

Abstract
Despite progress in the treatment of acute myelogenous leukaemia (AML) the outcome often remains poor. Tumour necrosis factor related apoptosis-inducing ligand (TRAIL) is a promising therapeutic agent in many different types of tumours, but AML cells are relatively insensitive to TRAIL-induced apoptosis. Here we show that TRAIL-induced apoptosis in AML cells is predominantly mediated by death receptor 4 (DR4) and not DR5. Therefore, we constructed a variant of TRAIL (rhTRAIL-C3) that is a strong inducer of DR4-mediated apoptosis. TRAIL-C3 demonstrated much stronger pro-apoptotic activity than wild-type (WT) TRAIL in a panel of AML cell lines as well as in primary AML blasts. The higher pro-apoptotic potential was further enhanced when the TRAIL mutant was used in combination with BMS-345541, a selective inhibitor of inhibitor-κB kinases. It illustrates that combination of this TRAIL variant with chemotherapeutics or other targeted agents can kill AML with high efficacy. This may represent a major advantage over the currently used therapies that have serious toxic side effects. The high efficacy of rhTRAIL-C3 containing therapies may enable the use of lower drug doses to reduce the toxic side effects and improve patient outcome. Our findings suggest that the rational design of TRAIL variants that target DR4 potentiate the death-inducing activity of TRAIL and offer a novel therapeutic strategy for the treatment of AML.
Keywords: apoptosis, death receptor 4 (DR4), DR5, tumour necrosis factor-related apoptosis-inducing ligand (TRAIL), acute myelogenous leukaemia (AML), receptor-selective TRAIL variant, primary AML blast
Introduction
Acute myeloid leukaemia (AML) is a heterogeneous group of diseases characterized by uncontrolled proliferation of clonal neoplastic haematopoietic precursor cells and impaired production of normal haematopoiesis leading to neutropenia, anaemia and thrombocytopenia [1]. The 5 year survival rate of AML patients has remained at 20–30% since the 1970s [2]. The outcome for AML patients depends on a variety of factors, including the age of the patient, the intensity of post-remission therapy and the biological characteristics of the disease [3–5]. Thus there remains a need for rational, minimally toxic but effective therapies for AML [6].
Tumour necrosis factor related apoptosis-inducing ligand (TRAIL) is a promiscuous cytokine with the ability to bind to five different receptors. TRAIL-R1 death receptor (DR)4 and TRAIL-R2 (DR5), the two apoptosis-inducing TRAIL receptors, contain a conserved cytoplasmic death domain motif that undergoes a conformational change upon binding of TRAIL and results in the recruitment of the adaptor molecule Fas-associated via death domain (FADD) [7–9]. This facilitates caspase-8 recruitment and activation, which activates the apoptotic machinery. Decoy receptor 1 (DcR1/TRAIL-R3), decoy receptor 2 (DcR2/TRAIL-R4) and the soluble receptor osteoprotegerin lack or have a truncated cytoplasmic death domain and thus these receptors act to prevent apoptosis by sequestering available TRAIL or by interfering with the formation of a DR4 or DR5 signalling complex [10, 11].
A number of haematological malignancies are sensitive to TRAIL either as a single agent or in combination with chemotherapeutics [12–14]. Chronic lymphocytic leukaemia (CLL) cells undergo TRAIL-induced apoptosis by activation of DR4 rather than DR5 [15], contrasting with the suggestion that DR5 may contribute more than DR4 to TRAIL-induced apoptosis in cancer cells expressing both TRAIL receptors [16]. Furthermore, primary mantle cell lymphoma cells were also found to be sensitive to TRAIL and similar to CLL cells, the TRAIL-death signal was almost exclusively transmitted through DR4 [12]. Interestingly, a study of 72 primary AML blasts revealed that while many AML blasts expressed DR4 (48.6%) and DR5 (12.5%), the AML cells generally showed low sensitivity to TRAIL [17].
A number of mechanisms have been associated with TRAIL resistance in AML. It has been suggested that high levels of decoy receptors were associated with TRAIL insensitivity [17]. Overexpression of cellular FADD-like Interleukin-1 converting enzyme (ICE)-like inhibitory protein (c-FLIP) has been reported as another mechanism of resistance of AML cells to TRAIL [18]. High c-FLIP expression has also been recently reported to be linked to inferior outcome in AML patients with normal karyotype and intermediate risk [19]. Therefore based on these observations, we suggested that by increasing the affinity of TRAIL for DR4 a higher efficacy may be achieved, thus making engineered TRAIL suitable for the treatment of AML and other haematological malignancies.
Experimental procedures
Site-directed mutagenesis, expression and purification of rhTRAIL variants
Construction and purification of wild-type (WT) TRAIL and variants was performed as described before [20]. The amino acid alterations in rhTRAIL-C3 did not change the homotrimeric structure of the molecule as the gel filtration elution profile of the mutant was not different from that of WT rhTRAIL (Fig. S2).
Determination of receptor binding by surface plasmon resonance (SPR)
Binding experiments were performed with a SPR-based biosensor Biacore 3000 (GE Healthcare, Munich, Germany). Research grade CM5 sensor chips, N-hydroxysuccimide, N-ethyl-N′-(3-diethylaminopropyl) carbodiimide, ethanolamine-HCl and standard buffers, e.g. HBS-N and HBS-EP were purchased from the manufacturer. All the buffers were filtered and degassed. Immobilization of DR4-Ig and DR5-Ig receptors (R&D Systems, Minneapolis, MN, USA) on the sensor surface of a Biacore CM5 sensor chip was performed following a standard amine coupling procedure according to the manufacturer’s instructions. Receptors were coated at a level of 600–1000 response units (RU). Reference surfaces consisted of activated CM dextran, subsequently blocked with ethanolamine. 50 μl aliquots of WT rhTRAIL or variants were injected in 3-fold at concentrations ranging from 0.5 to 250 nM at 70 μl/ml and at 37°C using HBS-N supplemented with 0.005% surfactant P20 (Biacore, Uppsala, Sweden) as running and sample buffer. Binding of ligands to the receptors was monitored in real time. Pre-steady state data was obtained by recording the responses 30 sec. after the end of injections for all concentrations. The response data as a function of TRAIL concentration were fitted by using a four-parameter equation to give apparent affinity constants. Between injections, the receptor/sensor surface was regenerated using 1:1 (10 mM glycine 1.5 M NaCl pH 2 : ethylene glycol) and a contact time of 35 sec.
Determination of receptor binding by competitive ELISA
Nunc Maxisorb plates were coated for 2 hrs with DR4-Ig (100 ng/well) in 0.1 M sodium carbonate/bicarbonate buffer (pH 8.6) remaining binding sites were subsequently blocked with 2% bovine serum albumin (BSA) for 1 hr. Serial dilutions of soluble recombinant receptor proteins DR4, DR5, DcR1 and DcR2 (Ig fusion proteins, 0–500 ng/well) were pre-incubated with WT rhTRAIL or rhTRAIL-C3 (10 ng/well) in phosphate-buffered saline (PBS; pH 7.4) for 1 hr at room temperature, added to the wells and incubated for an additional 1 hr. After washing for six times with Tris-buffered saline with Tween (TBST), a 1:200 dilution of anti-TRAIL antibody (R&D Systems) was added and incubated for 1 hr at room temperature, and, after washing six times with TBST, subsequently incubated with a 1:25,000 dilution of a horseradish peroxidase-conjugated swine–anti-goat antibody (BioSource International, Invitrogen, Carlsbad, CA, USA). After washing six times with TBST, 100 μl of 1-step Turbo TMB solution (Pierce, Thermo Fisher Scientific, Rockford, IL, USA) was added for 20 min., the reaction was stopped with 100 μl 1 M sulphuric acid. The absorbance was measured at 450 nM on a microplate reader (Thermo Fisher Scientific, Rockford, IL, USA). Binding of WT rhTRAIL or rhTRAIL-C3 to immobilized DR4-Ig with 0 ng/well of the soluble receptors was taken as 100%, and binding at other concentrations of soluble receptors was calculated relative to 0 ng/well of soluble receptor.
Cell lines and treatment
NHEK primary normal human epidermal keratinocytes (NHEK, PromoCell, Heidelberg, Germany) were maintained in serum free keratinocyte growth medium (PromoCell, C-12003), supplemented with bovine pituitary extract (0.004 ml/ml), epidermal growth factor (0.125 ng/ml), insulin (5 μg/ml), hydrocortisone (0.33 μg/ml), epinephrine (0.39 μg/ml), holo-transferrin (10 μg/ml) and CaCl2 (0.06 mM) (all from PromoCell). Primary human dermal fibroblasts (a kind gift from Dr. Linda Howard, REMEDI, National University of Ireland, always) were cultured in low glucose (100 mg/l) Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% foetal bovine serum (FBS), 50 U/ml penicillin and 50 μg/ml streptomycin. Keratinocytes and fibroblasts were seeded at a density of 50,000 cells/ml, 2 days before treatment. HL60 [German Collection of Microorganisms and Cell Cultures (DSMZ), ACC 3], EM-2 (DSMZ, ACC 135) and A2780 cells were maintained in RPMI medium supplemented with 10% FBS, 2 mM glutamine, 50 U/ml penicillin and 50 mg/ml streptomycin. HL60, EM-2 and A2780 cells were seeded at 5 × 105, 3 × 105 and 3.5 × 105 cells/ml, respectively, 24 hrs prior to treatment. The ML-1 cell line was maintained in RPMI medium supplemented with 20% FBS, 2 mM glutamine, 50 U/ml penicillin and 50 mg/ml streptomycin and seeded at 3 × 105 cells/ml 24 hrs prior to treatment. MOLM-13 cells (DSMZ, ACC 554) were maintained in RPMI medium supplemented with 10% FBS, 2 mM glutamine, 1 mM sodium pyruvate, 1% non-essential amino acids (NEA), 50 U/ml penicillin and 50 mg/ml streptomycin and seeded at 1 × 106 cells/ml 24 hrs prior to treatment. OCI-AML3 cells (DSMZ, ACC 582) were maintained in Modified Eagle Medium (MEM) supplemented with 20% FBS, 2 mM glutamine, 1 mM sodium pyruvate, 1% NEA, 50 U penicillin and 50 mg/ml streptomycin and seeded at 7.5 × 105 cells/ml 24 hrs prior to treatment. All cells were cultured at 37°C with 5% CO2 in a humidified incubator. All reagents were from Sigma-Aldrich unless otherwise stated. To examine the pro-apoptotic potential of the DR4-selective TRAIL variants, as a measure of their receptor selectivity and/or biological activity, cells were treated with increasing concentrations (0–250 ng/ml) of rhTRAILN199R/K201H, rhTRAILG131R/N199R/K201H (rhTRAIL-C3), rhTRAILD269H/E195R or rhTRAIL-WT.
Isolation and culture of primary AML blasts:
BM MNCs were isolated by density gradient centrifugation and cryopreserved in 10% dimethyl sulphoxide (DMSO) freezing medium. Following a rapid thawing and centrifugation to remove DMSO of the freezing medium, cells were cultured in RPMI containing 10% FBS, 2 mM glutamine, 1 mM sodium pyruvate, 50 U/ml penicillin and 50 mg/ml streptomycin and seeded at 1 × 106 cells/ml. Cell viability was assessed at the time of thawing the cells and experiments were only carried out in samples that showed at least 75% viability. In addition, samples that showed high rate of spontaneous death and the percentage of live cells reduced to 25% or less at time of harvesting were also excluded from further analysis. The cells were treated with 250 ng/ml of WT rhTRAIL or rhTRAIL-C3 alone for 24 hrs or in combination with 5 μM BMS-345541 added simultaneously.
Annexin V staining
Externalization of phosphatidyl serine on the plasma membrane of apoptotic cells was detected using annexin V-fluoro isothiocyanate (FITC) (IQ Corporation, Groningen, The Netherlands) as previously described [21].
Determination of surface TRAIL receptor expression
Cells were removed from culture dishes, harvested by centrifugation and washed twice with 1% BSA in PBS. Cells were incubated with 1:100 dilution of primary antibodies (DR4 and DR5: neutralizing mouse monoclonal antibodies, Alexis, DcR1 and DcR2: neutralizing goat polyclonal antibodies, R&D Systems) in 1% BSA in PBS for 40 min. on ice. After two washes in 1% BSA/PBS, cells were resuspended in 1:50 dilution of FITC-labelled secondary antibody and incubated for 40 min. on ice. Excess secondary antibody was removed by washing first in 1% BSA in PBS and then PBS. Cells were fixed in 1% formaldehyde/PBS before analysis by flow cytometry (FacsCalibur, Becton Dickinson, Franklin Lakes, NJ, USA).
Western blotting
Polyacrylamide gel electrophoresis and Western blotting was carried out as previously described [22]. For antigen detection membranes were incubated with antibodies to actin (1:500; Sigma), caspase-3 and -8 (1:1000; Cell Signaling Technology, Danvers, MA, USA) overnight at 4°C followed by 2 hrs incubation at room temperature with secondary antibodies (1:5000; Pierce). Protein bands were visualized using Supersignal Ultra Chemiluminescent Substrate (Pierce) on X-ray film (Agfa, Mortsel, Belgium).
Crosslinking of agonistic anti-DR4 and -DR5 antibodies
Agonistic DR4 and DR5 antibodies (200 nM; Novartis, Basel, Switzerland) were incubated with goat anti-human Fc antibody (crosslinking antibody, 600 nM; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) in serum free cell culture media for 30 min. at room temperature then added to the cell culture for 24 hrs.
7-aminoactinomycin D staining
Following treatment, cells were removed from culture dishes, harvested by centrifugation and washed with PBS. To fix, cells were resuspended in 70% ethanol and incubated overnight at 4°C. Cells were recovered by centrifugation, resuspended in PBS containing 0.3 μg/ml 7-aminoactinomycin D (7-AAD) and incubated for 10 min. at room temperature in the dark. Cells were immediately analyzed by flow cytometry (FacsCalibur, Beckton Dickinson).
Measurement of mitochondrial transmembrane potential (ΔΨm)
ΔΨm was measured using the fluorescent dye tetramethylrhodamine ethyl ester perchlorate (TMRE). Cells (500 μl) were transferred from wells to fluorescence-assisted cell sorter (FACS) tubes and TMRE was added to a final concentration of 100 nM. Cells were incubated for 30 min. at room temperature in the dark followed by immediate analysis by flow cytometry (FacsCalibur flow cytometer, Beckton Dickinson). As a positive control for mitochondrial depolarization, cells were treated for 2 hrs with 10 μM carbonyl cyanide 3-chlorophenylhydrazone.
Results
Design and biochemical characterization of rhTRAIL-C3
Although approximately 50% of AMLs express at least one death-inducing TRAIL receptor on the cell surface, AML blasts display a low sensitivity to TRAIL [17]. In AML blasts, TRAIL fails to activate pro-caspase-8, indicating that inadequate receptor activation by TRAIL may be the cause of the low sensitivity [17]. Since DR4 is more frequently expressed on AML blasts than DR5, it was suggested that a high affinity, DR4-selective TRAIL variant would be able to trigger AML cell apoptosis.
In the search for an rhTRAIL variant that can efficiently induce apoptosis in AML blasts, we have combined mutation D218H (replacement of the aspartate at position 218 to histidine) with mutation G131R (glycine at position 131 to arginine) that, respectively, reduces the affinity of TRAIL to DR5 [23] or increases the apoptotic potential of rhTRAIL (Reis et al.). However, combination of G131R with D218H did not result in higher biological activity compared to WT rhTRAIL (Fig. S1). Scanning the receptor binding interface of TRAIL with the protein design algorithm FoldX [24, 25] identified additional mutations which potentially improved DR4 selectivity. A N199R/K201H double mutation (replacement of the asparagine at position 199 to arginine and the lysine at position 201 to histidine) was predicted to improve DR4 binding (Predicted ΔΔGinteraction– 0.44 kcal/mol) and combination of these mutations with the G131R mutation was feasible from a structural point of view. Moreover, the combined effect of these mutations was predicted to minimally affect DR5 binding (Predicted ΔΔGinteraction < 0.1 kcal/mol). The TRAIL G131R/N199R/K201H mutant (rhTRAIL-C3) was constructed by site directed mutagenesis and purified as a trimeric protein as described previously [26]. The retained homotrimeric structure of rhTRAIL-C3 was confirmed by the unchanged gel filtration elution profile in comparison to WT rhTRAIL (Fig. S2).
Receptor binding of rhTRAIL-C3 to DR4 and DR5 was assessed in real time with SPR (Fig. S3) and apparent dissociation constants (KD) were calculated from pre-steady state response values (Fig. S4). rhTRAIL-C3 showed a 3-fold increase in apparent affinity for DR4-Ig (Table 1) and a very modest reduction in affinity to DR5-Ig when compared to WT rhTRAIL (Table 1, Fig. S3). Intriguingly, when looking at the sensograms for DR5-Ig binding of WT rhTRAIL and rhTRAIL-C3, an initial off-rate has been observed at high concentrations of rhTRAIL-C3 (Fig. S3D). This initial off-rate seems to represent the dissociation of trimeric rhTRAIL-C3 from one or two receptor molecules, suggesting a lower affinity of rhTRAIL-C3 towards DR5 than of WT rhTRAIL. However, this difference in the affinity could not be reflected in the pre-steady state affinity determination.
Table 1.
SPR measurements of DR4 and DR5 binding affinities and competitive ELISA for DR4 and DR5, and decoy receptors, DcR1 and DcR2. Apparent KA and EC50 ratios were calculated relative to WT rhTRAIL.
| Apparent KA ratio rhTRAIL-C3 / WT rhTRAIL by SPR | |
|---|---|
| DR4 | DR5 |
| 2.7 ± 0.1 | 0.83 ± 0.07 |
| EC50 ratio rhTRAIL-C3/WT rhTRAIL binding to DR4-Ig by competitive ELISA | |
| DR4-Ig competition | DR5-Ig competition |
| 7.1 ± 0.3 | 1.1 ± 0.3 |
| DcR1-Ig competition | DcR2-Ig competition |
| 3.5 ± 0.6 | 1.1 ± 0.2 |
To determine the receptor preference of rhTRAIL-C3 when multiple TRAIL receptors are present, competitive ELISA assays were performed. Plates were coated with DR4-Ig. Soluble DR4-, DR5-, DcR1- or DcR2-Ig were pre-incubated as competitors with WT rhTRAIL or rhTRAIL-C3 for 30 min. and added to the DR4-coated wells. Binding of WT rhTRAIL or rhTRAIL-C3 to immobilized DR4-Ig was measured. rhTRAIL-C3 showed a 7-fold reduction in binding to immobilized DR4-Ig in the presence of soluble DR4-Ig when compared to WT rhTRAIL (Table 1, Fig. S4C), indicating a significant increase in affinity for DR4. In contrast, WT rhTRAIL and rhTRAIL-C3 showed similar behaviour when soluble DR5-Ig or DcR2-Ig was the competitor (Table 1, Fig. S4D and F). DcR1-Ig also reduced binding of rhTRAIL-C3 to immobilized DR4-Ig by 3.5-fold when compared to WT rhTRAIL (Table 1, Fig. S4E). In summary, rhTRAIL-C3 has a higher affinity for DR4 and a moderate increase in affinity for DcR1. The binding preference of the variant to DR5 and DcR2 remained unchanged.
rhTRAIL-C3 has a DR4-selective, agonistic activity
Receptor selectivity and receptor activating ability (agonistic activity) of rhTRAIL-C3 was initially tested on tumour cells which responded exclusively to either DR4- or DR5-activation (EM-2 chronic myeloid leukaemia cells and A2780 ovarian carcinoma cells, respectively) [23]. The two cell types were treated with WT rhTRAIL, rhTRAIL-C3 or D269H/E195R, a DR5-selective TRAIL variant [20]. Induction of apoptosis was measured by annexin V assay. As reflected by the ED50 values, D269H/E195R demonstrated very weak apoptosis-inducing activity in EM-2 cells, but appeared to be the strongest inducer of apoptosis of the three rhTRAIL variants in A2780 cells, indicative of the DR4- or DR5 sensitivity of the two cell lines, respectively (Fig. 1). Conversely, the rhTRAIL-C3 variant displayed the highest pro-apoptotic activity in the DR4-responsive cell line, EM-2 (ED50 = 3.78 ng/ml for rhTRAIL-C3 versus 17.53 ng/ml for WT rhTRAIL), and only marginal activity in the DR5-responsive A2780 cells (5.65, 121.7 and 1972.3 ng/ml ED50 values for D269H/E195R, WT rhTRAIL and rhTRAIL-C3, respectively, Fig. 1A). These results indicate that rhTRAIL-C3 is an agonistic TRAIL variant that preferentially activates DR4.
Fig 1.
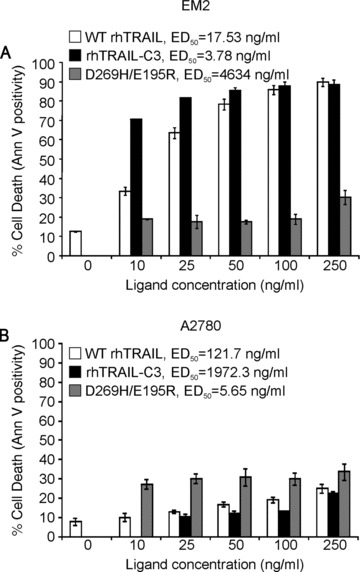
DR4-selectivity of rhTRAIL-C3. DR4-responsive EM-2 chronic myelogenous leukaemia cells (A) and DR5-responsive A2780 ovarian carcinoma cells (B) were treated with increasing doses of rhTRAIL-C3 (G131R/N199R/K201H), WT rhTRAIL or DR5-selective rhTRAIL variant (D269H/E195R) for 24 hrs. Induction of apoptosis was determined using annexin V assay. The graphs show the percentage of dead cells ± S.E.M. determined with flow cytometric analysis of annexin V positivity in three independent experiments.
Similar testing was carried out with the double mutant N199R/K201H, with predicted increased DR4-selectivity. N199R/K201H induced apoptosis in the DR4-responsive ML-1 (Fig. S1A) and EM-2 (Fig. S1B) cells, but not A2780 cells (Fig. S1C) confirming that the mutations increase DR4-selectivity.
The DR4-mediated action of rhTRAIL-C3 was further assessed in BJAB cells overexpressing DR4 or DR5 variants lacking the C-terminal tail involved in FADD binding (ΔCT-DR4-BJAB, ΔCT-DR5-BJAB). These DR4 and DR5 mutant receptors are not functional and likely to act in a dominant negative fashion by interfering with proper DISC formation on trimerized receptors [27]. rhTRAIL-C3 triggered apoptosis more efficiently than WT rhTRAIL in WT-BJAB cells, GFP overexpressing BJAB cells and ΔCT-DR5-BJAB cells (Fig. 2A, B and D). However, rhTRAIL-C3 failed to kill ΔCT-DR4-BJAB cells, confirming its DR4-selective pro-apoptotic action (Fig. 2C). This was reflected in the ED50 values, as it stayed nearly unchanged in WT, GFP and ΔCT-DR5 cells, but increased by 13.49-fold in the ΔCT-DR4 cells (Fig. 2). WT-rhTRAIL on the other hand showed a 2.94-fold increase in the ED50 value on ΔCT-DR5 BJAB cells and a 10.38-fold increase on the ΔCT-DR4 cells, indicating its dual specificity towards DR4 and DR5 (Fig. 2).
Fig 2.
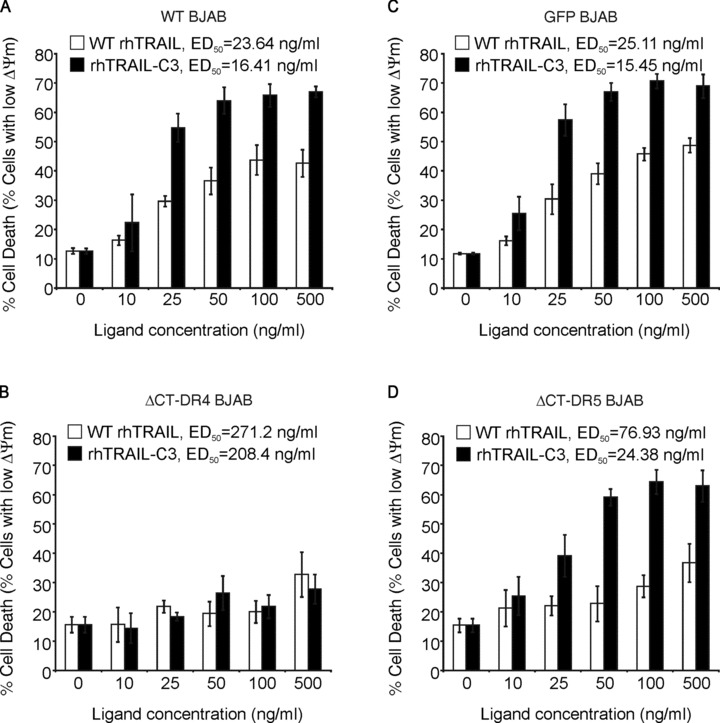
Deficiency of DR4 blocks rhTRAIL-C3 induced apoptosis. WT BJAB cells (A), BJAB cells overexpressing GFP (B), non-functional, C-terminal deletion mutants of DR4 (C) or DR5 (D) were treated with WT rhTRAIL and rhTRAIL-C3. Induction of apoptosis was determined by measuring loss of mitochondrial membrane potential (ΔΨm).
Fig 3.
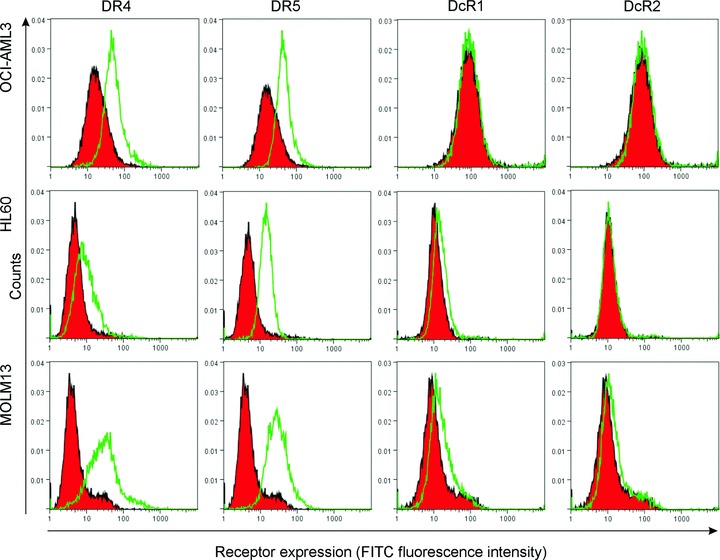
Cell surface expression of TRAIL receptors in OCI-AML3, HL-60 and MOLM-13 cells. Cell surface expression of DR4, DR5, DcR1 and DcR2 was measured by immunostaining and detected with flow cytometry (FacsCanto II, Beckton Dickinson). The histograms are representatives of three independent experiments. Red filled peaks: isotype control, green open peaks: TRAIL receptor stained sample.
rhTRAIL-C3 is a potent inducer of apoptosis in AML
The ability of rhTRAIL-C3 to induce AML cell death was assessed in ML-1, MOLM-13, OCI-AML3 and HL-60 AML cell lines. According to the French–American–British (FAB) classification, HL-60 cells are type M3 acute promyelocytic leukaemia cells, ML-1 and OCI-AML3 are M4 AMLs and MOLM-13 is type M5a [28–31]. All four cell lines express both DR4 and DR5 on their surface (Fig. 3).
Cells were treated with 10–250 ng/ml of WT rhTRAIL, rhTRAIL-C3 or D269H/E195R (DR5-selective TRAIL variant) for 24 hrs and induction of apoptosis was quantified with annexin V staining. rhTRAIL-C3 appeared to be a much more potent apoptosis-inducer than WT rhTRAIL in HL-60, ML-1 and MOLM13 cells (Fig. 4). Like WT rhTRAIL, rhTRAIL-C3 could not induce apoptosis in OCI-AML3 cells (Fig. 4D). OCI-AML3 cells are completely resistant to TRAIL (Fig. 4) probably due to high expression of X-linked inhibitor of apoptosis protein (XIAP), Bcl-XL and Mcl-1 (data not shown). On the other hand, the DR5-selective TRAIL variant was a very weak inducer of apoptosis in all four cell lines. This is despite the fact that D269H/E195R is substantially more potent than WT rhTRAIL in DR5-responsive cell lines, such as A2780 or the colon carcinoma cell line Colo205 [20, 32].
Fig 4.
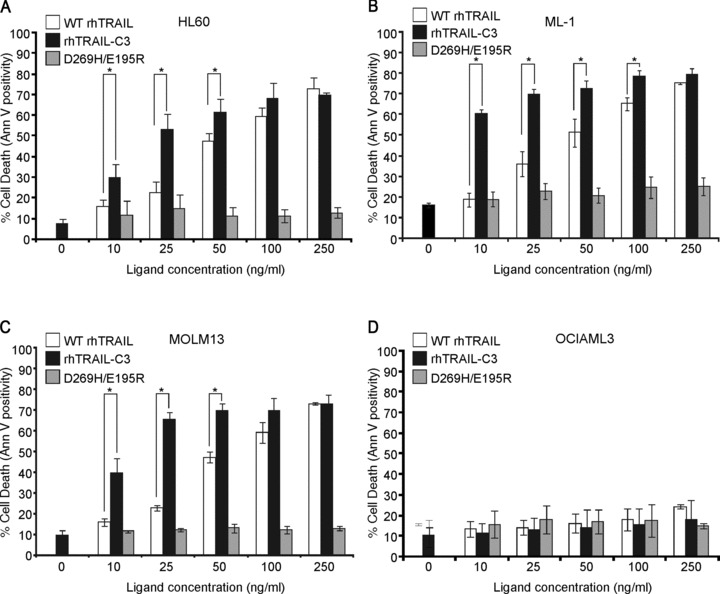
Pro-apoptotic potential of rhTRAIL-C3 in AML cell lines. HL-60 (A), ML-1 (B), MOLM-13 (C) and OCI-AML-3 (D) were treated with increasing doses of WT rhTRAIL, rhTRAIL-C3 and D269H/E195R for 24 hrs and induction of apoptosis was determined using annexin V assay. The graphs show the average percentage of dead cells ± S.E.M. determined with flow cytometric analysis of annexin V positivity. Stars (*) indicated significant difference between connected bar pairs (P < 0.05, determined with paired t-test).
We have previously reported that in ML-1 cells only DR4 is functional [20]. The functionality of DR4 and DR5 in HL-60, ML-1, MOLM-13, OCI-AML3 and EM-2 cells was determined using agonistic anti-DR4 and anti-DR5 antibodies (Novartis). The cells were treated with 5–20 nM of the agonistic antibodies with or without previous crosslinking of the antibodies through their Fc region using a secondary antibody. After 24 hrs of treatment, induction of apoptosis was determined with 7-AAD staining. Results demonstrated that in HL-60, ML-1 and EM-2 cells DR4 was the primary mediator of TRAIL-induced apoptosis (Fig. 5A, B and E), in OCI-AML3 cells signalling through DR4 and DR5 was blocked and in MOLM-13 cells, while both receptors were functional, DR5 required activation using a tetravalent, crosslinked antibody (Fig. 5C and D).
Fig 5.
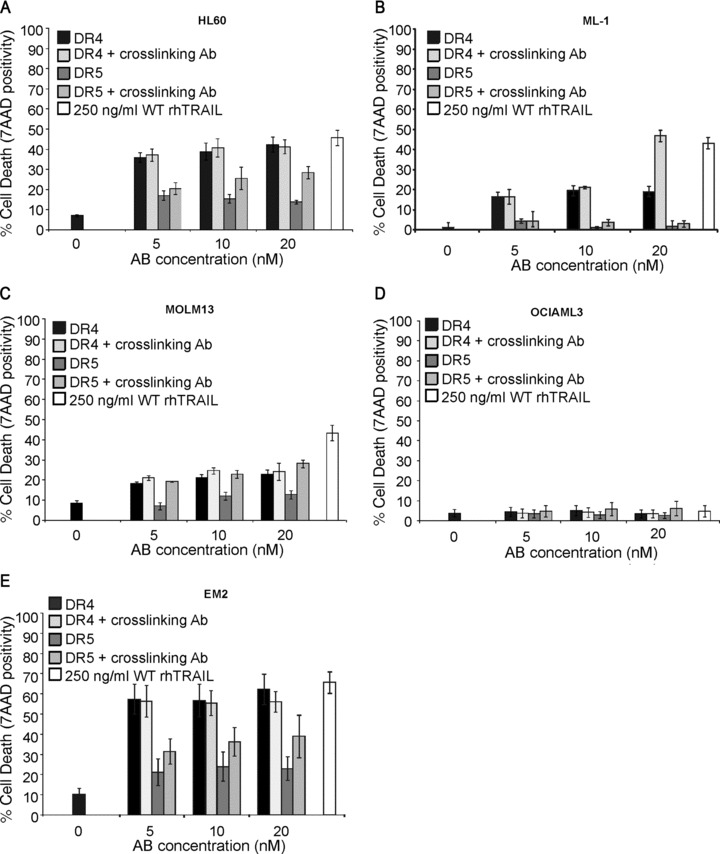
Functionality of DR4 and DR5 in AML cells. HL60 (A), ML-1 (B), MOLM-13 (C) OCI-AML3 (D) and EM-2 cells were treated with WT rhTRAIL, agonistic anti-DR4 and anti-DR5 antibodies for 24 hrs with or without previous crosslinking of the antibodies. Induction of cell death, as a measure of receptor functionality, was measured with 7-aminoactinomycin D staining and flow cytometric analysis. Graphs demonstrate the average percentage of dead cells (±S.E.M.) from three independent experiments.
To examine whether the high affinity of rhTRAIL-C3 enhanced DR4 activation, HL-60 cells, which have defective DR5 signalling, were selected. Two apical events of DR activation, i.e. pro-caspase-8 activation and NF-κB activation were monitored over time as a measure of DR4 activation. Cells were treated with 100 ng/ml of WT rhTRAIL or rhTRAIL-C3 for 5–120 min. Cells were then harvested for detection of pro-caspase-8 processing into its active fragments, phosphorylation and degradation of IκBα. To examine the kinetics of apoptosis induction, as another measure or receptor activation, at the end of the incubation time the unbound ligands were removed from the cells by a wash step and the cells were resuspended in normal growth medium. The cells were then further incubated in total for 12 hrs to allow the execution of the apoptotic programme. As a positive control, ligands were left on the cells for 12 hrs.
rhTRAIL-C3 induced earlier and stronger processing of pro-caspase-8 than WT rhTRAIL, indicating faster and more efficient DR4 activation by the TRAIL variant (Fig. 6A). The stronger and faster DR4 activation by rhTRAIL-C3 was also reflected by the pattern of IκBα phosphorylation and resulting degradation (Fig. 6B). The faster and more robust receptor activation induced by rhTRAIL-C3 was finally mirrored in the level and kinetics of apoptosis induction (Fig. 6C). rhTRAIL-C3 did not only induce higher level of cell death than WT rhTRAIL (67% and 53%, respectively, for full dosage please refer to Fig. 4), but it also required only 14 min. to reach its half-maximal effect, while WT rhTRAIL required 73 min. for the same (Fig. 6C).
Fig 6.
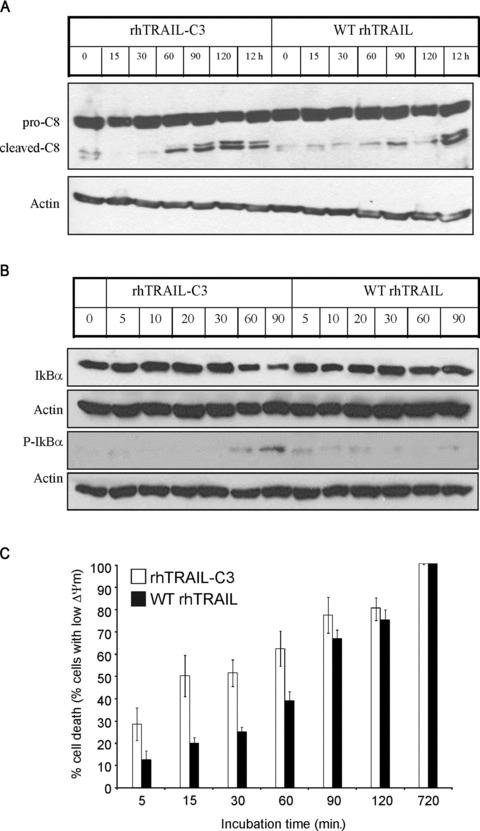
rhTRAIL-C3 activates DR4 more efficiently than WT rhTRAIL. The kinetics of receptor activation was measured by determining (A) pro-caspase-8 cleavage (B) IκB phosphorylation and degradation by Western blot analysis, or (C) apoptosis induction with TMRE staining. HL-60 cells were treated with 100 ng/ml of WT rhTRAIL or rhTRAIL-C3 for the times indicated, after which it was removed from the cells and the cells washed. To detect induction of apoptosis after washing the ligands out, the cells were resuspended in normal growth medium and incubated further for 12 hrs in total. The graph shows induction of apoptosis relative to maximum apoptosis observed at 12 hrs (100%). β-actin was used in the Western blot analyses as a protein loading control.
rhTRAIL-C3 is a potent inducer of apoptosis in primary AML blasts
To test whether rhTRAIL-C3 is a more potent apoptosis inducer than WT rhTRAIL in AML, primary AML blasts were analyzed for TRAIL receptor expression and sensitivity to WT rhTRAIL or rhTRAIL-C3, alone or in combination with the inhibitor κB kinase (IKK) inhibitor, BMS-345541. NF-κB has been shown to regulate the expression of a number of anti-apoptotic genes, including cellular FADD-like Interleukin-1 converting enzyme (FLICE)-inhibitory protein (c-FLIP) and XIAP, two molecules that have been associated with TRAIL resistance of AML and other tumour types [18, 33]. Accordingly, inhibition of NF-κB have been shown to restore TRAIL sensitivity in a range of tumour types [34, 35]. Further supporting the viability of the combination of an NF-κB inhibitor and TRAIL, we have found that HL-60 cells can be greatly sensitized to TRAIL by simultaneous inhibition of NF-κB activation (Fig. S5).
Most samples expressed the TRAIL receptor at a detectable level, and many displayed a high level of decoy receptor (Table 2) expression. The clinical information of the analyzed samples is summarized in Table 3. As expected, the AML samples had generally low TRAIL sensitivity (Fig. 7A). However, analysis of the samples that showed some level of TRAIL sensitivity revealed that rhTRAIL-C3 had a significantly higher pro-apoptotic activity than WT rhTRAIL (Fig. 7A, P= 0.009; paired Student’s t-test).
Table 2.
TRAIL receptor cell surface expression in primary AML blasts
| DR4 | DR5 | DcR1 | DcR2 | |
|---|---|---|---|---|
| 1 | + | − | + | − |
| 2 | + | + | − | +++ |
| 3 | + | + | + | − |
| 4 | − | + | − | ++ |
| 5 | + | + | − | + |
| 6 | + | ++ | − | +++ |
| 7 | − | +++ | ++ | +++ |
| 8 | + | + | + | + |
| 9 | + | ++ | − | ++ |
| 10 | − | − | − | + |
| 11 | + | + | − | + |
| 12 | − | − | + | − |
| 13 | + | + | + | − |
| 14 | ++ | ++ | + | + |
| 15 | + | − | ++ | ++ |
| 16 | − | + | + | − |
| 17 | − | − | − | − |
| 18 | ++ | + | + | ++ |
| 19 | − | + | − | − |
| 20 | + | ++ | − | + |
| 21 | + | +++ | ++ | +++ |
+: Low level of expression (geometric mean on histogram is 120–200% of isotype control); ++: medium level of expression (geometric mean is 200–300% of isotype control); +++: high level of expression (geometric mean is more than 300% of isotype control).
Table 3.
Clinical parameters for patient samples studied for TRAIL receptor
| Sample number | Sex | Age | Primary/secondary? | WBC | Karyotype | Cytogenetic risk group | FABsubtype | Treatment intensity | Clinical Outcome |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 52 | Primary | 21.5 | 45,X,−Y,t(8;21)(q22;q22)[10] | Favourable | M4 | Intensive | CR |
| 2 | M | 51 | Primary | 91.8 | 50,XY,+6,+8,+13,inv(16)(p13q22),+22[10] | Favourable | M4e | Intensive | CR |
| 3 | M | 33 | Primary | 71 | 46,XY,inv(16)(p13q22)[10] | Favourable | M4 | Intensive | CR |
| 4 | M | 77 | Primary | 8.8 | Unknown | Non-intensive | Resistant disease | ||
| 5 | M | 42 | Secondary | 107.4 | 46,XY,inv(16)(p13q22)[10] | Favourable | M4e | Intensive | CR |
| 6 | F | 41 | Primary | 47.1 | 46,XX[20] | Intermediate | M1 | Intensive | CR |
| 7 | M | 81 | Primary | 40 | 46,XY[20] | Intermediate | Unknown | Non-intensive | Induction death |
| 8 | F | 69 | Primary | 73.5 | 45,XX,−7[13] | Adverse | M4 | Non-intensive | Resistant disease |
| 9 | M | 85 | Primary | 45.8 | 46,XY[20] | Intermediate | Unknwon | Non-intensive | CR |
| 10 | F | 80 | Secondary | 89.9 | Unknown | Non-intensive | Induction death | ||
| 11 | M | 77 | Primary | 130 | 46,XY[20] | Intermediate | M1 | Non-intensive | CR |
| 12 | M | 56 | Primary | 31.1 | 46,XY,t(6;9)(p23;q34)[9]/46,XY[1] | Intermediate | M1 | Intensive | CR |
| 13 | M | 60 | Primary | 21 | 46,XY,t(13;19)(q14;p11)[10] | Intermediate | M4 | Intensive | CR |
| 15 | F | 72 | Secondary | 104 | 46,XX[20] | Intermediate | M4 | Non-intensive | Resistant disease |
| 16 | M | 48 | Primary | 171.5 | 46,XY[20] FLT3ITD positive | Intermediate | M1 | Intensive | Resistant disease |
| 17 | M | 62 | Primary | 69.5 | 46,XY,t(9;11)(p21;q23)[10]— | Intermediate | M5a | Intensive | . |
| 18 | M | 68 | Secondary | 20.4 | 45,XY,inv(3)(q21q26),−7[10] | Adverse | Other | Intensive | . |
| 19 | M | 69 | Primary | 35.3 | 47,XY,+21[13]/50,idem,+8,+9,+14[3]/46,XY[4] | Adverse | M4 | Intensive | CR |
| 20 | F | 36 | Primary | 148.9 | 46,XX[20] | Intermediate | M1 | Intensive | CR |
| 21 | M | 85 | Secondary | 69.2 | . | RAEB | Non-intensive | Induction death |
M: male; F: female; WBC: white blood cell count; CR: complete remission; FAB: French–American–British classification.
Fig 7.
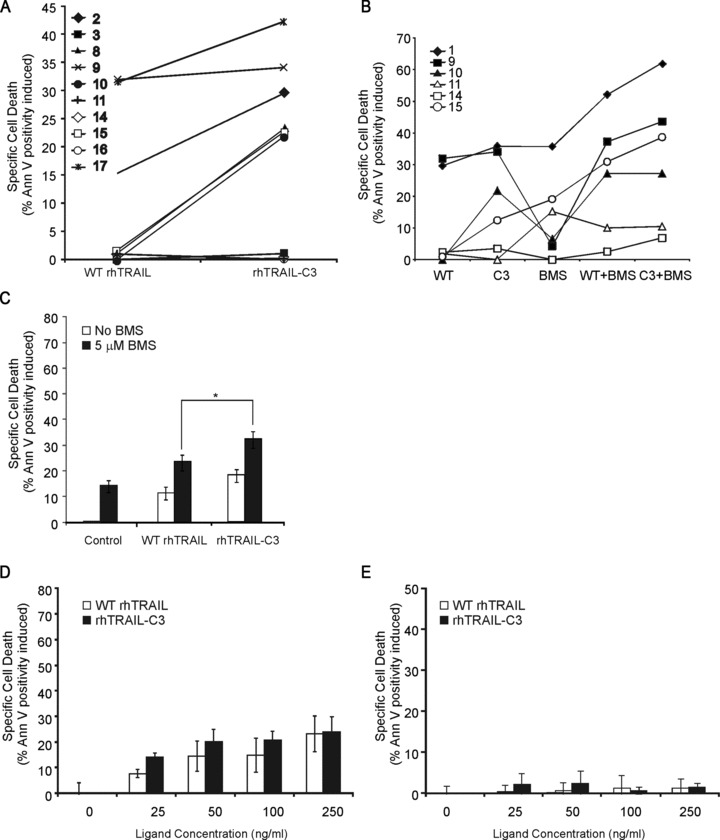
rhTRAIL-C3 is a more potent inducer of AML blast apoptosis than WT rhTRAIL. Primary AML blasts were treated with 250 ng/ml of WT rhTRAIL (WT) or rhTRAIL-C3 (C3) as single agents (A), or in combination with 5 μM BMS-345541 (BMS) for 24 hrs (B). Induction of apoptosis was measured with annexin V staining and expressed as percentage apoptosis induced in the live population. (C) shows the averaged effect of WT rhTRAIL, rhTRAIL-C3 or their combination with 5 μM BMS-345541 in the six samples shown in (B). The * denotes significant differences, P < 0.05 determined with paired t-test. (D, E) rhTRAIL-C3 does not show increased toxicity in non-transformed cells. (D) Primary human dermal fibroblasts and (E) primary normal human epidermal keratinocytes were treated with the indicated doses of WT rhTRAIL or rhTRAIL-C3 for 24 hrs. Induction of cell death was quantified with annexin V assay and flow cytometry. The graphs show the average ± S.E.M. values from at least three independent repeats. There was no significant difference between WT rhTRAIL- and rhTRAIL-C3-induced apoptosis at any of the concentrations tested (P > 0.05 with paired Student’s t-test).
The effect of BMS-345541 on WT rhTRAIL/rhTRAIL-C3 sensitivity was tested in 6 AML samples, including both rhTRAIL sensitive and resistant samples (Fig. 7B, samples 10, 11, 14, 15 and samples 1, 9, respectively). The response of the individual AML samples ranged from no sensitization (samples 11, 14) through additive effect (samples 1, 9, 10) to even synergistic effect (sample 15) to both WT rhTRAIL and rhTRAIL-C3. Additionally, the combination of rhTRAIL-C3 with BMS-345541 resulted in a stronger cytotoxic effect than WT rhTRAIL (Fig. 7C, P= 0.012 for 5 μM BMS-345541 + WT rhTRAIL and 5 μM BMS-345541 + WT rhTRAIL).
While rhTRAIL-C3 showed high activity on primary AML cells, it was not toxic to non-transformed, normal cells (Fig. 7D and E). Human primary dermal fibroblasts as an abundant somatic cell type were tested for sensitivity to rhTRAIL-C3. Additionally, NHEK were also tested as keratinocytes have been reported to be sensitive to rhTRAIL-induced apoptosis [36]. In line with the literature, NHEK cells showed a low level of sensitivity to WT rhTRAIL (Fig. 7E). rhTRAIL-C3 did not induce any cell death in fibroblasts and did not induce significantly more cell death than WT rhTRAIL in keratinocytes (Fig. 7D and E).
Discussion
TRAIL is a promising anti-cancer agent as it selectively induces apoptosis in cancer cells while leaving normal cells unharmed. However, some tumour types, such as AML, are relatively insensitive to TRAIL-induced apoptosis [37–40].
In AML blasts, despite the fact that approximately 50% of AMLs express at least one death-inducing TRAIL receptor on the cell surface, TRAIL fails to activate pro-caspase-8, indicating that inadequate receptor activation by TRAIL contributes to the low sensitivity [17]. Using agonistic antibodies specific to DR4 or DR5, we found that similar to some other leukemic cell types, such as CLL [12], AML cells were more sensitive to DR4 than to DR5 stimulation. At least half of the AML blasts examined here expressed DR4 and approximately 75% expressed DR5 (12 and 16 out of 21, respectively) on the cell surface, however, DR5 expression was associated with high DcR2 expression in 11 out of 16 cases (69%). DcR2 has been shown to associate with and inhibit the function of DR5 [41]. The study by Riccioni et al., using 72 primary AML blasts, demonstrated a strong correlation between decoy receptor expression and TRAIL sensitivity. As in our study, this study also identified that most DR5 expressing blasts also expressed DcR2 (eight of nine samples, 88%) [17]. Overall these results strongly indicate that the death-inducing TRAIL receptors are a major point of control of AML TRAIL sensitivity.
To increase the response of AML cells to TRAIL receptor stimulation, we have engineered a TRAIL variant using computational design. This variant, rhTRAIL-C3 possesses three mutations: a replacement of glycine to arginine at position 131, a replacement of asparagine to arginine at position 199 and a lysine to histidine mutation at position 201. These mutations generated high DR4-specific agonistic activity. This TRAIL mutant (rhTRAIL-C3) displayed significantly stronger pro-apoptotic activity in a range of AML cell lines. rhTRAIL-C3 activated DR4 more efficiently than WT rhTRAIL, resulting in a faster and more robust pro-caspase-8 activation, loss of mitochondrial membrane potential and cell death.
To date there is very little information about the differential regulation of DR4 and DR5. Rossin and colleagues have reported that translocation of DR4 to the lipid rafts and induction of apoptosis required palmitoylation in the membrane proximal, intracellular region of the receptor, while this event was not required for DR5 [42]. As palmitoylation of N-Ras has been shown to be necessary for leukogenic transformation [43], there is a possibility that the same mechanism that activates the Ras pathway also selectively enhances DR4 functionality.
In addition to mechanisms selectively activating DR4, it is also possible that other mechanisms selectively inhibit DR5. There is accumulating evidence that the decoy receptors have differential effects on DR4 and DR5, with DcR2 preferentially regulating DR5 activity [41, 44]. Furthermore, we have previously shown that the short isoform of cellular FLICE-like inhibitory protein (c-FLIPs) preferentially binds to and inhibits apoptotic signalling from the DR5 DISC and its expression can be controlled by early growth response-1 (Egr-1) [45]. Finally, a recent report has shown that similar to our findings on AML, in pancreatic tumours despite the expression of both DR4 and DR5, only DR4 is functional [46]. The study found that protein kinase C activity can selectively inhibit DR5 function [46]. Protein kinase C (PKC) isoforms regulate haemopoietic cell growth, differentiation and activation and thus, PKC-mediated DR5 inhibition may be another explanation why AML cells rarely have functional DR5 receptors.
Having observed an increased induction of apoptosis in several AML cell lines, we tested whether rhTRAIL-C3 also has an enhanced activity in primary AML blasts. rhTRAIL-C3 was indeed a better activator of apoptosis then rhTRAIL WT in primary AML blasts. It should be noted that approximately half of the primary AML samples showed complete resistance to WT rhTRAIL and many of these samples remained resistant to rhTRAIL-C3, but rhTRAIL-C3 showed enhanced activity in all samples which had some sensitivity to WT rhTRAIL.
It has to be noted that a significant proportion of AML cells do not express DR4 on the cell surface. It means that if rhTRAIL-C3 enters therapy, it should be used either in combination with drugs that induce DR4 expression in AML cells, or identify the patient population where the blast cells express DR4. Riccioni and colleagues have reported a comprehensive study of TRAIL receptor expression in primary AML [17]. Similarly to our results, they show that DR4 expression associates with AMLs displaying monocytic or to a lesser extent, myelomonocytic phenotype. Thus, the M5a/M5b FAB classified AML types are potential targets for the rhTRAIL-C3 variant. Additionally, DR4/DR5 typing of blast samples using flow cytometry could be utilized in the clinic to identify patient suitability.
Histone deacetylase inhibitors, such as valproic acid and LAQ824 as well as irradiation have been reported to induce DR4 expression in erythroleukaemias and in certain AMLs and sensitize these leukaemias to TRAIL [47, 48]. Combination of such therapeutics with rhTRAIL-C3 could increase the spread of leukaemias where rhTRAIL-C3 could be efficaceous.
Thus, rhTRAIL-C3 may only work as a single agent only in cases where resistance mechanisms are not present downstream of the TRAIL receptor (e.g. c-FLIP expression, low caspase-8 levels, or overexpression of Mcl-1 and XIAP). There is a strong rationale to combine receptor-selective TRAIL variants with agents that can target such mechanisms [33, 49, 50]. As an example, here we show that combination of WT rhTRAIL and rhTRAIL-C3 with the IKK inhibitor BMS-345541 resulted in increased sensitivity to WT rhTRAIL. However, similar to the single agent studies, the combination of rhTRAIL-C3 with BMS-345541 was more efficient than WT rhTRAIL. It exemplifies that combination of this TRAIL variant with chemotherapeutics or other targeted agents can kill AML with high efficacy. This could improve patient outcome and reduce the toxic side effects typical of the currently applied drug treatments by enabling the use of lower drug doses.
In conclusion, in AML cell lines TRAIL mediates apoptosis primarily via DR4. rhTRAIL-C3 is a novel and more potent TRAIL molecule which has superior DR4-activating potential compared to WT rhTRAIL. The activity of this variant could be further enhanced by inhibiting the DR4-mediated activation of NF-κB indicating that TRAIL variants with strong DR4-specific agonistic activity may represent a novel approach for treating AML either as a single agent, or in combination therapy.
Acknowledgments
This work was financially supported by the EU FP5 and FP6 programs (QLK3-CT-2001–00498, LSHC-CT-2006–037686) A.M.S was partially supported by a Juan de la Cierva grant of the Spanish ministry of Science and Education. R.H.C. was partially supported by the Dutch Technology Foundation STW, applied science division of NWO and the technology program of the Ministry of Economic Affairs.
Conflict of interest
Please note that A. Samali, L. Serrano and W. Quax are founders of Triskel Therapeutics Ltd. and members of its scientific advisory board and R. H. Cools is employed by Triskel Therapeutics Ltd.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1 DR4-selectivity and pro-apoptotic potential ofN199R/K201H and G131R/D218H TRAIL mutants. (A)DR4-responsive ML-1 acute myelogenous leukaemia cells (B)and EM-2 chronic myelogenous leukaemia cells (C) as well asDR5-responding A2780 ovarian carcinoma cells (C) weretreated with increasing doses of WT rhTRAIL, rhTRAIL-C3,G131R/D218H and N199R/K201H for 24 hrs. Induction of apoptosis wasdetermined using annexin V assay and flow cytometric analysis. Thegraphs presented demonstrate the percentage of dead cells ± S.E.M. as determined from three independent experiments.
Fig. S2 Gel filtration elution profile of WT rhTRAIL(A) and rhTRAIL-C3 (B) from Hiload Superdex 75 16/60column.
Fig. S3 Time versus response SPR sensorgrams.Receptor binding of WT rhTRAIL (A, C) and rhTRAIL-C3(B, D) towards DR4-Ig (A, B) and DR5-Ig(C, D). Receptor chimeras were coated at a level of∽600--1000 RU. Purified WT rhTRAIL and rhTRAIL-C3 were injected in3-fold at concentrations ranging from 250 to 2 nM at 70 μl/min.flow rate using HBS-P (Biacore) as running and sample buffer.Binding of ligands to the receptors was monitored in real time at37°C.
Fig. S4 Receptor binding of WT rhTRAIL and rhTRAIL-C3variant determined by SPR and competitive ELISA. Pre-steady statereceptor binding curves of WT rhTRAIL and rhTRAIL-C3 to DR4-Ig(A), or to DR5-Ig (B) as determined by SPR. Theresponse at each concentration was recorded 30 sec. after the endof the injections. Competitive ELISA assay of WT rhTRAIL andrhTRAIL-C3 for TRAIL receptors using immobilized DR4-Ig receptorand soluble DR4-Ig as competitor (C), soluble DR5-Ig ascompetitor (D), soluble DcR1-Ig as competitor (E) orsoluble DcR2-Ig as competitor (F). Binding was calculatedrelative to the value measured in the presence of 0 ng/well ofsoluble receptor.
Fig. S5 Inhibition of NF-κB activity with the IKKinhibitor BMS-345541 (Calbiochem) enhanced the biological activityof both WT rhTRAIL and rhTRAIL-C3. HL-60 cells were treated with100 ng/ml (A) or 10 ng/ml (B) of WT rhTRAIL orrhTRAIL-C3 in the presence or absence of the indicatedconcentrations of BMS-345541 for 12 hrs. Induction of cell deathwas determined by measuring the loss of mitochondrial membranepotential with TMRE. The graphs show average percentage of cellswith low mitochondrial membrane potential (ΔΨm).
References
- 1.Ashkenazi A, Pai RC, Fong S, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–62. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCulloch EA. Stem cells in normal and leukemic hemopoiesis (Henry Stratton Lecture, 1982) Blood. 1983;62:1–13. [PubMed] [Google Scholar]
- 3.Bennett JM, Young ML, Andersen JW, et al. Long-term survival in acute myeloid leukemia: the Eastern Cooperative Oncology Group experience. Cancer. 1997;80:2205–9. [PubMed] [Google Scholar]
- 4.Byrd JC, Mrozek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461) Blood. 2002;100:4325–36. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- 5.Grimwade D, Walker H, Oliver F, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood. 1998;92:2322–33. [PubMed] [Google Scholar]
- 6.Kaufmann SH, Steensma DP. On the TRAIL of a new therapy for leukemia. Leukemia. 2005;19:2195–202. doi: 10.1038/sj.leu.2403946. [DOI] [PubMed] [Google Scholar]
- 7.Bodmer JL, Holler N, Reynard S, et al. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol. 2000;2:241–3. doi: 10.1038/35008667. [DOI] [PubMed] [Google Scholar]
- 8.Kischkel FC, Lawrence DA, Chuntharapai A, et al. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. 2000;12:611–20. doi: 10.1016/s1074-7613(00)80212-5. [DOI] [PubMed] [Google Scholar]
- 9.Sprick MR, Weigand MA, Rieser E, et al. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity. 2000;12:599–609. doi: 10.1016/s1074-7613(00)80211-3. [DOI] [PubMed] [Google Scholar]
- 10.LeBlanc HN, Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003;10:66–75. doi: 10.1038/sj.cdd.4401187. [DOI] [PubMed] [Google Scholar]
- 11.Kimberley FC, Screaton GR. Following a TRAIL: update on a ligand and its five receptors. Cell Res. 2004;14:359–72. doi: 10.1038/sj.cr.7290236. [DOI] [PubMed] [Google Scholar]
- 12.MacFarlane M, Kohlhaas SL, Sutcliffe MJ, et al. TRAIL receptor-selective mutants signal to apoptosis via TRAIL-R1 in primary lymphoid malignancies. Cancer Res. 2005;65:11265–70. doi: 10.1158/0008-5472.CAN-05-2801. [DOI] [PubMed] [Google Scholar]
- 13.Snell V, Clodi K, Zhao S, et al. Activity of TNF-related apoptosis-inducing ligand (TRAIL) in haematological malignancies. Br J Haematol. 1997;99:618–24. doi: 10.1046/j.1365-2141.1997.4393250.x. [DOI] [PubMed] [Google Scholar]
- 14.Walczak H, Miller RE, Ariail K, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5:157–63. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 15.MacFarlane M, Inoue S, Kohlhaas SL, et al. Chronic lymphocytic leukemic cells exhibit apoptotic signaling via TRAIL-R1. Cell Death Differ. 2005;12:773–82. doi: 10.1038/sj.cdd.4401649. [DOI] [PubMed] [Google Scholar]
- 16.Kelley RF, Totpal K, Lindstrom SH, et al. Receptor-selective mutants of apoptosis-inducing ligand 2/tumor necrosis factor-related apoptosis-inducing ligand reveal a greater contribution of death receptor (DR) 5 than DR4 to apoptosis signaling. J Biol Chem. 2005;280:2205–12. doi: 10.1074/jbc.M410660200. [DOI] [PubMed] [Google Scholar]
- 17.Riccioni R, Pasquini L, Mariani G, et al. TRAIL decoy receptors mediate resistance of acute myeloid leukemia cells to TRAIL. Haematologica. 2005;90:612–24. [PubMed] [Google Scholar]
- 18.Suh WS, Kim YS, Schimmer AD, et al. Synthetic triterpenoids activate a pathway for apoptosis in AML cells involving downregulation of FLIP and sensitization to TRAIL. Leukemia. 2003;17:2122–9. doi: 10.1038/sj.leu.2403112. [DOI] [PubMed] [Google Scholar]
- 19.McLornan P, McMullin MF, Mills KI, et al. The prognostic role of c-FLIP in AML. 14th Congress of the European Hematology Association, June 4–7, 2009. Haematologica. 2009:108. . p. abs. 0272. [Google Scholar]
- 20.van der Sloot AM, Tur V, Szegezdi E, et al. Designed tumor necrosis factor-related apoptosis-inducing ligand variants initiating apoptosis exclusively via the DR5 receptor. Proc Natl Acad Sci USA. 2006;103:8634–9. doi: 10.1073/pnas.0510187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holohan C, Szegezdi E, Ritter T, et al. Cytokine-induced beta-cell apoptosis is NO-dependent, mitochondria-mediated and inhibited by BCL-XL. J Cell Mol Med. 2008;12:591–606. doi: 10.1111/j.1582-4934.2007.00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahalingam D, Keane M, Pirianov G, et al. Differential activation of JNK1 isoforms by TRAIL receptors modulate apoptosis of colon cancer cell lines. Br J Cancer. 2009;100:1415–24. doi: 10.1038/sj.bjc.6605021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tur V, van der Sloot AM, Reis CR, et al. DR4-selective tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) variants obtained by structure-based design. J Biol Chem. 2008;283:20560–8. doi: 10.1074/jbc.M800457200. [DOI] [PubMed] [Google Scholar]
- 24.Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320:369–87. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 25.Schymkowitz JW, Rousseau F, Martins IC, et al. Prediction of water and metal binding sites and their affinities by using the fold-X force field. Proc Natl Acad Sci USA. 2005;102:10147–52. doi: 10.1073/pnas.0501980102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Sloot AM, Mullally MM, Fernandez-Ballester G, et al. Stabilization of TRAIL, an all-beta-sheet multimeric protein, using computational redesign. Protein Eng Des Sel. 2004;17:673–80. doi: 10.1093/protein/gzh079. [DOI] [PubMed] [Google Scholar]
- 27.Thomas LR, Johnson RL, Reed JC, et al. The C-terminal tails of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas receptors have opposing functions in Fas-associated death domain (FADD) recruitment and can regulate agonist-specific mechanisms of receptor activation. J Biol Chem. 2004;279:52479–86. doi: 10.1074/jbc.M409578200. [DOI] [PubMed] [Google Scholar]
- 28.Collins SJ, Gallo RC, Gallagher RE. Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature. 1977;270:347–9. doi: 10.1038/270347a0. [DOI] [PubMed] [Google Scholar]
- 29.Matsuo Y, MacLeod RA, Uphoff CC, et al. Two acute monocytic leukemia (AML-M5a) cell lines (MOLM-13 and MOLM-14) with interclonal phenotypic heterogeneity showing MLL-AF9 fusion resulting from an occult chromosome insertion, ins(11;9) (q23;p22p23) Leukemia. 1997;11:1469–77. doi: 10.1038/sj.leu.2400768. [DOI] [PubMed] [Google Scholar]
- 30.Ohyashiki K, Ohyashiki JH, Sandberg AA. Cytogenetic characterization of putative human myeloblastic leukemia cell lines (ML-1, -2, and -3): origin of the cells. Cancer Res. 1986;46:3642–7. [PubMed] [Google Scholar]
- 31.Quentmeier H, Martelli MP, Dirks WG, et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia. 2005;19:1760–7. doi: 10.1038/sj.leu.2403899. [DOI] [PubMed] [Google Scholar]
- 32.Duiker EW, de Vries EG, Mahalingam D, et al. Enhanced antitumor efficacy of a DR5-specific TRAIL variant over recombinant human TRAIL in a bioluminescent ovarian cancer xenograft model. Clin Cancer Res. 2009;15:2048–57. doi: 10.1158/1078-0432.CCR-08-1535. [DOI] [PubMed] [Google Scholar]
- 33.Carter BZ, Mak DH, Schober WD, et al. Triptolide sensitizes AML cells to TRAIL-induced apoptosis via decrease of XIAP and p53-mediated increase of DR5. Blood. 2008;111:3742–50. doi: 10.1182/blood-2007-05-091504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braeuer SJ, Buneker C, Mohr A, et al. Constitutively activated nuclear factor-kappaB, but not induced NF-kappaB, leads to TRAIL resistance by up-regulation of X-linked inhibitor of apoptosis protein in human cancer cells. Mol Cancer Res. 2006;4:715–28. doi: 10.1158/1541-7786.MCR-05-0231. [DOI] [PubMed] [Google Scholar]
- 35.Jeremias I, Kupatt C, Baumann B, et al. Inhibition of nuclear factor kappaB activation attenuates apoptosis resistance in lymphoid cells. Blood. 1998;91:4624–31. [PubMed] [Google Scholar]
- 36.Leverkus M, Neumann M, Mengling T, et al. Regulation of tumor necrosis factor-related apoptosis-inducing ligand sensitivity in primary and transformed human keratinocytes. Cancer Res. 2000;60:553–9. [PubMed] [Google Scholar]
- 37.Eggert A, Grotzer MA, Zuzak TJ, et al. Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res. 2001;61:1314–9. [PubMed] [Google Scholar]
- 38.Fulda S, Kufer MU, Meyer E, et al. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene. 2001;20:5865–77. doi: 10.1038/sj.onc.1204750. [DOI] [PubMed] [Google Scholar]
- 39.Hinz S, Trauzold A, Boenicke L, et al. Bcl-XL protects pancreatic adenocarcinoma cells against CD95- and TRAIL-receptor-mediated apoptosis. Oncogene. 2000;19:5477–86. doi: 10.1038/sj.onc.1203936. [DOI] [PubMed] [Google Scholar]
- 40.Jones DT, Ganeshaguru K, Mitchell WA, et al. Cytotoxic drugs enhance the ex vivo sensitivity of malignant cells from a subset of acute myeloid leukaemia patients to apoptosis induction by tumour necrosis factor receptor-related apoptosis-inducing ligand. Br J Haematol. 2003;121:713–20. doi: 10.1046/j.1365-2141.2003.04340.x. [DOI] [PubMed] [Google Scholar]
- 41.Clancy L, Mruk K, Archer K, et al. Preligand assembly domain-mediated ligand-independent association between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced apoptosis. Proc Natl Acad Sci USA. 2005;102:18099–104. doi: 10.1073/pnas.0507329102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossin A, Derouet M, Abdel-Sater F, et al. Palmitoylation of the TRAIL receptor DR4 confers an efficient TRAIL-induced cell death signalling. Biochem J. 2009;419:185–92. doi: 10.1042/BJ20081212. [DOI] [PubMed] [Google Scholar]
- 43.Cuiffo B, Ren R. Palmitoylation of oncogenic NRAS is essential for leukemogenesis. Blood. 2010;115:3598–605. doi: 10.1182/blood-2009-03-213876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Merino D, Lalaoui N, Morizot A, et al. Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol Cell Biol. 2006;26:7046–55. doi: 10.1128/MCB.00520-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahalingam D, Natoni A, Keane M, et al. Early growth response-1 is a regulator of DR5-induced apoptosis in colon cancer cells. Br J Cancer. 2010;102:754–64. doi: 10.1038/sj.bjc.6605545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lemke J, Noack A, Adam D, et al. TRAIL signaling is mediated by DR4 in pancreatic tumor cells despite the expression of functional DR5. J Mol Med. 2010;88:729–40. doi: 10.1007/s00109-010-0619-0. [DOI] [PubMed] [Google Scholar]
- 47.Guo F, Sigua C, Tao J, et al. Cotreatment with histone deacetylase inhibitor LAQ824 enhances Apo-2L/tumor necrosis factor-related apoptosis inducing ligand-induced death inducing signaling complex activity and apoptosis of human acute leukemia cells. Cancer Research. 2004;64:2580–9. doi: 10.1158/0008-5472.can-03-2629. [DOI] [PubMed] [Google Scholar]
- 48.Iacomino G, Medici MC, Russo GL. Valproic acid sensitizes K562 erythroleukemia cells to TRAIL/Apo2L-induced apoptosis. Anticancer Research. 2008;28:855–64. [PubMed] [Google Scholar]
- 49.Rosato RR, Almenara JA, Coe S, et al. The multikinase inhibitor sorafenib potentiates TRAIL lethality in human leukemia cells in association with Mcl-1 and cFLIPL down-regulation. Cancer Res. 2007;67:9490–500. doi: 10.1158/0008-5472.CAN-07-0598. [DOI] [PubMed] [Google Scholar]
- 50.Tazzari PL, Tabellini G, Ricci F, et al. Synergistic proapoptotic activity of recombinant TRAIL plus the Akt inhibitor Perifosine in acute myelogenous leukemia cells. Cancer Res. 2008;68:9394–403. doi: 10.1158/0008-5472.CAN-08-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 DR4-selectivity and pro-apoptotic potential ofN199R/K201H and G131R/D218H TRAIL mutants. (A)DR4-responsive ML-1 acute myelogenous leukaemia cells (B)and EM-2 chronic myelogenous leukaemia cells (C) as well asDR5-responding A2780 ovarian carcinoma cells (C) weretreated with increasing doses of WT rhTRAIL, rhTRAIL-C3,G131R/D218H and N199R/K201H for 24 hrs. Induction of apoptosis wasdetermined using annexin V assay and flow cytometric analysis. Thegraphs presented demonstrate the percentage of dead cells ± S.E.M. as determined from three independent experiments.
Fig. S2 Gel filtration elution profile of WT rhTRAIL(A) and rhTRAIL-C3 (B) from Hiload Superdex 75 16/60column.
Fig. S3 Time versus response SPR sensorgrams.Receptor binding of WT rhTRAIL (A, C) and rhTRAIL-C3(B, D) towards DR4-Ig (A, B) and DR5-Ig(C, D). Receptor chimeras were coated at a level of∽600--1000 RU. Purified WT rhTRAIL and rhTRAIL-C3 were injected in3-fold at concentrations ranging from 250 to 2 nM at 70 μl/min.flow rate using HBS-P (Biacore) as running and sample buffer.Binding of ligands to the receptors was monitored in real time at37°C.
Fig. S4 Receptor binding of WT rhTRAIL and rhTRAIL-C3variant determined by SPR and competitive ELISA. Pre-steady statereceptor binding curves of WT rhTRAIL and rhTRAIL-C3 to DR4-Ig(A), or to DR5-Ig (B) as determined by SPR. Theresponse at each concentration was recorded 30 sec. after the endof the injections. Competitive ELISA assay of WT rhTRAIL andrhTRAIL-C3 for TRAIL receptors using immobilized DR4-Ig receptorand soluble DR4-Ig as competitor (C), soluble DR5-Ig ascompetitor (D), soluble DcR1-Ig as competitor (E) orsoluble DcR2-Ig as competitor (F). Binding was calculatedrelative to the value measured in the presence of 0 ng/well ofsoluble receptor.
Fig. S5 Inhibition of NF-κB activity with the IKKinhibitor BMS-345541 (Calbiochem) enhanced the biological activityof both WT rhTRAIL and rhTRAIL-C3. HL-60 cells were treated with100 ng/ml (A) or 10 ng/ml (B) of WT rhTRAIL orrhTRAIL-C3 in the presence or absence of the indicatedconcentrations of BMS-345541 for 12 hrs. Induction of cell deathwas determined by measuring the loss of mitochondrial membranepotential with TMRE. The graphs show average percentage of cellswith low mitochondrial membrane potential (ΔΨm).