

Abstract
Objective
Many forms of epilepsy are associated with aberrant neuronal connections, but the relationship between such pathological connectivity and the underlying physiological predisposition to seizures is unclear. We sought to characterize the cortical excitability profile of a developmental form of epilepsy known to have structural and functional connectivity abnormalities.
Methods
We employed transcranial magnetic stimulation (TMS) with simultaneous EEG recording in eight patients with epilepsy from periventricular nodular heterotopia (PNH) and matched healthy controls. We used connectivity imaging findings to guide TMS targeting and compared the evoked responses to single-pulse stimulation from different cortical regions.
Results
Heterotopia patients with active epilepsy demonstrated a relatively augmented late cortical response that was greater than that of matched controls. This abnormality was specific to cortical regions with connectivity to subcortical heterotopic gray matter. Topographic mapping of the late response differences showed distributed cortical networks that were not limited to the stimulation site, and source analysis in one subject revealed that the generator of abnormal TMS-evoked activity overlapped with the spike and seizure onset zone.
Interpretation
Our findings indicate that patients with epilepsy from gray matter heterotopia have altered cortical physiology consistent with hyperexcitability, and that this abnormality is specifically linked to the presence of aberrant connectivity. These results support the idea that TMS-EEG could be a useful biomarker in epilepsy in gray matter heterotopia, expand our understanding of circuit mechanisms of epileptogenesis, and have potential implications for therapeutic neuromodulation in similar epileptic conditions associated with deep lesions.
Introduction
Epilepsy is one of the most common, disabling, and costly neurological disorders in the world. In many forms of epilepsy, both acquired and developmental, aberrant connections involving cortical neurons appear to be pathogenically important.1-3 Such circuitry has been associated in animal models with both local disturbances of cortical excitability as well as functional alterations in larger brain networks.4-5 Unfortunately, our ability to investigate these physiological changes in patients with epilepsy is limited. Intracranial electrode recordings have shown signs of hyperexcitability within epileptogenic cortex as well as excessive synchrony between aberrantly connected regions of gray matter.2 However, these studies require neurosurgical implantation and do not allow for significant experimental control or manipulation.
The unique developmental brain malformation of periventricular nodular heterotopia (PNH) offers an opportunity to study epileptogenic circuits and focal hyperexcitability in an anatomically well-characterized and often genetically determined disorder that leads to a seizure disorder only after an average latency of 20 years from birth.6-11 PNH is a commonly encountered epileptic brain malformation, has a distinct radiological appearance that facilitates its initial diagnosis (Fig 1),12 and generally presents with the clinical triad of epilepsy, reading disability, and normal intelligence.13,14
Figure 1. Anatomy and functional connectivity in patients with PNH.

T1-weighted MR brain images show unilateral posterior gray matter heterotopia along the wall of the lateral ventricle in one patient, subject 3 in Table 1 (sagittal image in A), and diffuse bilateral periventricular heterotopic nodules in another patient, subject 7 (axial image in B), as indicated by red arrows. BOLD images acquired in these patients reveal discrete regions of cerebral cortex that demonstrate aberrant functional connectivity with the heterotopic gray matter during the task-free resting state (green areas show significant functional activation in C and D); these regions were then chosen as cortical targets for TMS in our experimental design.
We previously demonstrated that the deep nodules of heterotopic gray matter in PNH are structurally and functionally connected to discrete foci of overlying cerebral cortex, and that the strength of this abnormal connectivity is higher among patients with longer durations of epilepsy.15 In addition, we showed systematically that periventricular nodules can be metabolically co-activated with cortical regions during the performance of specific cognitive tasks, supporting the notion that these heterotopic nodules become integrated into functional cortical circuits.16
We hypothesized that epileptogenesis in this disorder is related specifically to focal hyperexcitability in cortical regions that have aberrant connectivity to the deep heterotopia, based on results from functional imaging, intracranial electroencephalography (EEG), and surgical outcome studies suggesting that particular areas of cortex might be critical to the generation of an epileptic state.7,9-11,17 Proof of this hypothesis could potentially have broad mechanistic and therapeutic implications for similar forms of epilepsy, particularly those with a long latency to seizure onset, abnormal circuitry demonstrable by neuroimaging, or deep lesions inaccessible to noninvasive forms of antiepileptic brain modulation. A demonstration of cortical hyperexcitability that is spatially restricted to regions of abnormal functional connectivity would also expand our understanding of resting-state functional connectivity, as connectivity abnormalities have been identified in numerous neurological and psychiatric conditions but direct evidence that they have electrophysiologic significance is limited.18,19
In this study, we employed single-pulse transcranial magnetic stimulation (TMS) with simultaneous scalp EEG recording to investigate the cortical excitability profile of patients with PNH. TMS-EEG is a safe and noninvasive technique that has been used to probe cortical activity in a variety of seizure disorder subtypes.20-22 By using connectivity imaging findings to guide TMS targeting in a novel experimental design, we sought to determine the physiological implications of aberrant connectivity in this epileptic brain disorder.
Methods
Subject recruitment
Patients with epilepsy related to PNH were drawn from a research database of individuals with malformations of cortical development who had participated in prior studies, and whose initial connectivity imaging results were previously reported by us.15 Patients with a neuroimaging-confirmed diagnosis of PNH based on the presence of at least two visible nodules of heterotopic gray matter adjacent to the lateral ventricle, each seen on more than one plane of sequence and on at least two consecutive images in one of those planes, were eligible to be enrolled. Those with prior brain surgery, inability to tolerate magnetic resonance imaging (MRI), or a specific MRI or TMS contraindication (including pregnancy) as set forth in standard institutional research protocols were excluded. Healthy control subjects, recruited from an existing research database and through advertisements, were matched to the PNH subjects by age (within 5 years), gender, and handedness. All control subjects were free of neurological symptoms and had normal anatomical brain MR images. All study participants provided written informed consent in accordance with research protocols approved by the institutional review boards of Beth Israel Deaconess Medical Center and the Massachusetts Institute of Technology.
Brain image acquisition
Anatomical images and functional connectivity (fc) MRI images were acquired in all PNH subjects as previously described15 on a Siemens 3-Tesla Magnetom Trio Tim system using a commercial 12-channel matrix head coil (Siemens Medical Solutions, Erlangen, Germany) and tetrahedron-shaped foam pads to minimize head movement. Sagittal localizer scans were aligned to a multisubject atlas to derive automatic slice prescription for consistent head position across subjects. High-resolution structural whole-brain images were acquired using a T1-weighted sequence with 128 slices per slab, a 256 · 256 matrix, a field of view (FOV) of 256 mm, a slice thickness of 1.33 mm with 0.63 mm interslice gap, repetition time (TR) of 2,530 milliseconds (ms), inversion time of 1,100 ms, echo time (TE) of 3.39 ms, and flip angle of 7 degrees. Resting-state functional image acquisition was performed while subjects were asked to rest quietly (acquisition time 6.4 min), using an echo-planar sequence sensitive to blood oxygenation level–dependent (BOLD) contrast with TR of 6,000 ms, TE of 30 ms, FOV of 256 mm, voxel size of 2.0 · 2.0 · 2.0 mm, and flip angle of 90 degrees.
Functional connectivity analysis
fcMRI analyses were performed on the functional image data acquired during the task-free resting state with an in-house software toolbox, using methods previously described.23,24 Functional images were realigned and coregistered to anatomic images for each PNH subject, without normalization. Images were segmented and BOLD signal was extracted. A band-pass filter (0.01 Hz < f < 0.1 Hz) was applied, and Gaussian smoothing was performed (6 mm full width at half maximum). Several possible confounding sources of noise were identified and removed.25 For each subject, multiple regions of interest (ROI), which together encompassed the entire volume of heterotopic gray matter, were manually outlined in native space using MRICroN software26 and served as seed regions for analysis. Color-coded functional connectivity maps were created for each seed ROI, showing correlations between the average BOLD signal time series of the ROI and that of every voxel in the brain, subject to a voxel-wise statistical threshold of p < 0.001 and cluster thresholding with an intensity cutoff as previously described.15
Stimulation target creation
For each PNH subject, two regions of interest (the “connected region” and the “non-connected region”) were determined based on the functional connectivity results. The connected region was manually outlined in MRICroN as a discrete region of cortex that demonstrated significant functional connectivity to a gray matter heterotopia ROI in the subject, based on the analyses described above (Fig. 1). The non-connected region served as a control site and was manually outlined as a separate, discrete region of cortex that met the following criteria: 1) located within the same hemisphere as the connected region but at least 2.5 cm away from it across the cortical surface (to avoid neighborhood stimulation effects during TMS); 2) contained the same volume of brain tissue as the connected region; 3) demonstrated no evidence of significant functional connectivity (as identified by the methods described) to any gray matter heterotopia ROI in the subject. Targets for neuronavigated TMS were placed in the center of the connected and non-connected regions. The exact MNI coordinates for each target in each PNH subject were then used for creation of the corresponding target in the matched healthy control subject. Targets were transferred directly into the TMS navigation software for visualization during the stimulation paradigm.
TMS-EEG experimental setup
TMS was performed with a Nexstim eXimia stimulator with real-time MRI neuronavigation (Nexstim NBS software version 3.2.1, Nexstim Ltd, Helsinki, Finland) to ensure accurate stimulation of the targets defined by resting-state fcMRI. Monophasic pulses of TMS were administered via figure-of-eight coils (mean diameter 59 mm, outer diameter 70 mm), with the coil handle oriented posteriorly, perpendicular to the long axis of the target gyrus. Electroencephalography (EEG) was recorded with a 60-channel TMS-compatible system (eXimia EEG, Nexstim Ltd), which utilizes a sample-and-hold circuit that holds the amplifier input constant from 100 μs pre-stimulus to 2 ms post-stimulus to avoid amplifier saturation by TMS.27 EEG signals were referenced to an additional electrode on the forehead, filtered (0.1 – 500 Hz) and sampled at 1450 Hz with 16 bit resolution. Two extra sensors recorded electrooculogram (EOG). Electrode impedance was kept below 5 kΩ at all times. Resting motor threshold (RMT) was determined via surface electromyography (EMG) recorded using pre-gelled disposable Ag/AgCl electrodes, with the active electrode over the first dorsal interosseus (FDI) muscle, the reference electrode over the metacarpophalangeal joint, and the ground electrode over the wrist (Table 1).
Table 1.
Characteristics of patients with periventricular nodular heterotopia.
| Subject | Sex | Age (years) |
Epilepsy duration (years) |
Seizure frequency |
Interval since last seizure (days) |
AEDs | Heterotopia (number, laterality) |
Interictal EEG findings |
Resting motor threshold, patient/control (% of MSO) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 42 | 34 | ~1 seizure/week | 7 | levetiracetam, lorazepam, oxcarbazepine, pregabalin | 3 right 3 left |
normal | 80%/58% |
| 2 | M | 23 | 15 | ~1 seizure/month | 21 | valproate, ezogabine, lacosamide | 3 right | right frontal and temporal spikes | 62%/75% |
| 3 | F | 20 | 6 | ~1 seizure/month | 41 | lamotrigine | 2 right ~3 left |
normal | 92%/62% |
| 4 | F | 30 | 5 | ~1 seizure/month | 14 | lamotrigine, levetiracetam | ~7 right | normal | 75%/78% |
| 5 | M | 22 | 5 | ~2 seizures/year | 216 | oxcarbazepine | 1 right ~5 left |
normal | 67%/56% |
| 6 | F | 28 | 6 | no seizures in 7 years | 2484 | none | 2 right | right fronto-temporal slowing | 57%/43% |
| 7 | F | 34 | 10 | no seizures in 10 years | 3726 | carbamazepine | ~8 right ~5 left |
normal | 78%/37% |
| 8 | F | 43 | 20 | no seizures in 14 years | 5110 | lamotrigine | 2 right ~4 left |
right temporal spikes | 81%/48% |
Abbreviations: M = male; F = female; AED = antiepileptic drug; EEG = electroencephalography; MSO = maximal stimulator output. Data in columns 3-7 were recorded at the time of transcranial magnetic stimulation. Resting motor thresholds are reported for each heterotopia subject and his or her matched healthy control.
TMS-EEG data acquisition
Subjects were seated in a comfortable chair, and were asked to keep their eyes open, stare straight ahead and maintain a relaxed state during stimulation. They were continually monitored for signs of drowsiness. All subjects wore earplugs to minimize risk of acoustic trauma.28 At the beginning of each experiment, the RMT was determined by applying single pulses of TMS to motor cortex ipsilateral to the connected target site while the coil was placed at the optimal position for eliciting motor evoked potentials (MEPs) from the contralateral FDI muscle. The RMT was conventionally defined as the minimum stimulus intensity that elicited a MEP of at least 50 μV in at least five out of ten trials.29 Up to 80 single pulses of TMS at an intensity of 120% RMT were then administered every 5-6 seconds to the connected target and the non-connected target, with simultaneous EEG recording. The order of stimulation of the target sites was randomized across subjects. In one subject (number 3 in Table 1), 120% RMT was greater than maximum stimulator output (MSO); in this subject, as well as in the matched control, single pulses of TMS were administered at 100% RMT (92% MSO).
TMS-EEG data preprocessing
TMS-EEG data were processed offline using the EEGLab toolbox30 and custom scripts running in MATLAB R2012b (The Mathworks, Inc., Natick, MA, USA). To minimize the effects of large-amplitude early TMS artifacts on subsequent preprocessing steps, and for visualization purposes, EEG datapoints during the period from 0 ms to 40 ms after each stimulation pulse were replaced by interpolating between the potentials recorded at these two time points with a half-Gaussian curve with ends matched to the signal at 0 and 40 ms. Since our analyses were restricted to the time period from 100 ms after the pulse onward, this had no impact on our analyses. The TMS-EEG data were then downsampled to 1000 Hz, bandpass filtered between 1 and 100 Hz, and notch filtered at 60 Hz using a zero-phase finite impulse response (FIR) filter. The continuous data were epoched from 1000 ms before to 2000 ms after each TMS pulse. Epochs were corrected with respect to the TMS-free pre-stimulus baseline period (−1000 ms to −100 ms). Each epoch was manually reviewed (by multiple researchers independently), and electrodes and epochs contaminated by significant muscle, movement, bad signal quality or high-amplitude artifacts were removed. The data were then re-referenced to an average reference. Independent component analysis (ICA) was subsequently performed to identify and remove components reflecting residual muscle activity, eye movements, blink-related activity, and residual stimulation-related artifacts. Components were identified as artifactual based on their topography, time courses, spectral characteristics, and association with EOG and/or stimulation pulse. After ICA cleaning, deleted electrodes were recomputed using a spherical spline interpolation, and the resulting datasets were low-pass filtered at 50 Hz. The average (across epochs) TMS-evoked potential was then calculated at each electrode for each subject. To minimize the effects of any residual early TMS-evoked artifacts on our results, we focused our subsequent analysis on the time period from 100 to 1000 ms after each stimulation pulse.
TMS-EEG data analysis
Global Mean Field Potential Analysis
For each subject, the Global Mean Field Potential (GMFP)31 was calculated as a function of time. The GMFP has been used as a measure of the global brain response to TMS,32,33 and is calculated using the equation:
where K is the number of electrodes, Vi(t) is the voltage measured at electrode i at time t, and Vmean(t) is the mean voltage across electrodes at time t.
In order to test our hypothesis that focal cortical hyperexcitability exists in patients with PNH and epilepsy, we segmented the TMS-evoked response into four time periods for analysis, based on prior studies demonstrating the normal response of primary motor cortex to single-pulse stimulation,32 and studies reporting specific abnormalities of stimulation-evoked potentials (recorded by EMG and EEG) in patients with epilepsy.21,34,35 The time periods were: (1) 100 – 225 ms after the TMS pulse, corresponding to the period during which evoked activity is normally present (at least with stimulation of primary motor cortex); (2) 225 – 400 ms after the TMS pulse, corresponding to the period that showed the greatest differences between epilepsy patients and healthy controls in prior TMS-EMG studies of primary motor cortex;32 (3) 400 – 700 ms after the TMS pulse; and (4) 700 – 1000 ms after the TMS pulse, to evaluate for abnormal delayed activity, as a previous TMS-EEG study suggested might be present in patients with epilepsy.21
For each subject, we calculated the area under the curve (AUC) of the baseline-corrected GMFP (AUC-GMFP) produced by stimulation of the connected target and the non-connected target during each time period. The absolute magnitude of the evoked response can vary widely between individuals because of factors independent of cortical physiology (such as skull-cortex distance and individual brain anatomy), and as a function of brain region. To enable comparative assessments of the late elements of the evoked response independent of the absolute magnitude, we calculated the ratios of the delayed response to the early response. Specifically, we divided the AUC-GMFP during the three later time periods (225-400 ms, 400-700 ms, and 700-1000 ms) by the AUCGMFP during the initial time period after TMS stimulation (100 – 225 ms). We refer to the resulting values as the “normalized AUC-GMFP.” To obtain another measure of the evoked response, in the time periods showing significant differences on the normalized AUC-GMFP measures, we also identified the maximum amplitude of the largest peak in the GMFP produced by stimulation of the connected target and the non-connected target for each subject. The amplitudes of the largest peaks in these time periods were subsequently normalized by the amplitude of the largest peak in the 100 – 225 ms time period.
Group comparisons
We evaluated differences in the TMS-evoked activity between all PNH subjects (regardless of epilepsy status) and their matched controls. For both the connected targets and the non-connected targets, we assessed the significance of differences in the raw AUC-GMFP and in the normalized AUC-GMFP between all subjects with PNH and their matched controls using paired t-tests. For the raw AUC-GMFP, as there were four time periods being compared for each stimulation site, differences were defined as significant at a p-value of less than 0.0125 (Bonferroni corrected p < 0.05). For the normalized AUC-GMFP, as there were three time periods being compared, the significance threshold was set at a p-value of less than 0.0167 (Bonferroni corrected p < 0.05). For the time periods that demonstrated significant group differences in AUC-GMFP measurements, we also evaluated group differences in the normalized amplitude of the maximum GMFP peaks, using paired t-tests with a significance threshold of Bonferroni-corrected p < 0.05. Finally, to further evaluate the time course of the evoked response, we also determined the mean GMFP amplitudes (across subjects) over time, and conducted paired t-tests at each time point. To correct for multiple comparisons, we used a false discovery rate (FDR) threshold of < 0.0536, as this has been suggested to be optimal for exploratory studies of evoked potential data.37 Since our specific hypothesis predicted that abnormalities in cortical excitability would be most prominent in patients with active epilepsy, we repeated these analyses in the subset of five PNH subjects with at least one seizure in the past year (and their matched controls).
Similarly, to assess whether there were systemic differences in the evoked response produced by stimulation of the pathologically connected versus non-connected target sites, we examined differences in the raw AUC-GMFP and normalized AUC-GMFP produced by stimulation of the connected versus non-connected targets in PNH subjects using paired t-tests, correcting for multiple comparisons done at each target. We also determined the mean GMFP amplitudes over time, as above, using paired t-tests and a FDR threshold < 0.05. To ensure that any resulting differences were not simply due to intrinsic differences in excitability between the cortical regions represented among the connected and non-connected targets, we determined these values with stimulation of the connected and non-connected sites in the matched control subjects as well.
Spatial distribution of abnormal evoked activity
The GMFP analysis provides a measure of the global response to TMS. We also examined the spatial topography of the abnormalities in evoked activity uncovered by GMFP analysis. For each electrode, the root mean square voltage (RMSV) during each time period was determined in each subject:
where Vi(t) is the voltage measured at electrode i at time t, ttp is the beginning of the time period (100, 225, 400 and 700 ms) and Ttp represents the ending of the time period (225, 400, 700 and 1000 ms). As in the normalized AUC-GMFP analyses, the single-electrode RMSV during the later time periods (225-400 ms, 400-700 ms, 700-1000 ms) was normalized by dividing by the RMSV during the initial time period (100-225 ms), and the resulting normalized RMSV was subsequently log transformed for variance stabilization and to facilitate graphical representation of changes in the relative amplitude of evoked activity.
To isolate the regions that showed differential delayed-activity between PNH subjects with active epilepsy and their matched controls, the log-transformed normalized RMSV from the healthy controls were subtracted from the values obtained from stimulation of the same site in the corresponding PNH subjects. A positive result indicates that the normalized RMSV was greater in the PNH subject than in the matched control subject, whereas a negative result indicates the converse.
Analysis of local evoked activity
To further evaluate whether abnormalities in evoked activity were due solely to changes in local (versus distributed) activity, we examined the TMS-evoked responses in the region of stimulation. For each subject, the neighborhood of electrodes immediately surrounding the site of stimulation was defined as the region of interest. For each time point, the mean of the absolute value of the voltages across electrodes (in this region of interest) was determined (referred to as Vl):
where Vi(t) is the voltage measured at electrode i at time t and Kn is the set of neighboring electrodes.
As in the AUC-GMFP analysis, the AUC of the mean absolute local voltage (AUC-local) was determined for each time period (100-225 ms, 225-400 ms, 400-700 ms, and 700-1000 ms), and the AUC-local during the later three time periods was normalized by dividing by the AUC-local during the initial time period. Significant differences between PNH subjects with active epilepsy and their matched healthy controls were assessed via paired t-tests, adjusted for multiple comparisons.
Electrical source imaging of evoked activity
One PNH subject (subject 2 in Table 1) had interictal epileptiform discharges and ictal recordings available on conventional scalp EEG (with 19 channels and a 10-20 montage). For this subject, we investigated whether abnormal TMS-evoked EEG activity had generator sources that spatially coincided with the known epileptogenic zone by comparing the electrical source analysis of these TMS-EEG peaks with that of the two most prominent available interictal discharges and the two available ictal onsets.
Source analysis of EEG data was performed using minimum norm estimate (MNE) software38,39 with anatomical surfaces reconstructed using the Freesurfer package.40 MNE provides a distributed source estimate of cortical currents incorporating constraints from a subject's MRI, transforming the data to brain space without requiring heuristic choices or strong assumptions about the sources. Electrode location for this subject was not available (as an interpretable digitized file); thus, in order to find the electrode location, a file from an alternate patient with similar approximate head size was used. The subject's cortical surface was reconstructed from T1-weighted magnetization-prepared rapid acquisition gradient-echo (MPRAGE) data.40 Electrode coordinates were aligned using the nasion and auricular points as fiducial markers. Head modeling utilized a three-layer boundary element method (BEM) model that was generated using the reconstructed cortical surface and fast low-angle shot (FLASH) MRI data, composed of the scalp, skull and brain with electrical conductivities of 0.33 S/m, 0.0042 S/m and 0.33 S/m, respectively.38 A three-dimensional grid with 5 mm spacing was used to form the solution space. The forward solution was calculated by using the BEM. The inverse operator was computed from the forward solution with a loose orientation constraint of 0.6 to eliminate implausible sources and 2 μV as the estimate of EEG noise. The resulting source data was thresholded to identify the region of maximal activity.
Results
Study population
Eight patients with PNH were recruited (Table 1), along with eight age-, gender-, and handedness-matched healthy control individuals. All PNH subjects had a history of at least one seizure in their lifetime; five (62%) had active epilepsy, with at least one seizure in the year prior to the TMS study visit. Seizure frequencies in these five subjects ranged from approximately one seizure per week to approximately one seizure every six months; interictal epileptiform discharges (IEDs) on EEG were present in one of these five. The remaining three PNH subjects (38%) had well-controlled epilepsy, with no seizures in the preceding five years; one of these three had IEDs on EEG. All PNH subjects had at least two heterotopic gray matter nodules; five (62%) had bilateral nodules.
Stimulation targets
A target site functionally connected to a region of periventricular heterotopic gray matter (“connected target”) and another target site within the same hemisphere without abnormal functional connectivity (“non-connected target”) were defined for each PNH subject as described in Methods. The targets were right hemispheric in five subjects (62%) and left hemispheric in three. The connected targets were located in the frontal (25%), temporal (25%) and parietal (50%) lobes (Fig. 2). The non-connected targets were located in the frontal (50%), parietal (38%) and occipital (12%) lobes.
Figure 2. Connectivity-derived targets for cortical stimulation in patients with PNH.
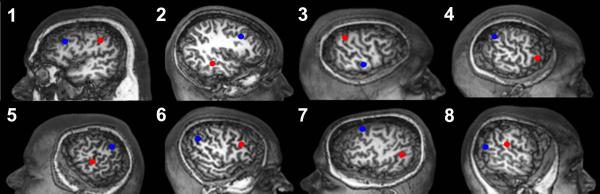
Cortical regions that demonstrate significant resting-state functional connectivity to periventricular gray matter nodules in eight patients with heterotopia, labeled according to subject number as listed in Table 1 and shown on surface brain MR renderings, were identified. Connected targets (red) for neuronavigation-guided TMS were then placed within these regions in each case. Non-connected cortical targets (blue), in regions without such connectivity, were also identified within the same hemisphere for each subject. Each control subject had neuronavigation-guided TMS of the same targets as his or her matched heterotopia subject.
TMS-evoked potentials and GMFP analyses
TMS-evoked potentials
In both PNH subjects and matched controls, TMS evoked a time-locked response characterized by recurrent waves of activity that evolved in amplitude and spatial distribution, lasting several hundred milliseconds (e.g., Fig. 3 A-E). The precise pattern of activity varied as a function of the stimulation site, and thus varied between the connected and non-connected targets, and between subject pairs (for example, note the different patterns evoked by stimulation of a different site in another PNH subject and healthy control pair in Fig. 4 A-B; see also Supplemental Fig. S1 for the raw GMFPs provoked by stimulation in all subjects).
Figure 3. Augmented late TMS-evoked cortical responses in patients with PNH and active epilepsy.

A. The TMS-evoked response produced by stimulation of the connected target site in subject 2. Note the relatively large evoked potential at 289 ms as compared to earlier potentials. B. The GMFP between 100 and 700 ms after stimulation of the connected target in a PNH patient, subject 2 (solid black line), and his matched control (dashed red line). The pink region corresponds to the first time period (100 – 225 ms after the pulse), the blue to the second (225- 400 ms), and the yellow to the third (400 – 700 ms). C. The TMS-evoked response produced by stimulation of the same site (as in Panel A) in the matched control for subject 2. Note that the potential at 167 ms is substantially larger than later potentials. D. The ratio of the normalized AUC-GMFP after stimulation of the connected target for each of the 5 PNH subjects with active epilepsy to the equivalent measure in their matched healthy controls, during the 225-400 ms and 400-700 ms time periods. A ratio greater than 1 indicates an augmented late cortical response in the epilepsy patient (E) as compared to the healthy control (HC). E. The mean GMFP over time, across subjects, evoked by stimulation of the connected target site for the 5 PNH subjects with active epilepsy (thick black line) and their matched controls (thick dashed red line). The gray and red bands indicate +/− standard error of the mean. F. The normalized AUC-GMFP averaged across all PNH subjects with active epilepsy and their matched healthy controls during the same two time periods as in Panel D. Asterisks indicate significant differences (Bonferroni corrected p < 0.05). Note that the raw evoked potentials in A and C are not plotted on a uniform scale.
Figure 4. Persistent TMS-evoked activity in a patient with PNH and active epilepsy, but with a normal interictal EEG.

A. The TMS-evoked response produced by stimulation of the connected target site in subject 1. B. The TMS-evoked response produced by stimulation of the same site in the matched healthy control subject. The later peaks (>225 ms) in the PNH subject are of the same size as or larger than earlier peaks, whereas the later peaks are smaller than the earlier peaks in the matched healthy control subject. Notably, subject 1 had entirely normal interictal findings on prolonged continuous EEG monitoring. Note that the evoked potentials are not plotted on a uniform scale.
Despite the variability in the TMS-evoked potentials between stimulation targets within subjects and between subject pairs, some prominent features were apparent. In healthy controls, stimulation resulted in an evoked response that was maximal within the first 225 ms and then attenuated (Fig. 3 C, E; Fig 4 B). In contrast, in PNH subjects with active epilepsy, the response evoked by stimulation of the abnormally connected target was sustained (Fig. 4 A) or even increased (Fig. 3 A) at later time points. This was true even for those subjects with normal interictal EEG findings, notably including one (subject 1; Fig. 4 A) who had no interictal epileptiform discharges (IEDs) recorded during 11 days of continuous EEG monitoring (that did capture three electrographic seizures). Topographic mapping of these delayed responses demonstrated evolving spatial distributions of evoked activity throughout the later time periods.
GMFP analysis – PNH subjects compared to healthy controls
There was a significant difference (Bonferroni corrected p < 0.05) between PNH subjects with active epilepsy and their matched healthy controls in the normalized AUC-GMFP with stimulation of the connected target (Fig. 3 B, D, F; Supplemental Fig. S1). Specifically, the normalized AUC-GMFP produced by stimulation of the connected target was significantly greater in those with active epilepsy for the 225 – 400 ms as well as the 400 – 700 ms periods; there was no significant difference in the 700 – 1000 ms period. In contrast, there were no significant differences in the normalized AUC-GMFP with stimulation of the non-connected target.
When comparing all PNH subjects (regardless of epilepsy status) with their matched controls, there were no significant differences (Bonferroni corrected p > 0.05) in either the normalized AUC-GMFP after stimulation of either the connected target or the non-connected target in any time period. There were also no significant differences between PNH subjects (or the subset with active epilepsy) and matched healthy controls in the raw AUC-GMFP for any time period with stimulation of either target.
There was a significant difference between PNH subjects with active epilepsy and their matched controls in the normalized amplitude of the largest peaks in the GMFP evoked by stimulation of the connected target during the 225 – 400 ms and 400 – 700 ms time periods (Table 2; Bonferroni-adjusted p < 0.05), again consistent with a relative increase in late activity with stimulation of the connected target. There were no significant differences between these subjects and their matched controls in normalized maximal peak amplitudes with stimulation of the non-connected site, or between all PNH subjects and their matched controls with stimulation of either site.
Table 2.
Maximum peak amplitudes and ratios in the global mean field potential after transcranial magnetic stimulation of cortical targets with aberrant connectivity.
| Subject | Period 1 Peak (100 – 225 ms) (μV) | Period 2 Peak (225 – 400 ms) (μV) | Period 2 Peak / Period 1 Peak | Period 3 Peak (400 – 700 ms) (μV) | Period 3 Peak / Period 1 Peak |
|---|---|---|---|---|---|
| 1 | 6.700 | 8.969 | 1.339 | 5.950 | 0.888 |
| C1 | 3.560 | 2.305 | 0.647 | 1.712 | 0.481 |
| 2 | 1.836 | 2.742 | 1.493 | 1.160 | 0.632 |
| C2 | 6.096 | 2.621 | 0.430 | 2.016 | 0.331 |
| 3 | 3.418 | 1.526 | 0.446 | 1.542 | 0.451 |
| C3 | 2.668 | 0.885 | 0.332 | 0.5422 | 0.203 |
| 4 | 1.962 | 1.933 | 0.985 | 1.088 | 0.555 |
| C4 | 3.845 | 1.115 | 0.290 | 0.9312 | 0.242 |
| 5 | 3.236 | 2.946 | 0.910 | 1.952 | 0.603 |
| C5 | 5.831 | 2.854 | 0.489 | 2.514 | 0.431 |
PNH subjects with active epilepsy are numbered as in Table 1. C1-C5 represent the matched healthy controls for subjects 1-5, respectively. Columns 2, 3, and 5 show the maximum amplitudes of the largest peak evoked by stimulation of the connected target in each of the three time periods, for the PNH subjects and their matched controls. Columns 4 and 6 show the ratios of the maximum amplitudes during the late versus early time periods.
There were no significant differences in the GMFP amplitudes at any individual time point with stimulation of either target, although the pattern of the activity evoked by stimulation of the connected target appeared different between PNH subjects with active epilepsy and their matched controls (Fig. 3 E).
GMFP analysis – Connected targets compared to nonconnected targets
There were differences in evoked activity with stimulation of the connected versus non-connected target site in PNH subjects and their matched controls (Fig. 5 A-D). There was a significant difference (Bonferroni corrected p < 0.05) in the normalized AUC-GMFP between stimulation of the connected versus non-connected target sites in all PNH subjects. Specifically, the normalized AUC-GMFP was significantly greater with stimulation of the connected target for the 225-400 ms and the 400-700 ms time periods; there was no significant difference for the 700-1000 ms time period. These differences were not simply due to inherent differences between the cortical regions represented by the connected and non-connected targets, as there were no significant differences in the normalized AUC-GMFP between stimulation of the two sites in the matched healthy controls (p > 0.05). A similar effect was seen in the subset of patients with active epilepsy, although the difference only reached significance (Bonferroni corrected p<0.05) for the 225-400 ms time period in this smaller subgroup.
Figure 5. Augmented late TMS-evoked cortical responses specific to stimulation of cortical regions with aberrant functional connectivity.

A. The mean GMFP over time, across all PNH subjects, evoked by stimulation of the connected target site (thick black line) and the non-connected target site (thick blue line). The gray and blue bands indicate +/− standard error of the mean. B. The ratio of the normalized AUC-GMFP after stimulation of the connected target to the equivalent measure after stimulation of the non-connected target in each of the PNH subjects, during the 225-400 ms and 400-700 ms time periods. A ratio greater than 1 indicates an augmented late cortical response after stimulation of the connected target (CT) as compared to the non-connected target (NCT). C. The mean GMFP over time, across all healthy control subjects, evoked by stimulation of the connected target site (thick dashed red line) and the non-connected target site (thick dashed purple line). The red and purple bands indicate +/− standard error of the mean. D. The normalized AUC-GMFP averaged across all connected and non-connected targets in the PNH subjects for the same two time periods as in Panel B. Asterisks indicate significant differences (Bonferroni corrected p < 0.05).
There was no significant difference in the raw AUC-GMFP evoked by stimulation of the connected versus non-connected target in any of the subject groups. There were no significant differences in the normalized amplitudes of the largest peaks with stimulation of the connected versus non-connected target in any group, except for a significant increase (p < 0.05) in the normalized maximum peak in the 400-700 time range with stimulation of the connected versus non-connected target in the PNH subjects with active epilepsy. There were no significant differences in the magnitude of the GMFP with stimulation of the connected versus non-connected target at any time point in either the PNH subjects or the matched controls (FDR > 0.05). .
Spatial distribution of abnormal evoked activity
We evaluated the spatial distribution of the abnormally increased delayed activity produced by stimulation of the connected targets in PNH subjects with active epilepsy relative to their matched controls. Topographic plots displaying the differences in the log-transformed RMSV (see Methods) with stimulation of the connected targets are shown (Fig. 6). Whereas in some subjects increased evoked activity was present at or immediately adjacent to the site of stimulation (e.g. subject pair 3), in others the focus of increased activity was spatially remote from the stimulation site (e.g. subject pair 1, left column). Notably, regions with increased activity were often located in the contralateral hemisphere. Furthermore, multiple discrete regions of abnormally increased activity were often present (e.g. subject pairs 1, 4, 5). There were no significant differences (Bonferroni corrected p > 0.05) between PNH subjects with active epilepsy and their matched controls in the measure of local evoked activity (see Methods) produced by stimulation of the connected targets for any time period.
Figure 6. Topography of differences in TMS-evoked activity between patients with PNH and active epilepsy and matched healthy controls.

The plots show the differences between PNH patients with active epilepsy (subjects 1 through 5) and their matched healthy controls in the log-transformed normalized RMSV at each electrode evoked by stimulation of the connected target. Warm colors indicate regions where the evoked activity was greater in the patient; cool colors indicate that the evoked activity was greater in the control. Asterisks indicate the site of stimulation (the connected target site). The left side of each image represents the left side of the brain.
Electrical source imaging
Subject 2 was the only PNH patient who had an epileptogenic zone determinable on available scalp EEG recordings. In order to identify whether the late TMS-EEG peaks (at 289 and 501 milliseconds after stimulation of the connected target; Fig. 3A) had generator sources that were spatially coincident with the spike and seizure focus, we performed source analysis on the two TMS-EEG peaks (Fig. 7 A-B), the two most prominent available interictal discharges (Fig. 7 C, E), and the two available periods of ictal onset on the subject's scalp EEG recordings (Fig. 7 D, F) using the MNE method as described.
Figure 7. Source analysis results of TMS-evoked activity, interictal epileptiform activity, and ictal onset activity.

In subject 2, the only PNH patient in this study with an identifiable epileptogenic zone on scalp EEG, electrical source imaging was performed on the two late TMS-EEG peaks seen after stimulation of the connected target (A,B), two prominent interictal epileptiform discharges seen on conventional scalp EEG recording (C,E), and the ictal onsets of the two recorded seizures available on scalp EEG (D,F). Warm colors represent positive maxima, while cool colors represent negative maxima. In all instances, results converge on an area of the right frontal lobe that was not the site of stimulation, but overlapped with a cortical region that demonstrated functional connectivity to an underlying heterotopic nodule.
In all six instances, source imaging results converged on a focus in the right frontal lobe. This region was not near the connected target of stimulation (as seen in Fig. 2), but instead overlapped with a cortical region that demonstrated functional connectivity to an underlying gray matter heterotopic nodule in the right hemisphere.
Discussion
Here we demonstrate that patients with epilepsy associated with the developmental brain malformation of PNH show evidence of altered cortical physiology, and that these changes detected by TMS-EEG appear to be limited to cortical regions with aberrant functional connectivity to deep regions of gray matter heterotopia. The nature of these abnormalities, which are specifically characterized by a relatively augmented late response that is widely distributed beyond the site of local stimulation, is most consistent with cortical hyperexcitability, and supports the hypothesis that aberrant gray matter connectivity in this disorder leads to epileptogenesis through alterations in cortical neurophysiology. In the one subject who had an epileptogenic zone determinable by scalp EEG, the generator source of the abnormal TMS-evoked activity was spatially coincident with the spike and seizure onset zone. The findings from our small sample support the potential utility of TMS-EEG as a novel and informative biomarker in certain forms of epilepsy, expand our mechanistic understanding of network changes as the pathobiological basis for seizures in gray matter heterotopia, provide support for the use of resting-state functional connectivity imaging in neuropsychiatric disorders, and raise the possibility of noninvasive therapeutic neuromodulation in similar seizure disorders associated with deep lesions.
TMS-EEG and cortical excitability in epilepsy
TMS has been increasingly used in recent years as a safe and noninvasive technique of probing cortical physiology in a number of neurological and psychiatric disorders.41,42 TMS has typically been coupled with the electromyographic recording of MEPs during stimulation of the motor cortex, and numerous TMS-MEP studies in epilepsy have shown alterations in motor cortex excitability, including in patients with both focal43 and generalized44 syndromes. The use of TMS with simultaneous scalp EEG, though technically challenging, allows for the determination of physiological responsiveness over more widespread regions of nonmotor cortex. The TMS-evoked cortical response as recorded on EEG consists of a highly reproducible sequence of early waveforms that provide information on the excitability and oscillatory properties of the stimulated cortical target; these are thought to reflect fluctuations of excitatory and inhibitory postsynaptic activity, particularly as modulated by GABAergic neurotransmission.45,46
A small number of TMS-EEG studies have shown that hyperexcitability and late oscillatory activity can be seen from nonmotor regions of cortex in epilepsy patients,21,22 suggesting augmented excitatory and/or diminished inhibitory local activity and a prolonged reverberation of cortical activity as contributors to the epileptic state. Our data confirm and extend these prior results. The analysis of TMS-evoked potentials allows us to determine cortical excitability “on demand”, rather than wait for spontaneous epileptiform activity. Indeed, 80% of our patients with active epilepsy had normal interictal EEG recordings, yet there were robust abnormalities in TMS-evoked responses in this group, indicating that TMS-EEG adds information above and beyond scalp EEG studies, even long-term recordings (as demonstrated in subject 1, who had no IEDs during 11 days of continuous recording). In our study, demonstration of the relative augmentation of late (>225 ms post-stimulation) responses, which takes the form of persistent oscillatory activity with widespread and shifting topography, appears to be most consistent with a reverberation of epileptic activity through large-scale neural circuits.
Aberrant connectivity and network changes in epileptogenesis
PNH is an uncommon disorder, estimated to be responsible for about 2% of medically refractory epilepsy.6 However, its unique characteristics make it well-suited as a model for circuit epileptogenesis, due to its well-defined and often genetically determined anatomical findings, its long latency to epilepsy onset, and its association with normal intelligence (which allows for cooperation with imaging, stimulation, and behavioral testing in the absence of confounding cognitive impairment). A longstanding question in PNH and related cortical malformations is how the onset of epilepsy can be delayed for years or decades after birth given the striking abnormalities of cerebral architecture that arise from defects in neuronal migration or other steps in fetal brain development. This study links the known alterations in structural and functional connectivity in PNH15 to specific changes in cortical physiology that may underlie the process of epileptogenesis. While some patients with PNH have bilateral widespread nodules, others have more anatomically limited regions of heterotopia. Our finding of a significant difference in the late TMS-evoked response between cortical regions with demonstrated aberrant connectivity and control regions without such connectivity, within the same hemispheres of the same individuals, demonstrates that hyperexcitability in PNH is not simply a diffuse phenomenon but is specifically linked to the presence of cortico-heterotopic circuitry. Indeed, in the one subject with an identifiable epileptogenic zone by scalp EEG, source modeling results for interictal spikes, ictal epileptiform activity, and abnormal late TMS-EEG activity all converged on a right frontal focus, where a region of aberrant functional connectivity to underlying heterotopia was seen.
A number of very common epileptic conditions are now thought to be associated with the gradual development of aberrant cortical connectivity over time,3 including epilepsy after head injury47, post-stroke epilepsy48, and epilepsy of medial temporal lobe onset.49 Similar studies in these disorders could identify whether pathologic circuit mechanisms analogous to those in gray matter heterotopia also lead to seizures in these conditions.
Neurophysiological markers of aberrant resting-state functional connectivity
A fundamental concept underlying resting-state fcMRI studies is that regions with coherent BOLD fluctuations interact in a neurophysiologically meaningful manner, and that alterations in the “normal” pattern of resting-state connectivity have pathologic significance.50 However, direct physiologic evidence for this concept is lacking. In this study, regions with abnormal resting-state connectivity were shown to have abnormal TMS-evoked EEG activity that correlated with disease state, whereas regions without such connectivity did not show such abnormalities. These results provide critical evidence that alterations in resting-state functional connectivity, which have been widely reported in a number of different conditions18, can be directly linked to pathologically important neurophysiological changes (at least in this epilepsy syndrome), and that TMS-EEG metrics can be used to assess the physiological significance of abnormal connectivity directly.
The potential for noninvasive therapeutic modulation based on connectivity imaging
Low-frequency repetitive TMS (rTMS) is known to down-modulate cortical excitability at the stimulation target, and in some clinical trials for epilepsy, has significantly reduced seizure frequency, particularly when ictal foci are neocortical and associated with visible anatomical lesions (and thus easily accessible for TMS targeting).51,52 Deep lesions, such as periventricular nodules or even mesial temporal sclerosis, however, cannot be selectively modulated by conventional TMS coils.53 Our results suggest that hyperexcitable cortical “partner” regions of deep lesions can be identified through structural and functional connectivity imaging, and could be accessible to neuromodulatory techniques that are successful at treating neocortical foci. This same principle underlies the use of rTMS in the approved clinical treatment of medication-refractory depression when targeted to the dorsolateral prefrontal cortex (DLPFC), since it is the functional connectivity of the DLPFC to deeper subgenual cingulate cortex that may mediate the mood effects of this stimulation treatment.54,55
Limitations
There are a number of limitations, both technical and subject-related, to address. First, there is less experience with TMS-EEG as a measure of cortical physiology than with TMS-MEP, and the use of RMT as the basis for defining stimulation intensities for nonmotor regions of cortex may not be ideal. However, our finding of the augmented late response and its interpretation as a marker of cortical hyperexcitability and persistent activity in epileptic circuits are consistent with the results of other such studies in seizure disorders. There are numerous technical challenges in TMS-EEG data acquisition, including muscle artifact within the first 100 ms post-stimulation depending on target location. The effects of this and other sources of artifact were minimized by manual or ICA-based removal. Auditory evoked potential activity related to the clicking sound generated by the TMS coil could be a potential confounding signal that was not removed. However, to contribute to the observed increase in the normalized late responses, auditory evoked activity would likely have to be systematically diminished in active epilepsy patients relative to controls, as the highest-amplitude activity would be expected to fall within the 100-225 ms range. In fact, we found no differences in raw TMS-evoked potentials between PNH subjects and controls during the relevant time window. While our study employed only single-pulse stimulation, paired-pulse TMSEEG is a powerful method of determining hyper- or hypo-excitability within neuronal circuits, and would likely provide useful results in a future study.
It is important to acknowledge our small patient sample. There is clearly interindividual variability in baseline responsiveness to single-pulse stimulation as well as variability within any given individual between different regions of cortex. To address this, we normalized our measures of the late components of the TMS-evoked response as already described. We cannot exclude the possibility that antiepileptic drugs (AEDs) affected our results; all but one of our PNH patients were taking AEDs at the time of TMS, and AED usage is known to alter measures of cortical excitability.56 A subject population free of AEDs or with standardization of medications and serum levels would be ideal, but this was not feasible in our setting. Our findings, however, were robust across subjects, despite variability in the specific AEDs being used, and in general the effect of AEDs is to reduce rather than augment measures of cortical excitability. Finally, PNH is an uncommon disorder and our findings may have limited applicability to epileptogenesis in the absence of visible anatomical lesions. Further work with a larger population of heterotopia patients will be needed to confirm and extend our results, and similar studies in a broader range of epileptic disorders will be needed to examine whether analogous findings are present in other conditions associated with aberrant cortical connectivity, and to determine whether TMS-EEG metrics are associated with seizure frequency.
Conclusions
As our understanding of epileptogenic mechanisms and markers moves beyond consideration of isolated seizure foci and spontaneous epileptiform discharges, respectively, new methods will be needed to explore pathological circuits and evaluate brain network excitability. Drawing on a small number of patients with a unique developmental form of epilepsy, our work with resting-state functional MRI-guided TMS-EEG demonstrates that such a multimodal methodological approach can be employed to investigate functional cortical changes in central nervous system disease, even when lesions are not apparent or are anatomically remote.
Supplementary Material
Acknowledgements
We thank our subjects, without whom this study could not have been performed. Linsey Walker assisted with subject recruitment. MMS was supported by a KL2/Catalyst Medical Research Investigator Training award from Harvard Catalyst/The Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health Award KL2 TR001100). MV was supported by the Fyssen Foundation. CJC was supported by a grant from the National Institutes of Health (5K12NS066225). MBW was supported by a grant from the American Brain Foundation. APL was supported in part by grants from the Sidney R. Baer Jr. Foundation, the National Institutes of Health (R01 HD069776, R01 NS073601, R21 MH099196, R21 NS082870, R21 NS085491, R21 HD07616), and Harvard Catalyst/The Harvard Clinical and Translational Science Center (NCRR and the NCATS, NIH UL1 RR025758). BSC was supported by the National Institute of Neurological Disorders and Stroke (R01 NS073601, with subcontract to JDEG).
The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centers, the National Institutes of Health or the Sidney R. Baer Jr. Foundation.
Footnotes
Conflict of interest disclosures.
MMS, MV, DK, CJC, KB, MEB, KR, MBW, JEC, JDEG, SWG and BSC report no potential conflicts of interest. APL serves on the scientific advisory boards for Nexstim, Neuronix, Starlab Neuroscience, Neuroelectrics, and Neosync, and is listed as an inventor on several issued and pending patents on the real-time integration of transcranial magnetic stimulation (TMS) with electroencephalography (EEG) and magnetic resonance imaging (MRI).
References
- 1.Scharfman HE. The neurobiology of epilepsy. Curr Neur Neurosci Rep. 2007;7:348–54. doi: 10.1007/s11910-007-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spencer SS. Neural networks in human epilepsy: evidence of and implications for treatment. Epilepsia. 2002;43:219–27. doi: 10.1046/j.1528-1157.2002.26901.x. [DOI] [PubMed] [Google Scholar]
- 3.Engel J, Jr, Thompson PM, Stern JM, et al. Connectomics and epilepsy. Curr Opin Neurol. 2013;26:186–94. doi: 10.1097/WCO.0b013e32835ee5b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin X, Prince DA, Huguenard JR. Enhanced excitatory synaptic connectivity in layer V pyramidal neurons of chronically injured epileptogenic neocortex in rats. J Neurosci. 2006;26:4891–900. doi: 10.1523/JNEUROSCI.4361-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prince DA, Parada I, Scalise K, et al. Epilepsy following cortical injury: cellular and molecular mechanisms as targets for potential prophylaxis. Epilepsia. 2009;50(Suppl 2):30–40. doi: 10.1111/j.1528-1167.2008.02008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Battaglia G, Granata T. Periventricular nodular heterotopia. Handb Clin Neurol. 2008;87:177–189. doi: 10.1016/S0072-9752(07)87011-4. [DOI] [PubMed] [Google Scholar]
- 7.Aghakhani Y, Kinay D, Gotman J, et al. The role of periventricular nodular heterotopia in epileptogenesis. Brain. 2005;128:641–51. doi: 10.1093/brain/awh388. [DOI] [PubMed] [Google Scholar]
- 8.Dubeau F, Tampieri D, Lee N, et al. Periventricular and subcortical nodular heterotopia: A study of 33 patients. Brain. 1995;118:1273–87. doi: 10.1093/brain/118.5.1273. [DOI] [PubMed] [Google Scholar]
- 9.Kothare SV, VanLandingham K, Armon C, et al. Seizure onset from periventricular nodular heterotopias: depth-electrode study. Neurology. 1998;51:1723–7. doi: 10.1212/wnl.51.6.1723. [DOI] [PubMed] [Google Scholar]
- 10.Li LM, Dubeau F, Andermann F, et al. Periventricular nodular heterotopia and intractable temporal lobe epilepsy: poor outcome after temporal lobe resection. Ann Neurol. 1997;41:662–8. doi: 10.1002/ana.410410516. [DOI] [PubMed] [Google Scholar]
- 11.Tassi L, Colombo N, Cossu M, et al. Electroclinical, MRI and neuropathological study of 10 patients with nodular heterotopia, with surgical outcomes. Brain. 2005;128:321–37. doi: 10.1093/brain/awh357. [DOI] [PubMed] [Google Scholar]
- 12.Barkovich AJ, Kjos BO. Gray matter heterotopias: MR characteristics and correlation with developmental and neurologic manifestations. Radiology. 1992;182:493–9. doi: 10.1148/radiology.182.2.1732969. [DOI] [PubMed] [Google Scholar]
- 13.Chang BS, Katzir T, Liu T, et al. A structural basis for reading fluency: white matter defects in a genetic brain malformation. Neurology. 2007;69:2146–54. doi: 10.1212/01.wnl.0000286365.41070.54. [DOI] [PubMed] [Google Scholar]
- 14.Chang BS, Ly J, Appignani B, et al. Reading impairment in the neuronal migration disorder of periventricular nodular heterotopia. Neurology. 2005;64:799–803. doi: 10.1212/01.WNL.0000152874.57180.AF. [DOI] [PubMed] [Google Scholar]
- 15.Christodoulou JA, Walker LM, Del Tufo SN, et al. Abnormal structural and functional brain connectivity in gray matter heterotopia. Epilepsia. 2012;53:1024–32. doi: 10.1111/j.1528-1167.2012.03466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christodoulou JA, Barnard ME, Del Tufo SN, et al. Integration of gray matter nodules into functional cortical circuits in periventricular heterotopia. Epilepsy Behav. 2013;29:400–6. doi: 10.1016/j.yebeh.2013.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Archer JS, Abbott DF, Masterton RA, et al. Functional MRI interactions between dysplastic nodules and overlying cortex in periventricular nodular heterotopia. Epilepsy Behav. 2010;19:631–4. doi: 10.1016/j.yebeh.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 18.Greicius M. Resting-state functional connectivity in neuropsychiatric disorders. Curr Opin Neurol. 2008;21:424–430. doi: 10.1097/WCO.0b013e328306f2c5. [DOI] [PubMed] [Google Scholar]
- 19.Guye M, Bartolomei F, Ranjeva J-P. Imaging structural and functional connectivity: towards a unified definition of human brain organization? Curr Opin Neurol. 2008;21:393–403. doi: 10.1097/WCO.0b013e3283065cfb. [DOI] [PubMed] [Google Scholar]
- 20.Rotenberg A. Prospects for clinical applications of transcranial magnetic stimulation and real-time EEG in epilepsy. Brain Topogr. 2010;22:257–66. doi: 10.1007/s10548-009-0116-3. [DOI] [PubMed] [Google Scholar]
- 21.Valentin A, Arunachalam R, Mesquita-Rodrigues A, et al. Late EEG responses triggered by transcranial magnetic stimulation (TMS) in the evaluation of focal epilepsy. Epilepsia. 2008;49:470–80. doi: 10.1111/j.1528-1167.2007.01418.x. [DOI] [PubMed] [Google Scholar]
- 22.Julkunen P, Saisanen L, Kononen M, et al. TMS-EEG reveals impaired intracortical interactions and coherence in Unverricht-Lundborg type progressive myoclonus epilepsy (EPM1). Epilepsy Res. 2013;106:103–12. doi: 10.1016/j.eplepsyres.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 23.Fox MD, Snyder AZ, Vincent JL, et al. The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc Natl Acad Sci USA. 2005;102:9673–8. doi: 10.1073/pnas.0504136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitfield-Gabrieli S, Thermenos HW, Milanovic S, et al. Hyperactivity and hyperconnectivity of the default network in schizophrenia and in first-degree relatives of persons with schizophrenia. Proc Natl Acad Sci USA. 2009;106:1279–84. doi: 10.1073/pnas.0809141106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Behzadi Y, Restom K, Liau J, Liu TT. A component based noise correction method (CompCor) for BOLD and perfusion based fMRI. Neuroimage. 2007;37:90–101. doi: 10.1016/j.neuroimage.2007.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rorden C, Brett M. Stereotaxic display of brain lesions. Behavioural Neurology. 2000;12:191–200. doi: 10.1155/2000/421719. [DOI] [PubMed] [Google Scholar]
- 27.Virtanen J, Ruohonen J, Naatanen R, Ilmoniemi RJ. Instrumentation for the measurement of electric brain responses to transcranial magnetic stimulation. Med Biol Eng Comput. 1999;37:322–326. doi: 10.1007/BF02513307. [DOI] [PubMed] [Google Scholar]
- 28.Rossi S, Hallett M, Rossini PM, et al. Safety, ethical considerations, and application guidelines for the use of transcranial magnetic stimulation in clinical practice and research. Clin Neurophysiol. 2009;120:2008–2039. doi: 10.1016/j.clinph.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossini PM, Barker At, Berardelli A, et al. Non-invasive electrical and magnetic stimulation of the brain, spinal cord, and roots: basic principles and procedures for routine clinical application. Report of an IFCN committee. Electroencephalogr Clin Neurophysiol. 1994;91:79–92. doi: 10.1016/0013-4694(94)90029-9. [DOI] [PubMed] [Google Scholar]
- 30.Delorme A, Makeig S. EEGLAB: an open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. J Neurosci Methods. 2004;134:9–21. doi: 10.1016/j.jneumeth.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 31.Lehmann D, Skrandies W. Reference-free identification of components of checkerboard-evoked multichannel potential fields. Electroencephalogr Clin Neurophysiol. 1980;48:609–621. doi: 10.1016/0013-4694(80)90419-8. [DOI] [PubMed] [Google Scholar]
- 32.Komssi S, Kahkonen S, Ilmoniemi RJ. The effect of stimulus intensity on brain responses evoked by transcranial magnetic stimulation. Hum Brain Mapp. 2004;21:154–164. doi: 10.1002/hbm.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kahkonen S, Wilenius J, Komssi S, Ilmoniemi RJ. Distinct differences in cortical reactivity of motor and prefrontal cortices to magnetic stimulation. Clin Neurophysiol. 2004;115:534–542. doi: 10.1016/j.clinph.2003.10.032. [DOI] [PubMed] [Google Scholar]
- 34.Badawy RA, Curatolo JM, Newton M, et al. Changes in cortical excitability differentiate generalized and focal epilepsy. Ann Neurol. 2007;61:324–331. doi: 10.1002/ana.21087. [DOI] [PubMed] [Google Scholar]
- 35.Badawy RA, Macdonell RA, Berkovic SF, et al. Predicting seizure control: cortical excitability and antiepileptic medication. Ann Neurol. 2010;67:64–73. doi: 10.1002/ana.21806. [DOI] [PubMed] [Google Scholar]
- 36.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 37.Groppe DM, Urbach TP, Kutas M. Mass univariate analysis of event-related brain potentials/fields I: a critical tutorial review. Psychophysiology. 2011;48:1711–25. doi: 10.1111/j.1469-8986.2011.01273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamalainen MS, Sarvas J. Realistic conductivity geometry model of the human head for interpretation of neuromagnetic data. IEEE Trans Biomed Eng. 1989;36:165–71. doi: 10.1109/10.16463. [DOI] [PubMed] [Google Scholar]
- 39.Gramfort A, Luessi M, Larson E, Engemann DA, Strohmeier D, Brodbeck C, Goj R, Jas M, Brooks T, Parkkonen L, Hamalainen M. MEG and EEG data analysis with MNE-Python. Front Neurosci. 2013;7:267. doi: 10.3389/fnins.2013.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fischl B. FreeSurfer. Neuroimage. 2012;62:774–81. doi: 10.1016/j.neuroimage.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kobayashi M, Pascual-Leone A. Transcranial magnetic stimulation in neurology. Lancet Neurol. 2003;2:145–56. doi: 10.1016/s1474-4422(03)00321-1. [DOI] [PubMed] [Google Scholar]
- 42.Hallett M. Transcranial magnetic stimulation: a primer. Neuron. 2007;55:187–99. doi: 10.1016/j.neuron.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 43.Badawy RA, Vogrin SJ, Lai A, Cook MJ. The cortical excitability profile of temporal lobe epilepsy. Epilepsia. 2013;54:1942–9. doi: 10.1111/epi.12374. [DOI] [PubMed] [Google Scholar]
- 44.Badawy RA, Vogrin SJ, Lai A, Cook MJ. Patterns of cortical hyperexcitability in adolescent/adult-onset generalized epilepsies. Epilepsia. 2013;54:871–8. doi: 10.1111/epi.12151. [DOI] [PubMed] [Google Scholar]
- 45.Rogasch NC, Fitzgerald PB. Assessing cortical network properties using TMSEEG. Human Brain Mapp. 2013;34:1652–1669. doi: 10.1002/hbm.22016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Premoli I, Castellanos N, Rivolta D, Belardinelli P, Bajo R, Zipser C, Espenhahn S, Heidegger T, Muller-Dahlhaus F, Ziemann U. TMS-EEG signatures of GABAergic neurotransmission in the human cortex. J Neurosci. 2014;34:5603–5612. doi: 10.1523/JNEUROSCI.5089-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, McDonald W, Parada I, et al. Targets for preventing epilepsy following cortical injury. Neurosci Lett. 2011;497:172–6. doi: 10.1016/j.neulet.2011.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bladin CF, Bornstein N. Post-stroke seizures. Handb Clin Neurol. 2009;93:613–621. doi: 10.1016/S0072-9752(08)93030-X. [DOI] [PubMed] [Google Scholar]
- 49.Bonilha L, Nesland T, Martz GU, et al. Medial temporal lobe epilepsy is associated with neuronal fibre loss and paradoxical increase in structural connectivity of limbic structures. J Neurol Neurosurg Psychiatry. 2012;83:903–909. doi: 10.1136/jnnp-2012-302476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fox MD, Greicius M. Clinical applications of resting state functional connectivity. Front Syst Neurosci. 2010;4:19. doi: 10.3389/fnsys.2010.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fregni F, Otachi PTM, do Valle A, et al. A randomized clinical trial of repetitive transcranial magnetic stimulation in patients with refractory epilepsy. Ann Neurol. 2006;60:447–455. doi: 10.1002/ana.20950. [DOI] [PubMed] [Google Scholar]
- 52.Sun W, Mao W, Meng X, et al. Low-frequency repetitive transcranial magnetic stimulation for the treatment of refractory partial epilepsy: a controlled clinical study. Epilepsia. 2012;53:1782–1789. doi: 10.1111/j.1528-1167.2012.03626.x. [DOI] [PubMed] [Google Scholar]
- 53.Wagner T, Gangitano M, Romero R, et al. Intracranial measurement of current densities induced by transcranial magnetic stimulation in the human brain. Neurosci Lett. 2004;354:91–94. doi: 10.1016/s0304-3940(03)00861-9. [DOI] [PubMed] [Google Scholar]
- 54.Fox MD, Buckner RL, White MP, et al. Efficacy of transcranial magnetic stimulation targets for depression is related to intrinsic functional connectivity with the subgenual cingulate. Biol Psychiatry. 2012;72:595–603. doi: 10.1016/j.biopsych.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Downar J, Daskalakis ZJ. New targets for rTMS in depression: a review of convergent evidence. Brain Stim. 2013;6:231–240. doi: 10.1016/j.brs.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 56.Badawy RA, Macdonell RA, Berkovic SF, et al. Predicting seizure control: cortical excitability and antiepileptic medication. Ann Neurol. 2010;67:64–73. doi: 10.1002/ana.21806. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.