, mutant superoxide dismutase 1 (mSOD1) and protein disulfide isomerase (PDI) are characteristics of the disease [95, 96].
Approximately 2% of ALS patients have a mutation in the SOD1 gene and transgenic rodents expressing the mSOD1 are the most commonly used model of study in ALS research [93]. mSOD1 misfolds, aggregates and induces the UPR in transgenic mSOD1 mice, causing apoptosis [97] and has been implicated in the development of ALS. The protein level of the ER chaperone, PDI, in particular was increased, and was shown to co-localize with aggregated mSOD1 protein [97]. In a longitudinal study using mSOD1 mouse models of ALS, vulnerable motoneurones were shown to be selectively prone to a UPR response and axonal degeneration, which could be attenuated or exacerbated by treatment protecting against or stimulating further ER stress, respectively [98]. As previously discussed, ERAD of misfolded proteins has been implicated in a range of neurodegenerative conditions, including ALS. Dysfunction of ERAD, causing ER stress has been shown to occur in mSOD1 containing motor neurones [99], through a mechanism involving Derilin-1, an ERAD-linked protein, subsequent ER stress–induced activation of the ASK1 pathway and ultimately apoptosis [99]. Specifically, mSOD1 was shown to interact with Derilin-1 causing dysregulation of ERAD leading to ER stress–induced ASK1 activation, apoptosis and disease progression (Fig. 3D).
Mutation of the vesicle-associated membrane protein/synaptobrevin-associated protein B (VAPB), which associates with intracellular membranes, such as ER, has been implicated in the development of late-onset ALS [100]. It has been proposed that development of ALS may occur due to the disruption of the UPR caused by the mutation in VAPB, resulting in accumulation of misfolded protein in the ER [101]. Native VAPB has been implicated in the UPR via the IRE1/XBP1[101], and ATF6 pathways [102], a function that is lost in mutants which contain abnormally highly ubiquitinated and misfolded VAPBP56S[101, 102]. It was found that both VAPB and VAPBP56S directly interact with ATF6 reducing its ability to promote transcription of XBP1 with the mutant having more potent activity as an ATF6 inhibitor [102].
In addition, mutations in the gene coding for the TAR DNA-binding protein (TARBP) also known as TDP-43 protein have recently been implicated in familial and sporadic ALS [103] and in frontotemporal lobar degeneration (FTLD) [104]. Abnormal and ubiquitinated TDP-43 has been identified as a key pathological hallmark of ALS and FTLD [105]. Mutations in a second, functionally-related DNA/RNA-binding protein, fused in sarcoma/translocation in liposarcoma (FUS/TLS) have also been implicated in familial ALS [106, 107] and result in abnormal cytoplasmic inclusions containing FUS protein in spinal cord motor neurones [107]. Despite the functional similarities of TDP-43 and FUS/TLS, whether they converge on the same pathogenic pathway remains to be clarified, although enhanced interaction between FUS/TLS and mutant TDP-43 has been reported [108].
More recently, evidence of induction of the UPR has been reported in sporadic human ALS [96]. UPR sensors IRE1, PERK and ATF6 show increased expression in spinal cord from sporadic human ALS patients [96]. However, conflicting evidence suggests that activation of the UPR may cause ER stress–induced apoptosis [96] or may actually be a neuroprotective response triggering increased levels of autophagy [12]. Therefore, it is likely that the cellular mechanisms influencing the balance between the protective response and cell death response of the UPR are crucial in these cells.
Prion diseases
Prion diseases such as new variant Creutzfeldt Jacob’s disease (CJD) are rare, fatal neurodegenerative diseases that are both inheritable and infectious. CJD causes a spongiform encephalopathy, reflecting neurodegeneration and an accumulation of abnormal protein aggregates in diffuse synaptic plaques containing prion protein and amyloid [84]. Prion protein (PrP) exists in at least two conformational states, the normal cellular form (PrPc) and an abnormal infective form (PrPSc). The abnormal PrPSc differs from the normal cellular form only in its three-dimensional conformation, having a higher β-sheet structure than the native protein [85]. PrPc undergoes post-translational modifications at the ER and the mature protein is expressed in lipid rafts in cell membranes, anchored to the membrane by a glycosylated phosphatidylinositol anchor [85]. Interaction of PrPSc with PrPc changes the normal host protein to the abnormal form, therefore amplifying the infectious agent. This amplification is difficult to achieve in vitro, but occurs readily in the presence of brain homogenate, suggesting the involvement of a co-factor in the conversion process [85]. The mature prion protein contains 209 amino acids, and the sequence of amino acids between residues 106–126 can trigger apoptosis [86]. The physiological function of PrPc is not well established. It has been linked to neuronal growth and survival [109] and cytoprotection against a number of cellular stresses including oxidative stress [110], TNF-induced cell death [111], BAX-induced cell death [112] and serum deprivation in a BAX-dependent manner [113]. Prion regulation of BAX is a plausible mechanistic link between the cytoprotective effects of prions against various stressors, although the details of this have yet to be elucidated. Possible mechanisms include prions signalling via an unknown pathway to prevent BAX oligomerization and translocation of prions may enhance the BAX–BCL-2 interaction [114]. A loss of the protective function of cellular prion protein due to genetic mutation could therefore lead to cell death in prion diseases. This hypothesis however, is likely to be overly simplistic as PrPc null mice are resistant to disease. In fact, these mice have no obvious phenotype and do not develop neurodegeneration [115]. It is now well-established that protein deposition and neurodegeneration occur in CJD and that both PrPc and PrPsc are needed to induce neurodegeneration associated with prion disease. However, there is a lack of a strong correlation between clinical symptoms and PrPSc levels, which has led to the suggestion that there could be a toxic intermediate produced during the conversion of PrPc monomers to the fibril PrPSc deposits [87].
PrPSc has been shown to result in accumulation of protein in intracellular compartments such as the ER and lead to ER stress–induced apoptosis (Fig. 3D) [116, 117]. The expression of ER stress markers such as GRP78, GRP94 and GRP58 is up-regulated in the cerebral cortex of CJD patients, suggesting an involvement of the ER stress response in the pathophysiology of this prion illness [12]. PrPSc purified from brains of scrapie-infected mice causes induction of the UPR and apoptosis [12]. This has also been shown in a cellular model where ER stress leads to misfolding of PrPc, which is more readily converted into PrPSc than wild-type protein thereby creating a cycle of ER stress.
Alteration of ER Ca2+ homeostasis and subsequent ER stress has also been implicated in the pathogenesis of prion diseases. PrPSc induces an increase of cystolic Ca2+ released mainly from the ER, which leads to loss of ΔΨm, increased ROS and cell death. This release of Ca2+ is dependent on the apoptosis triggering domain (residues 106–126) of prion protein. These effects could be inhibited by blocking release of Ca2+ from the ER or by addition of antioxidants [118]. Reticulon 3, an ER-localized protein that can cause the rapid depletion of ER Ca2+ stores, is up-regulated in the ME7/CV mouse scrapie model [12]. The resultant loss of Ca2+ from the ER would inhibit the activity of several ER chaperones and enzymes triggering ER stress. This provides a possible a link between PrpSc and ER stress. By inhibiting the ryanodine receptors and inositol trisphosphate receptor Ca2+ channels or by the addition of antioxidants, the effects on the mitochondrial membrane potential and cell death were significantly attenuated. These results strongly implicate the ER and specifically signalling between the ER and mitochondria in the neurodegeneration associated with prion protein infections [118].
Evidence that there is a mechanistic link between disease pathogenesis and cell death induced in both a PrPc and PrPSc-dependent manner was provided by Kang et al.[119]. PrPc has previously been shown to undergo pre-emptive quality control degradation [119]. During conditions of ER stress this mechanism is in place to prevent further accumulation of misfolded protein. However in prion diseases, excessive and prolonged ER stress, due to presence of PrPSc, leads to decreased translocation of PrPc to the ER. Using a PrP variant which cannot translocate to the ER, Kang et al. [119] showed development of PrP-associated neurodegeneration in both cell models and transgenic animals. The excessive degradation of PrP may exacerbate ER stress conditions as PrP loss leads to increased ROS levels. Also given the links between PrPc and BAX it is also possible that PrPSc infection indirectly leads to enhanced BAX oligomerization, translocation and eventually cell death.
Future perspectives
ER stress responses and in particular UPR is a fast emerging field of research. As reviewed here, there is evidence for the accumulation of misfolded proteins and also evidence for the involvement of the UPR in several human neurodegenerative conditions (Fig. 3). Dominant and recessive mutations predisposing to neurodegenerative conditions such as AD, PD and ALS have been identified. In particular, findings such as the effect of PS1 mutations on PERK and IRE1 functioning, causing a switch to pro-death signalling, implicates ER stress in the evolution of AD. Mutations in genes implicated in dominant and recessive forms of PD cause impairment of protein degradation pathways and apoptosis. Dysregulation of ERAD and induction of the UPR have been implicated in ALS pathophysiology and up-regulation of the expression of ER stress markers occurs in prion disease. However, although there is strong evidence for the occurrence of ER stress responses in neurodegenerative diseases, it is not clear how important ER stress and the UPR are in terms of the evolution of neurodegeneration. Is ER stress the cause or simply an effect of disease pathology? The elucidation of the exact role of ER stress in neurodegenerative disorders requires focused study on the individual arms of the UPR, namely PERK, IRE1 and ATF6. The first step will be to characterize how the UPR is affected using in vitro models of neurodegenerative disease, by assessing how knockdown of the individual arms of the UPR affects cell fate. The majority of work on the UPR has been performed with non-neuronal cells and therefore, it would be beneficial to explore this in neuronal cells given the tissue specific properties of the UPR. Animal models would facilitate determination of the phenotypic relevance of deregulated UPR functioning. If ER stress is found to cause neurodegeneration in these disorders, it raises the possibility for the development of a common neuroprotective therapy for the treatment of neurodegenerative conditions.
Acknowledgments
We thank Shane Deegan for his help and advice on the manuscript. D.K. is supported by a fellowship from NUIG College of Science. Our research is supported by Science Foundation Ireland (09/RFP/BIC2371, 09/RFP/BMT2153) and the Health Research Board (HRA/2009/59).
Conflict of interest
The authors declare there are no conflicts of interest.
References
- 1.Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci. 2003;4:49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- 2.Salminen A, Kauppinen A, Suuronen T, et al. ER stress in Alzheimer’s disease: a novel neuronal trigger for inflammation and Alzheimer’s pathology. J Neuroinflammation. 2009;6(41) doi: 10.1186/1742-2094-6-41. doi: 10.1186/1742-2094-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorman AM. Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling. J Cell Mol Med. 2008;12:2263–80. doi: 10.1111/j.1582-4934.2008.00402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams A, Jahreiss L, Sarkar S, et al. Aggregate-prone proteins are cleared from the cytosol by autophagy. Curr Top Dev Biol. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- 5.Gao H-M, Hong J-S. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 2008;8:357–65. doi: 10.1016/j.it.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 7.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 8.Boot-Handford R, Briggs M. The unfolded protein response and its relevance to connective tissue diseases. Cell Tissue Res. 2010;339:197–211. doi: 10.1007/s00441-009-0877-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calfon M, Zeng H, Urano F, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–6. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 10.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–6. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 11.Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–31. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol Cell. 2009;35:551–61. doi: 10.1016/j.molcel.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta SDA, Deegan S, Lisbona F, et al. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1a-XBP1 signaling through a physical interaction. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000410. : e1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–4. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 15.Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol. 2004;167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol. 2006;38:317–32. doi: 10.1016/j.biocel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 17.Haze K, Yoshida H, Yanagi H, et al. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–99. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu J, Rutkowski DT, Dubois M, et al. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13:351–64. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 19.Murakami T, Saito A, Hino S-I, et al. Signaling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat Cell Biol. 2009;11:1205–11. doi: 10.1038/ncb1963. [DOI] [PubMed] [Google Scholar]
- 20.Saito A, Hino S-I, Murakami T, et al. Regulation of endoplasmic reticulum stress response by a BBF2H7-mediated Sec23a pathway is essential for chondrogenesis. Nat Cell Biol. 2009;11:1197–204. doi: 10.1038/ncb1962. [DOI] [PubMed] [Google Scholar]
- 21.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–98. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olson M, Kornbluth S. Mitochondria in apoptosis and human disease. Curr Mol Med. 2001;1:91–122. doi: 10.2174/1566524013364239. [DOI] [PubMed] [Google Scholar]
- 23.Szegezdi E, MacDonald DC, Ni Chonghaile T, et al. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol. 2009;296:941–53. doi: 10.1152/ajpcell.00612.2008. [DOI] [PubMed] [Google Scholar]
- 24.Bao Q, Shi Y. Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ. 2006;14:56–65. doi: 10.1038/sj.cdd.4402028. [DOI] [PubMed] [Google Scholar]
- 25.Fribley A, Zhang K, Kaufman RJ. Regulation of apoptosis by the unfolded protein response. Methods Mol Biol. 2009;559:191–204. doi: 10.1007/978-1-60327-017-5_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalai M, Lamkanfi M, Denecker G, et al. Regulation of the expression and processing of caspase-12. J Cell Biol. 2003;162:457–67. doi: 10.1083/jcb.200303157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuzaki S, Hiratsuka T, Kuwahara R, et al. Caspase-4 is partially cleaved by calpain via the impairment of Ca2+ homeostasis under the ER stress. Neurochem Int. 2010;56:352–6. doi: 10.1016/j.neuint.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Upton JP, Austgen K, Nishino M, et al. Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol Cell Biol. 2008;28:3943–51. doi: 10.1128/MCB.00013-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta S, Cuffe L, Szegezdi E, et al. Mechanisms of ER stress-mediated mitochondrial membrane permeabilization. Int J Cell Biol. 2010 doi: 10.1155/2010/170215. doi: 10.1155/2010/170215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reimertz C, Kogel D, Rami A, et al. Gene expression during ER stress-induced apoptosis in neurons. J Cell Biol. 2003;162:587–97. doi: 10.1083/jcb.200305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szegezdi E, Logue SE, Gorman AM, et al. Mediators of endoplasmic stress-induced apoptosis. EMBO Rep. 2006;7:880–5. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zinszner H, Kuroda M, Wang X, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–95. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCullough KD, Martindale JL, Klotz L-O, et al. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–59. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fradejas N, Pastor MD, Burgos M, et al. Caspase-11 mediates ischemia-induced astrocyte death: involvement of endoplasmic reticulum stress and C/EBP homologous protein. J Neurosci Res. 2010;88:1094–105. doi: 10.1002/jnr.22280. [DOI] [PubMed] [Google Scholar]
- 36.Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 37.Nedelsky NB, Todd PK, Taylor JP. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim Biophys Acta. 2008;1782:691–9. doi: 10.1016/j.bbadis.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–57. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verfaillie T, Salazar M, Velasco G, et al. Linking ER stress to autophagy: potential implications for cancer therapy. Int J Cell Biol. 2010 doi: 10.1155/2010/930509. 930509. doi: 10.1155/2010/930509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–9. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 41.Ganley IG, Lam du H, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–94. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoyer-Hansen M, Jaattela M. AMP-activated protein kinase: a universal regulator of autophagy. Autophagy. 2007;3:381–3. doi: 10.4161/auto.4240. [DOI] [PubMed] [Google Scholar]
- 44.Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007;14:1576–82. doi: 10.1038/sj.cdd.4402200. [DOI] [PubMed] [Google Scholar]
- 45.Hoyer-Hansen M, Bastholm L, Szyniarowski P, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 46.Talloczy Z, Jiang W, Virgin HWT, et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci. 2002;99:190–5. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ding WX, Ni HM, Gao W, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–24. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei Y, Pattingre S, Sinha S, et al. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–88. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hetz C, Thielen P, Matus S, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294–306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shelbourne PF, Keller-McGandy C, Bi WL, et al. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet. 2007;16:1133–42. doi: 10.1093/hmg/ddm054. [DOI] [PubMed] [Google Scholar]
- 51.Kouroku Y, Fujita E, Tanida I, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–9. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 52.Rouschop KM, van den Beucken T, Dubois L, et al. The unfolded protein response protects human tumour cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127–41. doi: 10.1172/JCI40027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salazar M, Carracedo A, Salanueva IJ, et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest. 2009;119:1359–72. doi: 10.1172/JCI37948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winklhofer KF, Tatzelt J, Haass C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008;27:336–49. doi: 10.1038/sj.emboj.7601930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soto C, Estrada LD. Protein misfolding and neurodegeneration. Arch Neurol. 2008;65:184–9. doi: 10.1001/archneurol.2007.56. [DOI] [PubMed] [Google Scholar]
- 56.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 57.Tabira T, Chui DH, Kuroda S. Significance of the intracellular Ab42 accumulation in Alzheimer’s disease. Front Biosci. 2002;7:44–9. doi: 10.2741/tabira. [DOI] [PubMed] [Google Scholar]
- 58.Baba M, Nakajo S, Tu PH, et al. Aggregation of a-synuclein in lewy bodies of sporadic Parkinson’s disease and dementia with lewy bodies. Am J Pathol. 1998;152:879–84. [PMC free article] [PubMed] [Google Scholar]
- 59.Ferreiro E, Resende R, Costa R, et al. An endoplasmic-reticulum-specific apoptotic pathway is involved in prion and amyloid-beta peptides neurotoxicity. Neurobiol Dis. 2006;23:669–78. doi: 10.1016/j.nbd.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 60.Smith DH, Chen X-H, Iwata A, et al. Aβ accumulation in axons after traumatic brain injury in humans. J Neurosurg. 2003;98:1072–7. doi: 10.3171/jns.2003.98.5.1072. [DOI] [PubMed] [Google Scholar]
- 61.Hu BR, Martone ME, Jones YZ, et al. Protein aggregation after transient cerebral ischemia. J Neurosci. 2000;20:3191–9. doi: 10.1523/JNEUROSCI.20-09-03191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nordberg A. PET imaging of amyloid in Alzheimer’s disease. Lancet Neurol. 2004;3:519–27. doi: 10.1016/S1474-4422(04)00853-1. [DOI] [PubMed] [Google Scholar]
- 63.Lin JH, Li H, Yasumura D, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–9. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–5. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- 65.Irvine GB, El-Agnaf OM, Ganesh M, et al. Protein aggregation in the brain: the molecular basis for Alzheimer’s and Parkinson’s diseases. Mol Med. 2008;14:451–64. doi: 10.2119/2007-00100.Irvine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rochet JC. Novel therapeutic strategies for the treatment of protein-misfolding diseases. Expert Rev Mol Med. 2007;9:1–34. doi: 10.1017/S1462399407000385. [DOI] [PubMed] [Google Scholar]
- 67.Ishibashi K, Tomiyama T, Nishitsuji K, et al. Absence of synaptophysin near cortical neurons containing oligomer Abeta in Alzheimer’s disease brain. J Neurosci Res. 2006;84:632–6. doi: 10.1002/jnr.20952. [DOI] [PubMed] [Google Scholar]
- 68.Imaizumi K, Miyoshi K, Katayama T, et al. The unfolded protein response and Alzheimer’s disease. Biochim Biophys Acta. 2001;1536:85–96. doi: 10.1016/s0925-4439(01)00049-7. [DOI] [PubMed] [Google Scholar]
- 69.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 70.Cook DG, Sung JC, Golde TE, et al. Expression and analysis of presenilin 1 in a human neuronal system: localization in cell bodies and dendrites. Proc Natl Acad Sci USA. 1996;93:9223–8. doi: 10.1073/pnas.93.17.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 72.Qing Guo WF, Sopher BryceL, et al. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–6. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 73.Katayama T, Imaizumi K, Honda A, et al. Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer’s disease-linked presenilin-1 mutations. J Biol Chem. 2001;276:43446–54. doi: 10.1074/jbc.M104096200. [DOI] [PubMed] [Google Scholar]
- 74.Hoozemans JJM, van Haastert ES, Nijholt DAT, et al. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol. 2009;174:1241–51. doi: 10.2353/ajpath.2009.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Acosta-Alvear D, Zhou Y, Blais A, et al. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. 2007;27:53–66. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 76.Leissring MA, LaFerla FM, Callamaras N, et al. Subcellular mechanisms of presenilin-mediated enhancement of calcium signaling. Neurobiol Dis. 2001;8:469–78. doi: 10.1006/nbdi.2001.0382. [DOI] [PubMed] [Google Scholar]
- 77.Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer’s disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 78.Brundin P, Li JY, Holton JL, et al. Research in motion: the enigma of Parkinson’s disease pathology spread. Nat Rev Neurosci. 2008;9:741–5. doi: 10.1038/nrn2477. [DOI] [PubMed] [Google Scholar]
- 79.Lesage S, Brice A. Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18:R48–59. doi: 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- 80.Smith WW, Jiang H, Pei Z, et al. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet. 2005;14:3801–11. doi: 10.1093/hmg/ddi396. [DOI] [PubMed] [Google Scholar]
- 81.Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci. 2010;11:791–7. doi: 10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tong Y, Yamaguchi H, Giaime E, et al. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of a-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci USA. 2010;107:9879–84. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kabuta T, Furuta A, Aoki S, et al. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem. 2008;283:23731–8. doi: 10.1074/jbc.M801918200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shimura H, Hattori N, Kubo S-I, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–5. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 85.Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem. 2000;275:35661–4. doi: 10.1074/jbc.C000447200. [DOI] [PubMed] [Google Scholar]
- 86.Abrahante J, Daul A, Li M, et al. The Caenorhabditis elegans hunchback-like gene lin-57/hbl-1 controls developmental time and is regulated by microRNAs. Dev Cell. 2003;4:625–37. doi: 10.1016/s1534-5807(03)00127-8. [DOI] [PubMed] [Google Scholar]
- 87.Bouman L, Schlierf A, Lutz AK, et al. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011;18:769–82. doi: 10.1038/cdd.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–31. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 89.Gegg ME, Cooper JM, Schapira AH, et al. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS One. 2009;4:e4756. doi: 10.1371/journal.pone.0004756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yokota T, Sugawara K, Ito K, et al. Down regulation of DJ-1 enhances cell death by oxidative stress, ER stress, and proteasome inhibition. Biochem Biophys Res Commun. 2003;312:1342–8. doi: 10.1016/j.bbrc.2003.11.056. [DOI] [PubMed] [Google Scholar]
- 91.Yang W, Tiffany-Castiglioni E, Koh HC, et al. Paraquat activates the IRE1/ASK1/JNK cascade associated with apoptosis in human neuroblastoma SH-SY5Y cells. Toxi. Lett. 2009;191:203–10. doi: 10.1016/j.toxlet.2009.08.024. [DOI] [PubMed] [Google Scholar]
- 92.Holtz WA, O’Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem. 2003;278:19367–77. doi: 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- 93.Goodall EF, Morrison KE. Amyotrophic lateral sclerosis (motor neuron disease): proposed mechanisms and pathways to treatment. Expert Rev Mol Med. 2006;8:1–22. doi: 10.1017/S1462399406010854. [DOI] [PubMed] [Google Scholar]
- 94.Shi P, Wei Y, Zhang J, et al. Mitochondrial dysfunction is a converging point of multiple pathological pathways in amyotrophic lateral sclerosis. J Alzheimers Dis. 2010;20:S311–24. doi: 10.3233/JAD-2010-100366. [DOI] [PubMed] [Google Scholar]
- 95.Strong MJ, Kesavapany S, Pant HC. The pathobiology of amyotrophic lateral sclerosis: a proteinopathy. J Neuropathol Exp Neurol. 2005;64:649–64. doi: 10.1097/01.jnen.0000173889.71434.ea. [DOI] [PubMed] [Google Scholar]
- 96.Atkin JD, Farg MA, Walker AK, et al. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008;30:400–7. doi: 10.1016/j.nbd.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 97.Atkin JD, Farg MA, Turner BJ, et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281:30152–65. doi: 10.1074/jbc.M603393200. [DOI] [PubMed] [Google Scholar]
- 98.Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–36. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- 99.Nishitoh H, Kadowaki H, Nagai A, et al. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008;22:1451–64. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nishimura AL, Mitne-Neto M, Silva HCA, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–31. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kanekura K, Nishimoto I, Aiso S, et al. Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8) J Biol. Chem. 2006;281:30223–33. doi: 10.1074/jbc.M605049200. [DOI] [PubMed] [Google Scholar]
- 102.Gkogkas C, Middleton S, Kremer AM, et al. VAPB interacts with and modulates the activity of ATF6. Hum Mol Genet. 2008;17:1517–26. doi: 10.1093/hmg/ddn040. [DOI] [PubMed] [Google Scholar]
- 103.Pesiridis GS, Lee VM-Y, Trojanowski JQ. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet. 2009;18:R156–62. doi: 10.1093/hmg/ddp303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gitcho MA, Baloh RH, Chakraverty S, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63:535–8. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 106.Chio A, Schymick JC, Restagno G, et al. A two-stage genome-wide association study of sporadic amyotrophic lateral sclerosis. Hum Mol Genet. 2009;18:1524–32. doi: 10.1093/hmg/ddp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis Type 6. Science. 2009;323:1208–11. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ling S-C, Albuquerque CP, Han JS, et al. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci USA. 2010;107:13318–23. doi: 10.1073/pnas.1008227107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen S, Mangé A, Dong L, et al. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol Cell Neurosci. 2003;22:227–33. doi: 10.1016/s1044-7431(02)00014-3. [DOI] [PubMed] [Google Scholar]
- 110.Brown DR, Schulz-Schaeffer WJ, Schmidt B, et al. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp Neurol. 1997;146:104–12. doi: 10.1006/exnr.1997.6505. [DOI] [PubMed] [Google Scholar]
- 111.Diarra-Mehrpour M, Arrabal S, Jalil A, et al. Prion protein prevents human breast carcinoma cell line from tumour necrosis factor alpha-induced cell death. Cancer Res. 2004;64:719–27. doi: 10.1158/0008-5472.can-03-1735. [DOI] [PubMed] [Google Scholar]
- 112.Bounhar Y, Zhang Y, Goodyer CG, et al. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem. 2001;276:39145–9. doi: 10.1074/jbc.C100443200. [DOI] [PubMed] [Google Scholar]
- 113.Kuwahara C, Takeuchi AM, Nishimura T, et al. Prions prevent neuronal cell-line death. Nature. 1999;400:225–6. doi: 10.1038/22241. [DOI] [PubMed] [Google Scholar]
- 114.Roucou X, Giannopoulos PN, Zhang Y, et al. Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 2005;12:783–95. doi: 10.1038/sj.cdd.4401629. [DOI] [PubMed] [Google Scholar]
- 115.Büeler H, Aguzzi A, Sailer A, et al. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–47. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 116.Ivanova L, Barmada S, Kummer T, et al. Mutant prion proteins are partially retained in the endoplasmic reticulum. J Biol Chem. 2001;276:42409–21. doi: 10.1074/jbc.M106928200. [DOI] [PubMed] [Google Scholar]
- 117.Wang X, Shi Q, Xu K, et al. Familial CJD associated PrP mutants within transmembrane region induced Ctm-PrP retention in ER and triggered apoptosis by ER Stress in SH-SY5Y Cells. PLoS One. 2011;6:e14602. doi: 10.1371/journal.pone.0014602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ferreiro E, Oliveira CR, Pereira CMF. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol Dis. 2008;30:331–42. doi: 10.1016/j.nbd.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 119.Kang SW, Rane NS, Kim SJ, et al. Substrate-specific translocational attenuation during ER stress defines a pre-emptive quality control pathway. Cell. 2006;127:999–1013. doi: 10.1016/j.cell.2006.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sado M, Yamasaki Y, Iwanaga T, et al. Protective effect against Parkinson’s disease-related insults through the activation of XBP1. Brain Res. 2009;1257:16–24. doi: 10.1016/j.brainres.2008.11.104. [DOI] [PubMed] [Google Scholar]
- 121.Kudo T, Katayama T, Imaizumi K, et al. The unfolded protein response is involved in the pathology of Alzheimer’s disease. Ann N Y Acad Sci. 2002;977:349–55. doi: 10.1111/j.1749-6632.2002.tb04837.x. [DOI] [PubMed] [Google Scholar]