

Abstract
Novel therapies are urgently needed to prevent and treat tyrosine kinase inhibitor resistance in chronic myeloid leukaemia (CML). MLN8237 is a novel Aurora A kinase inhibitor under investigation in multiple phase I and II studies. Here we report that MLN8237 possessed equipotent activity against Ba/F3 cells and primary CML cells expressing unmutated and mutated forms of breakpoint cluster region-Abelson kinase (BCR-ABL). Notably, this agent retained high activity against the T315I and E255K BCR-ABL mutations, which confer the greatest degree of resistance to standard therapy. MLN8237 treatment disrupted cell cycle kinetics, induced apoptosis, caused a dose-dependent reduction in the expression of the large inhibitor of apoptosis protein Apollon, and produced a morphological phenotype consistent with Aurora A kinase inhibition. In contrast to other Aurora kinase inhibitors, MLN8237 did not significantly affect BCR-ABL activity. Moreover, inhibition of Aurora A with MLN8237 significantly increased the in vitro and in vivo efficacy of nilotinib. Targeted knockdown of Apollon sensitized CML cells to nilotinib-induced apoptosis, indicating that this is an important factor underlying MLN8237’s ability to increase the efficacy of nilotinib. Our collective data demonstrate that this combination strategy represents a novel therapeutic approach for refractory CML that has the potential to suppress the emergence of T315I mutated CML clones.
Keywords: apoptosis, cancer, BCR-ABL, CML, aurora kinase A, kinase inhibitors
Introduction
Imatinib targets the constitutively active BCR-ABL tyrosine kinase in chronic myeloid leukaemia (CML) and has become standard treatment based on excellent responses achieved in clinical trials [1–3]. However, imatinib resistance can occur through several mechanisms including BCR-ABL kinase domain mutations, amplification, overexpression and clonal evolution [4]. Successful strategies to overcome imatinib resistance include dose escalation or the use of second-generation BCR-ABL kinase inhibitors including nilotinib, dasatinib or bosutinib [5–7]. However, none of these agents are effective in CML cells harbouring the ‘gatekeeper’ T315I mutation at the base of the ATP binding pocket, which occurs in up to 20% of imatinib resistance cases.
The discovery that Aurora kinases were abnormally expressed in malignancies including leukaemia prompted the development of agents that inhibit their activity [8–10]. The pan-Aurora kinase inhibitors, MK-0457 and danusertib (PHA-739358) have shown pre-clinical and clinical activity against CML harbouring the BCR-ABL T315I mutation [10–13]. The anti-leukaemia efficacy of MK-0457 in CML was originally attributed to direct inhibition of BCR-ABL kinase activity [14, 15]. However, a recent study demonstrated that the efficacy of MK-0457 at clinically relevant doses in BCR-ABL+ cells was primarily due to inhibition of Aurora, rather than BCR-ABL, kinase activity [16]. The development of MK-0457 was ceased due to problems with cardiac toxicity observed in some patients during early phase clinical trials with the compound. In spite of this, the clinical responses achieved by MK-0457 in refractory CML patients have served to maintain interest in targeting Aurora kinases for CML therapy and a significant effort is currently being put forth to develop new agents that inhibit Aurora kinase activity and lack undesired cardiac side effects.
Aurora A kinase is a central mitotic regulator necessary for mitotic entry, mitotic spindle assembly and accurate chromosome separation [17–19]. The therapeutic potential of specifically targeting Aurora A kinase activity as an anticancer strategy has not been rigorously investigated because all of the agents previously designed to target Aurora kinases have significant off-target effects on other family members and/or BCR-ABL kinase activity. MLN8237 is a novel, highly selective ATP-competitive and reversible inhibitor of Aurora A kinase with an in vitro inhibition constant (Ki) of 0.43 nM [20]. It has a benzazepine core scaffold and is orally available. It is approximately 200-fold more selective for Aurora A kinase than the structurally related family member, Aurora B kinase. Moreover, MLN8237 is selective for Aurora A kinase when compared to most other kinases and receptors. It has shown broad-spectrum anticancer activity in preclinical models and is currently undergoing early clinical evaluation in solid tumours and heme-lymphatic malignancies.
We suggested that MLN8237-mediated inhibition of Aurora A kinase activity would abrogate the growth and survival of CML cells in a manner independent of BCR-ABL mutation status. Our results indicate that MLN8237 impairs growth, disrupts cell cycle kinetics, induces a cellular phenotype consistent with Aurora A kinase inhibition and triggers apoptosis in CML cell lines and primary human resistant CML cells including those bearing the drug resistance conferring T315I mutation. Furthermore, MLN8237 significantly increases the anticancer activity of the standard agent nilotinib through a mechanism involving down-regulation of the apoptotic and mitotic regulator, Apollon. Our collective data demonstrate that MLN8237 is a promising novel agent for the treatment of refractory CML that warrants further investigation.
Materials and methods
Cells and cell culture
Ba/F3 cells with wild-type (p210) BCR-ABL with and without stable shRNA p53 knockdown and T315I, E255K, H396P, Y253F, M351T and Q252H mutant forms of BCR-ABL, LAMA 84, K562 cells and imatinib-resistant K562 cells were maintained as previously described [21, 22]. Primary human CML cells were obtained from the peripheral blood of imatinib-resistant CML patients after obtaining informed consent in accordance with an approved institutional IRB protocol. Normal CD34+ bone marrow cells were purchased from Stem Cell Technologies (Vancouver, British Columbia, Canada).
Chemicals and reagents
Reagents were obtained from: MLN8237 (Millennium Pharmaceuticals, Cambridge, MA, USA), nilotinib (Novartis Oncology, East Hanover, NJ, USA), anti-actin, anti-active caspase-3, anti-phospho-Aurora A, anti-Aurora A phospho-BCR, phospho-CRKL, and CRKL antibodies (Cell Signaling, Beverly, MA, USA), anti-β tubulin (Sigma-Aldrich, St. Louis, MO, USA), anti-Apollon antibody (Bethyl Laboratories, Montgomery, TX, USA) and sheep antimouse-HRP and donkey anti-rabbit-HRP antibodies (Amersham, Piscataway, NJ, USA).
Enzyme assays
MLN8237 was screened against a subset of Invitrogen’s SelectScreen™ kinase panel at concentrations ranging between 10 and 0.00051 μM in 3-fold serial dilutions. The enzymes screened included ABL1, ABL1 E255K, ABL1 G250E, ABL1 T315I, ABL1 Y253F, ABL12 and Aurora A, each at the respective apparent ATP Km.
Analysis of cell cycle effects and apoptosis
Apoptosis was evaluated by propidium iodide/fluorescence activated cell sorting (PI/FACS) analysis of sub-G0/G1 DNA content as previously described [21, 23].
MTT assay
Cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously described [21].
Colony assays
K562, LAMA 84, normal CD34+ bone marrow or primary CML cells were treated for 24 hrs with the indicated concentrations of MLN8237 and nilotinib and then washed twice in PBS, seeded in Methocult methylcellulose containing medium (Stem Cell Technologies), incubated and scored as previously described [24].
Immunoblotting
CML cells were incubated with MLN8237, nilotinib or the combination for 24 hrs. Cells were then lysed and subjected to SDS-PAGE as previously described [24].
In vivo evaluation of MLN8237 and nilotinib
K562 and Ba/F3 cells were harvested, washed in PBS, and suspended in a mixture of HBSS and Matrigel (BD BioSciences, San Diego, CA, USA). An in vivo model of CML was generated by injecting K562 or Ba/F3 cells expressing wild-type (p210) or T315I mutant forms of BCR-ABL into the flanks of female nude mice. After tumour growth reached 150 mm3, mice were randomly assigned to receive MLN8237 20 mg/kg BID (n= 10), Nilotinib 50 mg/kg once daily (n= 10), vehicle control (n= 10) or both MLN8237 and Nilotinib (n= 10) for 14 days. Mice were monitored daily and tumour volumes were measured twice weekly. At the completion of the study, tumours were excised, formalin-fixed and paraffin-embedded for immunohistochemical analysis.
Immunohistochemistry
Paraffin-embedded tumour sections (4–6 μm thick) were mounted on slides and stained with haematoxylin and eosin as previously described [25].
Terminal deoxyribonucleotide-transferase–mediated dUTP nick-end labeling assay (TUNEL)
TUNEL staining and quantification were performed as previously described [25].
shRNA knockdown of p53
Ba/F3 p210 cells were infected with a retrovirus encoding a short hairpin RNA (shRNA) sequence specific for the knockdown of murine p53 or an empty vector control as previously described [26].
siRNA transfection
Apollon and Aurora kinase A SMARTpool or siCONTROL siRNA directed at luciferase (Dharmacon, Lafayette, CO, USA) were transfected into CML cells as previously described using the Nucleofector II according to the manufacturer’s instructions (Amaxa, Inc., Gaithersburg, MD, USA) [26]. Transfected cells were treated with the indicated concentrations of MLN8237 and nilotinib for 48 hrs. Drug-induced apoptosis was quantified by PI/FACS as described above.
Statistical analyses
Statistical significance of differences observed between samples was determined using the Tukey–Kramer comparison test or the Student’s t-test. Differences were considered significant in all experiments at P < 0.05.
Results
MLN8237 impairs growth, disrupts cell cycle kinetics and induces apoptosis in CML cell lines
Treatment with MLN8237 inhibited the in vitro growth and survival of the human K562 and LAMA 84 CML cell lines with IC50 values less than 100 nM (Fig. 1A). Considering that inhibition of the Aurora kinases results in mixed outcome, including polyploidy and G2/M growth arrest, we assessed the cell cycle distribution and apoptotic fraction (sub-G0/G1) of cells treated with MLN8237 by propidium iodide staining and flow cytometry. MLN8237 treatment disrupted cell cycle kinetics as demonstrated by the accumulation of cells in G2/M phase and cells with >4N DNA prior to the onset of apoptosis (sub-G0/G1) in a dose- and time-dependent manner (Fig. 1B, C).
Fig 1.
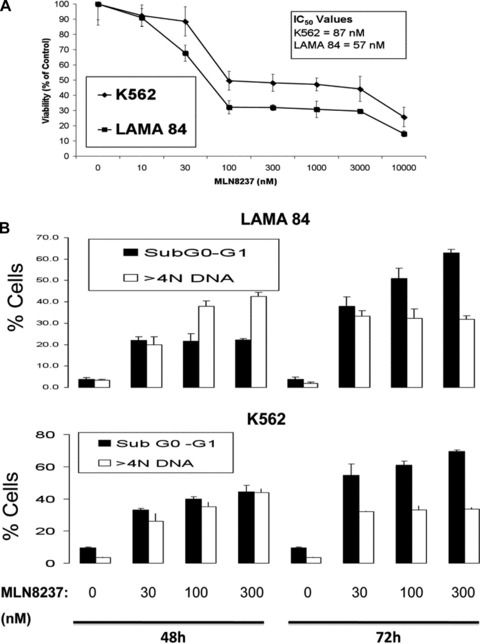
MLN8237 impairs growth, disrupts cell cycle kinetics and induces apoptosis in CML cell lines. (A) Effects of MLN8237 on the in vitro growth and survival of K562 and LAMA 84 human CML cell lines. Cells were treated with the indicated concentrations of MLN8237 for 96 hrs and viability was assessed by MTT assay. n= 3 ± S.D. (B)–(C) Time-dependent induction of DNA fragmentation. LAMA 84 and K562 cells were treated with 30, 100 or 300 nM MLN8237 for 48 hrs and 72 hrs. Percentages of cells with sub-G0-G1 DNA and >4N DNA were determined by PI/FACS. n= 3 ± S.D.
MLN8237 has in vitro and in vivo antiproliferative effects in imatinib-resistant cells and its activity is unaffected by impairment of p53 function
Loss or mutation of the tumour suppressor gene TP53 occurs in over 30% of cases of CML blast crisis and can impede the response to therapy [27–29]. We evaluated the potential impact of loss of p53 function on cellular sensitivity to MLN8237 by achieving stable shRNA-mediated p53 knockdown in Ba/F3 cells expressing p210 BCR-ABL. Cells were treated with the chemotherapeutic agent vincristine and immunoblotting analyses of the expression of p53 and its direct transcriptional target, p21, were conducted to confirm functional p53 knockdown. Impairment of p53 function did not significantly affect the anticancer activity of MLN8237, indicating that it may be an effective agent for patients with p53 defects (Fig. 2A). To investigate the potential impact of imatinib resistance on the efficacy of MLN8237, we treated Ba/F3 expressing unmutated BCR-ABL and the clinically relevant tyrosine kinase inhibitor resistant BCR-ABL mutants T315I, E255K, H396P, Y253F, M351T and Q252H as well as K562 cells that are sensitive and resistant to imatinib due to differential expression of unmutated BCR-ABL with this agent for 96 hrs. Notably, MLN8237 inhibited the viability of Ba/F3 cells expressing unmutated BCR-ABL and mutated BCR-ABL and imatinib-sensitive and –resistant K562 cells at similar concentrations (Fig. 2B) [22]. We next created an animal model of unmutated and T315I-mutated CML by injecting Ba/F3 cells expressing p210 or T315I-mutated BCR-ABL into the flanks of nude mice to investigate the in vivo efficacy of MLN8237 against CML cells bearing the T315I mutation. Consistent with our in vitro data, MLN8237 possessed equipotent in vivo activity against xenografts of Ba/F3 cells expressing unmutated and imatinib, nilotinib and dasatinib resistant T315I-mutated forms of BCR-ABL (Fig. 2C). We subsequently determined the anti-leukemic effects of MLN8237 against primary CML cells from three imatinib refractory patients, (one each from a patient in chronic phase CML, blast crisis CML and a patient in blast crisis harbouring the T315I mutation) and primary leukaemia cells from a patient with Philadelphia chromosome positive acute lymphocytic leukaemia. MLN8237 equally inhibited the viability of primary human CML cells from patients with unmutated and T315I-mutated BCR-ABL (Fig. 2D). As primary cells do not tend to actively proliferate in culture, higher concentrations of MLN8237 were needed to inhibit their viability compared to CML cell lines. Normal peripheral blood mononuclear cells cultured under the same conditions were less susceptible to the effects of MLN8237 compared to the primary CML cells, thus demonstrating the therapeutic selectivity of this agent (Fig. 2D). Collectively, these results suggest that the activity of MLN8237 in CML cells is unaffected by BCR-ABL mutational status or impairment of p53 function.
Fig 2.
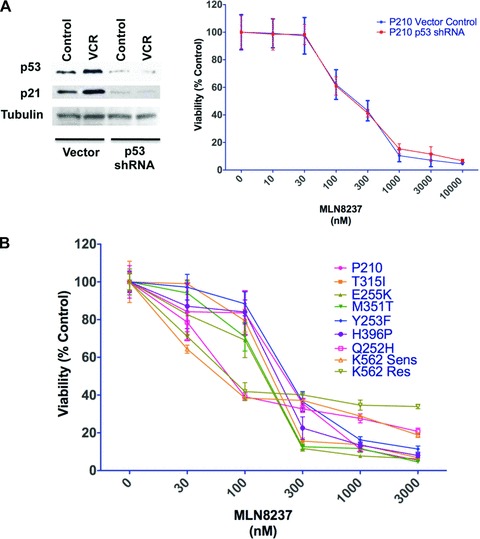

MLN8237 has in vitro and in vivo antiproliferative effects in Ba/F3 cells expressing unmutated and mutated BCR-ABL and its activity is unaffected by impairment of p53 function. (A) Left, Ba/F3 p210 BCR-ABL cells stably infected with p53 shRNA or vector control were treated with 100 nM vincristine (VCR) for 24 hrs and subjected to immunoblotting for p53 and p21 to confirm functional knockdown efficiency. Tubulin documented equal loading. Right, Ba/F3 p210 BCR-ABL cells stably infected with p53 shRNA or vector control were treated the indicated concentrations of MLN8237 for 96 hrs and viability was assessed by MTT assay. n= 3 ± S.D. (B) MLN8237 has activity in cells expressing unmutated and mutated BCR-ABL. Ba/F3 cells expressing p210 (unmutated) and T315I, E255K, H396P, Y253F, M351T and Q252H mutant forms of BCR-ABL and imatinib-sensitive and –resistant K562 cells were treated with the indicated concentrations of MLN8237 for 96 hrs and viability was assessed by MTT assay. n= 3 ± S.D. (C) In vivo efficacy of MLN8237. Immunodeficient mice bearing xenografts of P210 and T315I BCR-ABL expressing Ba/F3 cells were administered MLN8237 (20 mg/kg BID) daily or vehicle control. n= 10 ± S.D. (D) Activity of MLN8237 in primary CML cells and normal peripheral blood mononuclear cells. Cells from healthy donors or patients (4) with BCR-ABL+ leukaemia including 1 patient each with: unmutated BCR-ABL, T315I-mutated BCR-ABL, blast crisis CML and Ph+ ALL were treated with MLN8237 for 96 hrs and cell viability was assessed by MTT assay.
MLN8237 inhibits autophosphorylation of Aurora A without affecting BCR-ABL activity
We next determined the in vitro inhibitory effects of MLN8237 on Aurora A kinase. Exposure of cultured K562 cells to 30 nM MLN8237 reduced the phosphorylation of Aurora A kinase as demonstrated by reduced phosphorylation of Aurora A at Thr288 within its kinase activation loop without affecting the total levels of Aurora A (Fig. 3A). As mentioned earlier, there has been some controversy regarding whether the anti-leukemic activity of MK-0457, which is no longer being clinically developed, was due to inhibition of Aurora kinases, BCR-ABL or both. In contrast to MK-0457, MLN8237 is a potent inhibitor of the Aurora A enzyme (IC50= 2 nM) and demonstrated approximately 50–700-fold selectivity against various ABL isoform enzyme assays (Fig. 3B). To confirm that efficacious concentrations of MLN8237 do not significantly affect BCR-ABL activity, we evaluated its effect on BCR-ABL autophosphorylation and phosphorylation of the BCR-ABL direct substrate CRKL, which are accurate predictors of BCR-ABL kinase activity. Consistent with the in vitro enzyme assay data, immunoblotting analysis demonstrated that treatment of LAMA 84 and K562 cells with MLN8237 did not have a significant effect on the total levels of BCR-ABL or the levels of phospho-BCR-ABL at its Tyr177 autophosphorylation site (Fig. 3C). We conducted similar experiments to also confirm that the concentration of MLN8237 that we utilized for our experiments with primary patient cells did not significantly affect BCR-ABL kinase activity (Fig. 3D).
Fig 3.
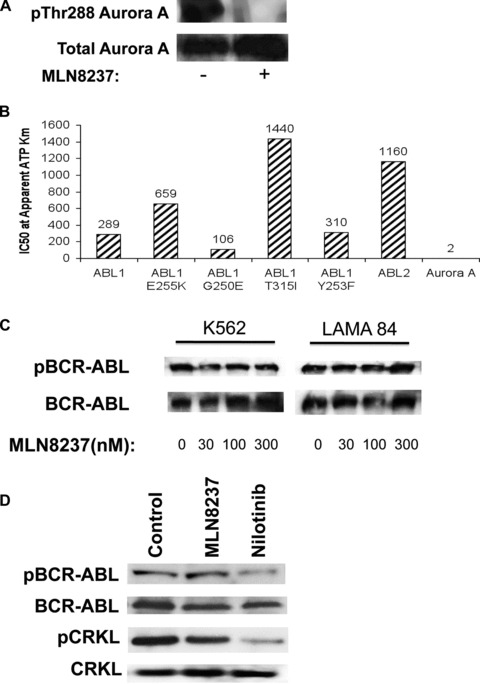
MLN8237 reduces autophosphorylation of Aurora A without significantly affecting BCR-ABL activity. (A) MLN8237 reduces Aurora kinase A phosphorylation. K562 cells were treated with 30 nM MLN8237 for 24 hrs. Protein lysates were subjected to SDS-PAGE, blotted, and probed with phospho-Aurora A (Thr288) and Aurora A antibodies. (B) Effects of MLN8237 on the activity of selected kinases. MLN8237 was screened against a kinase panel as described in ‘Materials and methods’. The ABL1 and the related ABL2 cytoplasmic tyrosine kinases share 89% sequence identity and have some overlapping functions, but are distinct in that ABL1 fuses with BCR to form the Philadelphia chromosome while ABL2 does not. (C) K562 and LAMA 84 cells were treated with MLN8237 for 24 hrs. Protein lysates were subjected to SDS-PAGE, blotted, and probed with phospho-BCR and c-Abl antibodies. (D) MLN8237 treatment does not significantly affect BCR-ABL kinase activity in primary CML cells. Primary CML cells obtained from a patient with unmutated BCR-ABL were treated with 10 μM MLN8237 for 24 hrs. BCR-ABL autophosphorylation and CRKL phosphorylation were assessed by immunoblotting. Nilotinib was used as a positive control for BCR-ABL inhibition.
Co-treatment with MLN8237 and nilotinib results in significantly greater apoptosis, growth inhibition and reduction in clonogenic survival than what is achieved by either agent alone
Patients with advanced stage CML have been suggested to benefit from non-cross-resistant combinations of tyrosine kinase inhibitors and agents effective against CML cells harbouring the T315I and E255K mutations [30, 31]. Considering that MLN8237 is active against cells expressing the E255K and T315I mutations, we investigated whether MLN8237 augmented the activity of nilotinib, an FDA-approved BCR-ABL inhibitor that is used in CML therapy. LAMA 84 and K562 cells were treated with 30 nM MLN8237, 10 nM nilotinib or the combination for 48 hrs. Percentages of cells with sub-G0-G1 DNA were quantified by PI/FACS. Co-treatment with MLN8237 and nilotinib resulted in significantly greater levels of apoptosis as determined by accumulation of sub-G0-G1 cells (Fig. 4A). Immunoblotting analysis showed that the combination of these two agents induced mitochondrial-dependent apoptosis as demonstrated by processing of caspases-9 and -3 to their active forms (Fig. 4B). The cytotoxic effects of the combination were also assessed by MTT assay, which demonstrated that inhibition of growth and survival were significantly increased by combination treatment (Fig. 4C). Finally, clonogenic assays were performed to evaluate the prolonged in vitro effects of MLN8237 and nilotinib on the growth and survival of normal CD34+ bone marrow cells, primary CML cells from patients in blast crisis, LAMA 84 and K562 cells (Fig. 4D). As expected, MLN8237 enhanced the ability of nilotinib to inhibit the clonogenic survival of CML cells, but had little effect on normal CD34+ cells. Collectively, these data suggest that MLN8237 significantly and selectively enhances the anti-leukaemia activity of nilotinib in human CML cells.
Fig 4.
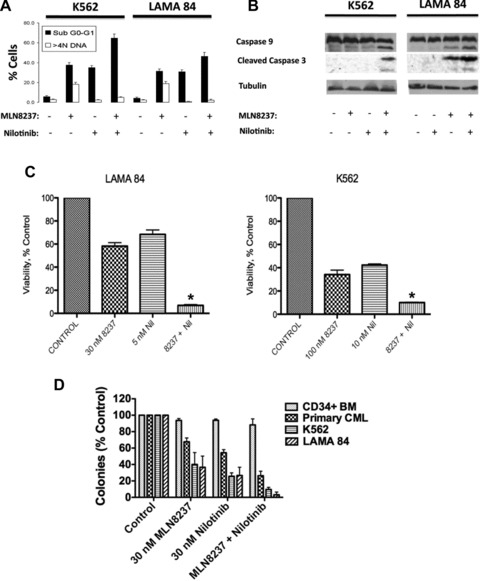
MLN8237 significantly increases the efficacy of nilotinib. (A) MLN8237 potentiates the pro-apoptotic effects of nilotinib. LAMA 84 and K562 cells were treated with 30 nM MLN8237, 10 nM nilotinib or the combination for 48 hrs. Percentages of cells with sub-G0-G1 DNA were determined by PI/FACS. n= 3 ± S.D. (B) The combination of MLN8237 and nilotinib induces mitochondrial-dependent apoptosis. K562 and LAMA 84 cells were treated with 100 nM MLN8237, 30 nM nilotinib or both for 24 hrs. Protein lysates were subjected to SDS-PAGE, blotted, and probed with active caspase-3 and caspase-9 antibodies. Anti β-tubulin was used as a loading control. (C) Co-treatment with MLN8237 and nilotinib results in significantly greater growth inhibition and reduction in survival than that achieved by either agent alone. Cells were treated with the indicated concentrations of MLN8237 for 96 hrs and viability was assessed by MTT assay. Error bars indicate the S.D. *P < 0.05. (D) Effects of MLN8237 and nilotinib on clonogenic survival. CD34+ normal bone marrow (n= 3), primary CML from patients in blast crisis (n= 3), K562 and LAMA 84 cells were treated with MLN8237, nilotinib or both drugs for 24 hrs. Cells were plated and scored as described in the ‘Materials and methods’.
MLN8237 cooperates with nilotinib to reduce tumour burden in K562 xenografts
K562 xenograft studies were carried out to investigate the in vivo therapeutic potential of the combination of MLN8237 and nilotinib. Both agents had substantial effects on tumour burden and the combination resulted in significantly greater tumour growth inhibition than what was achieved by either agent alone (Fig. 5A). Furthermore, the combination was well tolerated and only a modest, statistically insignificant loss in body weight was observed in the treated groups (Fig. 5A). Notably, the single agent in vivo effects of MLN8237 were more impressive in this K562 experiment than what we observed in our studies with the murine Ba/F3 models (Fig. 2C). Given that human and murine Aurora A genes share 79% sequence homology and that MLN8237 was specifically designed to target human Aurora A kinase activity, it is possible that species-specific differences in the potency of MLN8237-mediated kinase inhibition could have contributed to this phenomenon.
Fig 5.

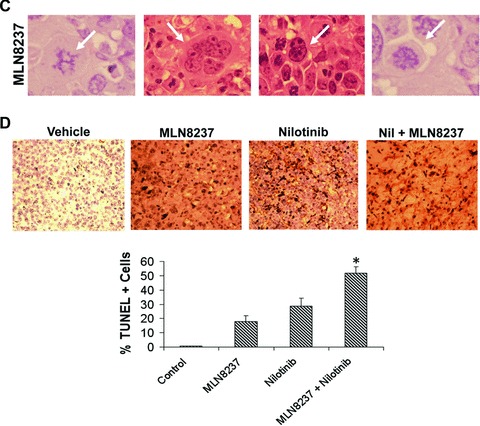
In vivo efficacy and tolerability of MLN8237 and nilotinib. (A) K562 cells were injected into the flanks of nude mice. Vehicle, MLN8237, nilotinib or both were administered for 14 days. n= 10 ± S.D. *P= 0.00028. (B) Immunohistochemistry. Tumours were stained with haematoxylin and eosin as described in ‘Materials and methods’. Representative images are shown from each treatment group. (C) Treatment with MLN8237 leads to a morphological phenotype consistent with Aurora A kinase inhibition. Tumours were stained with haematoxylin and eosin. Representative images from the MLN8237 treatment group are shown. Arrows indicate the following: an elevated number of cells with chromatin bridging (far left), mitotic slippage (second from left), cells with internuclear bridging (second from right) and monopolar mitotic spindles (far right). (D) Quantification of TUNEL+ cells. Positive cells were scored manually under 20× magnification. Mean ± S.D., n= 5. *P < 0.05.
Haematoxylin and eosin staining was used to visualize the architecture of tumours from each treatment group and revealed substantial differences in the morphology of single agent and combination-treated tumours. In particular, the tumours treated with the combination of MLN8237 and nilotinib displayed evidence of stromal disruption and high levels of cell death with very few intact CML cells remaining (Fig. 5B). This suggests that remaining tumours from combination-treated mice were largely comprised Matrigel and non-viable cells/tissue and also highlights the potential therapeutic benefit provided by the combination over single agent treatments.
Treatment with MLN8237 in vivo leads to a morphological phenotype consistent with inhibition of Aurora A kinase
Aurora A inhibition causes defects in centrosome segregation, spindle pole organization and chromosome congression. This can ultimately lead to tumour cell death via the development of deleterious aneuploidy [19]. Consistent with our in vitro data, the number of multi-nucleated cells visible in the MLN8237-treated tumours stained with haematoxylin and eosin was increased as compared with vehicle control indicating that MLN8237 causes cells to exit mitosis without completing cytokinesis, a process known as mitotic slippage (Fig. 5C, second from the left). Although this outcome is primarily associated with inhibition of Aurora B, cytokinesis failure can occur upon inhibition of Aurora A as well [18]. Other functional consequences of Aurora A inhibition that were prominent in the MLN8237-treated tumours (Fig. 5C) included an elevated number of cells with chromatin bridging (far left), mitotic slippage (second from left), cells with internuclear bridging (second from right) and monopolar mitotic spindles (far right). Taken together, these findings indicate that treatment with MLN8237 results in morphological changes in CML cells that are consistent with Aurora A kinase inhibition and indicative of deleterious aneuploidy.
MLN8237 augments the in vivo pro-apoptotic effects of nilotinib
TUNEL assays were carried out on xenograft tumour sections after completion of treatment to assess the degree of apoptosis induced by MLN8237 and nilotinib in vivo. The percentage of TUNEL+ cells was significantly greater in the combination group compared to treatment with either agent alone indicating that the two agents cooperate to provoke apoptosis in vivo (Fig. 5D).
Inhibition of Aurora A activity leads to reduced expression of the IAP and mitotic regulator Apollon and this effect sensitizes cells to nilotinib-induced apoptosis
Apollon (also known as BRUCE or BIRC6, baculovirus inhibitor of apoptosis protein (IAP) repeats (BIR)-containing protein 6) is a very large (528 kD) IAP that can be distinguished from other IAP family members by the presence of its ubiquitin conjugating enzyme (UCE) domains, which are not contained within any other IAPs [32]. The UCE domains within Apollon allow it to reduce the pro-apoptotic potential of cells by targeting Smac and caspase-9 for proteasomal degradation in addition to directly binding to caspases and other pro-apoptotic molecules via its BIR domain to prevent apoptosis in a manner similar to other IAPs [33, 34]. Apollon is overexpressed in leukaemia and other cancers and has been linked with resistance to chemotherapy [32, 35]. A recent investigation demonstrated that Apollon also has important functions during mitosis. It coordinates multiple events in cytokinesis and moves to the midbody ring during cell division where it serves as a platform for the membrane delivery machinery and mitotic regulators including the Aurora kinases [36]. Apollon depletion causes defective abscission and cytokinesis-associated apoptosis [36].
Considering that Apollon is associated with Aurora kinases and has roles in cell division and inhibition of apoptosis, we investigated whether abrogation of Aurora A kinase activity would affect Apollon expression. Immunoblotting analysis showed that MLN8237 treatment resulted in a dose-dependent reduction in the expression of Apollon and increased expression of the pro-apoptotic Apollon substrate, Smac (Fig. 6A). To confirm that this reduction in Apollon expression is a direct consequence of Aurora A kinase inhibition, we used siRNA to knockdown Aurora A expression. This led to a very significant reduction in the levels of Apollon (Fig. 6B). This effect does not appear to be a general feature of mitotic disruption as treatment with nocodazole and vincristine did not significantly decrease Apollon expression and therefore rather suggests a link between Aurora A activity and Apollon expression (Fig. 6C).
Fig 6.
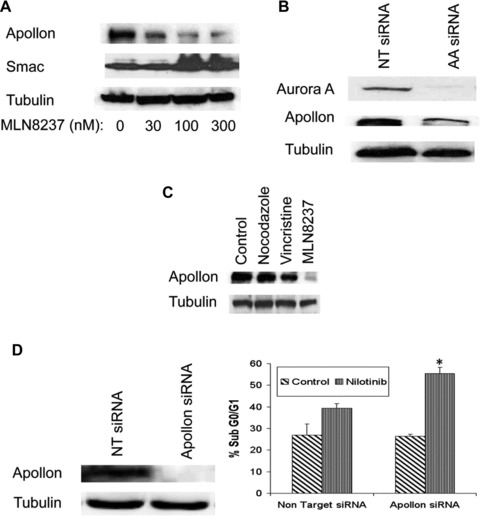
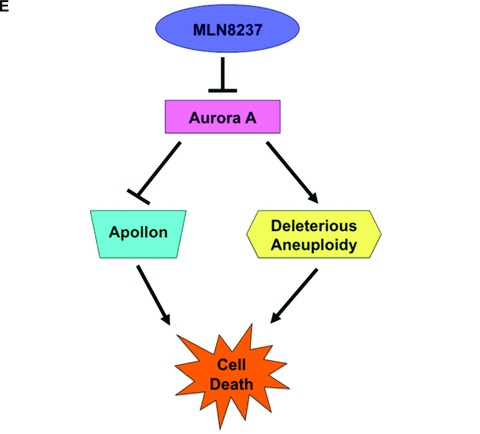
Targeting Apollon expression sensitizes CML cells to nilotinib-induced apoptosis. (A) MLN8237 treatment results in a dose-dependent reduction in the large IAP, Apollon and increased expression of its substrate, Smac. LAMA 84 cells were treated with MLN8237 for 24 hrs. Protein lysates were subjected to SDS-PAGE, blotted, and probed with Apollon and Smac antibodies. Tubulin documented equal loading. (B) Aurora A SMARTpool or siCONTROL siRNA directed at luciferase were transfected into LAMA 84 cells using the Nucleofector II. (C) General disruption of mitosis does not significantly affect Apollon expression. LAMA 84 cells were treated with nocodazole, vincristine or MLN8237 for 24 hrs. Protein lysates were subjected to SDS-PAGE, blotted, and probed with an Apollon antibody. Tubulin documented equal loading. (D) Apollon SMARTpool or siCONTROL siRNA directed at luciferase were transfected into LAMA 84 cells using the Nucleofector II. Tubulin was used as a loading control. LAMA 84 cells transfected with Apollon-targeted siRNA and non-targeted siRNA were treated with nilotinib for 48 hrs and the percentage of apoptotic cells were determined by PI/FACS analysis. n= 3 ± S.D., *P < 0.05. (E) Schematic depicting the multiple anti-leukaemia properties of the MLN8237. MLN8237 inhibits Aurora A kinase leading to deleterious aneuploidy, inhibition of the IAP, Apollon and cell death.
To determine whether inhibition of Apollon by MLN8237 significantly contributes to its ability to sensitize CML cells to nilotinib, we knocked down Apollon expression in LAMA 84 cells using siRNA (Fig. 6D). Nilotinib induced significantly greater levels of apoptosis in LAMA 84 cells treated with Apollon-targeted siRNA compared to non-targeted controls (Fig. 6D). These data suggest that inhibition of Apollon expression caused by MLN8237 treatment sensitizes CML cells to nilotinib-induced apoptosis and provides a rationale for the combination of these two agents in CML.
Discussion
Resistance to tyrosine kinase inhibitor therapy in CML continues to be a significant problem. In particular, the T315I and E255K mutations in BCR-ABL confer cross-resistance to imatinib, dasatinib and nilotinib [30]. Several investigational agents have demonstrated preclinical efficacy in T315I-mutated CML cells. The pan-Aurora kinase inhibitor MK-0457 has shown clinical activity against CML cells harbouring the T315I mutation [11]. Early in vitro competition binding assays revealed that MK-0457 bound to wild-type ABL1 and T315I ABL1 [37, 38]. This observation led to the hypothesis that the anti-leukemic efficacy of MK-0457 was due to inhibition of BCR-ABL, rather than Aurora, activity [14, 15]. However, a more recent investigation revealed that the efficacy of clinically relevant concentrations of MK-0457 was primarily due to inhibition of Aurora kinase activity [16]. How significantly Aurora kinase inhibition contributes to the activity of MK-0457 in CML remains somewhat controversial. This matter will not be definitively resolved for MK-0457 as its development has been stopped due to issues with cardiac toxicity observed in some early phase clinical trials and will have to be addressed in studies with other Aurora kinase inhibitors. MLN8237 is a highly selective inhibitor of Aurora A (IC50= 2 nM) and demonstrates little inhibition of various ABL isoforms in enzyme assays. Our data suggest that MLN8237 does not directly inhibit BCR-ABL activity, indicating that Aurora A kinase is a valid therapeutic target in CML.
The biological consequences of Aurora A kinase inhibition have been intensively investigated in recent years. Aurora A kinase plays an essential role in the assembly and function of the mitotic spindle and disruption of its activity affects spindle pole organization, centrosome separation and chromosome congression [39]. Ultimately, cells treated with Aurora A kinase inhibitors undergo cell death through the development of deleterious aneuploidy [19]. Our data show that MLN8237 treatment initially results in a significant degree of aneuploidy before cell death ensues (Figs 1B, C, 5C). The collective effects of MLN8237 in BCR-ABL+ cells are summarized in Table 1.
Table 1.
Effects of MLN8237 in BCR-ABL+ cells
| Apoptosis | Cell cycle |
|---|---|
| Activation of caspases-9 and -3 | Accumulation of aneuploid cells |
| Decreased expression of Apollon | Inhibition of colony formation |
| Increased expression of Smac | Mitotic slippage |
| Induction of TUNEL+ | Monopolar mitotic spindles |
| DNA fragmentation | Chromatin bridging |
Notably, MLN8237-induced apoptosis was associated with significant decreases in the expression levels of Apollon. Apollon has several functional domains, multiple binding partners and plays important roles in the regulation of apoptosis and cell division [36, 40]. Apollon can be distinguished from other IAP family members by its high molecular weight (528 kD) and unique E2/E3 ubiquitin conjugating/ubiquitin ligase functions. As Apollon overexpression has been associated with an unfavourable outcome and resistance to chemotherapy in leukaemia, we were particularly interested in examining whether the ability of MLN8237 to reduce Apollon expression could potentially sensitize CML cells to the standard of care agent nilotinib [35]. Our results show that targeted knockdown of Apollon significantly augments the pro-apoptotic effects of nilotinib, suggesting that suppression of Apollon expression by MLN8237 may contribute to its ability to heighten the anticancer activity of nilotinib. Furthermore, it is likely that the reduction in Apollon expression associated with MLN8237 is a direct consequence of Aurora inhibition as targeted knockdown of Aurora A kinase by siRNA was associated with a significant reduction in Apollon expression. These findings represent a novel mechanistic approach for improving the efficacy of tyrosine kinase inhibitor therapy in CML (Fig. 6E).
There are two potential important clinical translations of the results of the current study. We have shown that MLN8237 is effective against cell lines and primary patient CML cells expressing unmutated and mutated forms of BCR-ABL including the highly resistant T315I mutation and that it has activity independent of p53 function. Therefore, MLN8237 may be clinically active as a single agent in the setting of T315I and E255K mutations for which the currently available tyrosine kinase inhibitors are ineffective. Moreover, the combination of MLN8237 and nilotinib is effective and well tolerated in preclinical models of CML and represents a novel therapeutic strategy for advanced phase CML that is orally active and has the potential to suppress the emergence of CML clones expressing a range of resistant mutations including T315I and E255K.
The current treatment strategy for resistant CML involves sequential administration of tyrosine kinase inhibitors, which is associated with the development of compound mutations in BCR-ABL with increased oncogenic potency [31]. Patients with imatinib resistance have heightened genomic instability and in this setting combination treatment with an agent effective against cells harbouring the T315I mutation and a BCR-ABL kinase inhibitor could possibly prevent resistance caused by kinase domain mutations in CML [30, 41]. The combination of these two agents has the potential to eliminate BCR-ABL kinase domain mutation as a mechanism of resistance in CML, suppressing resistant disease and leading to sustained remissions in the vast majority of patients. Based on this promising preclinical data, a phase I/II study is warranted to investigate the safety and activity of MLN8237 in refractory CML.
Acknowledgments
The authors thank Dr. Charles L. Sawyers (Howard Hughes Medical Institute, Memorial Sloan-Kettering Cancer Center) and Dr. Brian Druker (Howard Hughes Medical Institute, Oregon Health Sciences University) for providing Ba/F3 cells expressing wild-type and mutant forms of BCR-ABL, and Dr. James Griffin (Dana-Farber Cancer institute) for providing imatinib-sensitive and imatinib-resistant K562 cells for this study. This work was supported by grants from LeukemiaTexas (J.S.C and F.J.G.), the CTRC at UTHSCSA Experimental and Developmental Therapeutics Program (J.S.C.), the Owens Biomedical Research Foundation (J.S.C.) and The AT&T Foundation (F.J.G.).
Conflict of interest
J.E. is an employee of Millennium Pharmaceuticals. The other authors have no conflicts of interest to declare.
References
- 1.Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–17. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 2.de Lavallade H, Apperley JF, Khorashad JS, et al. Imatinib for newly diagnosed patients with chronic myeloid leukemia: incidence of sustained responses in an intention-to-treat analysis. J Clin Oncol. 2008;26:3358–63. doi: 10.1200/JCO.2007.15.8154. [DOI] [PubMed] [Google Scholar]
- 3.Talpaz M, Silver RT, Druker BJ, et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood. 2002;99:1928–37. doi: 10.1182/blood.v99.6.1928. [DOI] [PubMed] [Google Scholar]
- 4.Volpe G, Panuzzo C, Ulisciani S, et al. Imatinib resistance in CML. Cancer letters. 2009;274:1–9. doi: 10.1016/j.canlet.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 5.Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer cell. 2005;7:129–41. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Shah NP, Tran C, Lee FY, et al. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 7.Puttini M, Coluccia AM, Boschelli F, et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer research. 2006;66:11314–22. doi: 10.1158/0008-5472.CAN-06-1199. [DOI] [PubMed] [Google Scholar]
- 8.Warner SL, Stephens BJ, Von Hoff DD. Tubulin-associated proteins: Aurora and Polo-like kinases as therapeutic targets in cancer. Curr Oncol Rep. 2008;10:122–9. doi: 10.1007/s11912-008-0020-0. [DOI] [PubMed] [Google Scholar]
- 9.Ikezoe T, Yang J, Nishioka C, et al. A novel treatment strategy targeting Aurora kinases in acute myelogenous leukemia. Molecular Cancer Ther. 2007;6:1851–7. doi: 10.1158/1535-7163.MCT-07-0067. [DOI] [PubMed] [Google Scholar]
- 10.Harrington EA, Bebbington D, Moore J, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–7. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 11.Giles FJ, Cortes J, Jones D, et al. MK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutation. Blood. 2007;109:500–2. doi: 10.1182/blood-2006-05-025049. [DOI] [PubMed] [Google Scholar]
- 12.Gontarewicz A, Balabanov S, Keller G, et al. Simultaneous targeting of Aurora kinases and Bcr-Abl kinase by the small molecule inhibitor PHA-739358 is effective against imatinib-resistant BCR-ABL mutations including T315I. Blood. 2008;111:4355–64. doi: 10.1182/blood-2007-09-113175. [DOI] [PubMed] [Google Scholar]
- 13.Cortes-Franco J, Dombret H, Schafhausen P, et al. Danusertib hydrochloride (PHA-739358), a multi-kinase Aurora inhibitor, elicits clinical benefit in advanced chronic myeloid leukemia and Philadelphia chromosome positive acute lymphoblastic leukemia ASH Annual Meeting. Washington: Blood J. 2009. p. 864.
- 14.Dai Y, Chen S, Venditti CA, et al. Vorinostat synergistically potentiates MK-0457 lethality in chronic myelogenous leukemia cells sensitive and resistant to imatinib mesylate. Blood. 2008;112:793–804. doi: 10.1182/blood-2007-10-116376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiskus W, Wang Y, Joshi R, et al. Cotreatment with vorinostat enhances activity of MK-0457 (VX-680) against acute and chronic myelogenous leukemia cells. Clin Cancer Res. 2008;14:6106–15. doi: 10.1158/1078-0432.CCR-08-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donato NJ, Fang D, Sun H, et al. Targets and effectors of the cellular response to aurora kinase inhibitor MK-0457 (VX-680) in imatinib sensitive and resistant chronic myelogenous leukemia. Biochem Pharmacol. 2010;79:688–97. doi: 10.1016/j.bcp.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 17.Fu J, Bian M, Jiang Q, et al. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res. 2007;5:1–10. doi: 10.1158/1541-7786.MCR-06-0208. [DOI] [PubMed] [Google Scholar]
- 18.Marumoto T, Honda S, Hara T, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem. 2003;278:51786–95. doi: 10.1074/jbc.M306275200. [DOI] [PubMed] [Google Scholar]
- 19.Hoar K, Chakravarty A, Rabino C, et al. MLN8054, a small-molecule inhibitor of Aurora A, causes spindle pole and chromosome congression defects leading to aneuploidy. Mol Cell Biol. 2007;27:4513–25. doi: 10.1128/MCB.02364-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sells T, Ecsedy J, Stroud S, et al. MLN8237: an orally active small molecule inhibitor of Aurora A kinase in phase I clinical trials. Philadelphia: American Association for Cancer Research. 2008;237 [Google Scholar]
- 21.Carew JS, Nawrocki ST, Krupnik YV, et al. Targeting endoplasmic reticulum protein transport: a novel strategy to kill malignant B cells and overcome fludarabine resistance in CLL. Blood. 2006;107:222–31. doi: 10.1182/blood-2005-05-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weisberg E, Griffin JD. Mechanism of resistance to the ABL tyrosine kinase inhibitor STI571 in BCR/ABL-transformed hematopoietic cell lines. Blood. 2000;95:3498–505. [PubMed] [Google Scholar]
- 23.Swords RT, Kelly KR, Smith PG, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115:3796–800. doi: 10.1182/blood-2009-11-254862. [DOI] [PubMed] [Google Scholar]
- 24.Nawrocki ST, Carew JS, Maclean KH, et al. Myc regulates aggresome formation, the induction of Noxa, and apoptosis in response to the combination of bortezomib and SAHA. Blood. 2008;112:2917–26. doi: 10.1182/blood-2007-12-130823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carew JS, Nawrocki ST, Reddy VK, et al. The novel polyamine analogue CGC-11093 enhances the antimyeloma activity of bortezomib. Cancer Res. 2008;68:4783–90. doi: 10.1158/0008-5472.CAN-07-6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carew JS, Nawrocki ST, Kahue CN, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110:313–22. doi: 10.1182/blood-2006-10-050260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Bacco A, Keeshan K, McKenna SL, et al. Molecular abnormalities in chronic myeloid leukemia: deregulation of cell growth and apoptosis. Oncologist. 2000;5:405–15. doi: 10.1634/theoncologist.5-5-405. [DOI] [PubMed] [Google Scholar]
- 28.Wendel HG, de Stanchina E, Cepero E, et al. Loss of p53 impedes the antileukemic response to BCR-ABL inhibition. Proc Natl Acad Sci USA. 2006;103:7444–9. doi: 10.1073/pnas.0602402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–15. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 30.O’Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110:2242–9. doi: 10.1182/blood-2007-03-066936. [DOI] [PubMed] [Google Scholar]
- 31.Shah NP, Skaggs BJ, Branford S, et al. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117:2562–9. doi: 10.1172/JCI30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Z, Naito M, Hori S, et al. A human IAP-family gene, Apollon, expressed in human brain cancer cells. Biochem Biophys Res Commun. 1999;264:847–54. doi: 10.1006/bbrc.1999.1585. [DOI] [PubMed] [Google Scholar]
- 33.Hao Y, Sekine K, Kawabata A, et al. Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat Cell Biol. 2004;6:849–60. doi: 10.1038/ncb1159. [DOI] [PubMed] [Google Scholar]
- 34.Qiu XB, Goldberg AL. The membrane-associated inhibitor of apoptosis protein, BRUCE/Apollon, antagonizes both the precursor and mature forms of Smac and caspase-9. J Biol Chem. 2005;280:174–82. doi: 10.1074/jbc.M411430200. [DOI] [PubMed] [Google Scholar]
- 35.Sung KW, Choi J, Hwang YK, et al. Overexpression of Apollon, an antiapoptotic protein, is associated with poor prognosis in childhood de novo acute myeloid leukemia. Clin Cancer Res. 2007;13:5109–14. doi: 10.1158/1078-0432.CCR-07-0693. [DOI] [PubMed] [Google Scholar]
- 36.Pohl C, Jentsch S. Final stages of cytokinesis and midbody ring formation are controlled by BRUCE. Cell. 2008;132:832–45. doi: 10.1016/j.cell.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 37.Young MA, Shah NP, Chao LH, et al. Structure of the kinase domain of an imatinib-resistant Abl mutant in complex with the Aurora kinase inhibitor VX-680. Cancer Res. 2006;66:1007–14. doi: 10.1158/0008-5472.CAN-05-2788. [DOI] [PubMed] [Google Scholar]
- 38.Carter TA, Wodicka LM, Shah NP, et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci USA. 2005;102:11011–6. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marumoto T, Zhang D, Saya H. Aurora-A – a guardian of poles. Nat Rev Cancer. 2005;5:42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- 40.Martin SJ. An Apollon vista of death and destruction. Nat Cell Biol. 2004;6:804–6. doi: 10.1038/ncb0904-804. [DOI] [PubMed] [Google Scholar]
- 41.Quintás-Cardama A, Gibbons DL, Kantarjian HM, et al. Mutational analysis of chronic phase chronic myeloid leukemia (CMLCP) clones reveals heightened BCR-ABL1 genetic instability in patients failing sequential imatinib and dasatinib therapy ASH Annual Meeting Abstracts. Washington: Blood J. 112:2114. [Google Scholar]