

Significance
The design of effective T-cell vaccines against pathogens and tumor antigens is challenged by the highly inefficient identification of the subset of peptides from a given antigen that effectively stimulate an immune response. Here we report that the relative hydrophobicity of T-cell receptor contact residues is markedly enriched in immunogenic major histocompatibility complex class I epitopes in both human and murine MHCs, and in both self and pathogen-derived immunogenic epitopes. Incorporating hydrophobicity into T-cell epitope prediction models increases the efficiency of epitope identification, which will manifest in the time and cost of T-cell vaccine development. Amino acid hydrophobicity may represent a biochemical basis by which T cells discriminate immunogenic epitopes within the background of self peptides.
Keywords: T cell, nonself, MHC class I, vaccine, epitope prediction, immunogenicity
Abstract
Despite the availability of major histocompatibility complex (MHC)-binding peptide prediction algorithms, the development of T-cell vaccines against pathogen and tumor antigens remains challenged by inefficient identification of immunogenic epitopes. CD8+ T cells must distinguish immunogenic epitopes from nonimmunogenic self peptides to respond effectively against an antigen without endangering the viability of the host. Because this discrimination is fundamental to our understanding of immune recognition and critical for rational vaccine design, we interrogated the biochemical properties of 9,888 MHC class I peptides. We identified a strong bias toward hydrophobic amino acids at T-cell receptor contact residues within immunogenic epitopes of MHC allomorphs, which permitted us to develop and train a hydrophobicity-based artificial neural network (ANN-Hydro) to predict immunogenic epitopes. The immunogenicity model was validated in a blinded in vivo overlapping epitope discovery study of 364 peptides from three HIV-1 Gag protein variants. Applying the ANN-Hydro model on existing peptide-MHC algorithms consistently reduced the number of candidate peptides across multiple antigens and may provide a correlate with immunodominance. Hydrophobicity of TCR contact residues is a hallmark of immunogenic epitopes and marks a step toward eliminating the need for empirical epitope testing for vaccine development.
The interaction of CD8+ T cells with peptide-major histocompatibility complex (MHC) complexes (pMHCs) is a key event in the development of cell-mediated immunity (1). MHC class I (MHC-I) molecules typically present 8- to 11-aa peptides derived predominantly from proteasomal degradation of intracellular proteins, either self peptides or infection-derived antigens (2). T-cell receptors (TCRs) from CD8+ T cells bind antigenic pMHC molecules, triggering a downstream signaling cascade that leads to T-cell activation, T-cell differentiation, and ultimately cytolysis of target cells presenting the same epitope (3). Vaccines and immunotherapies for the treatment of infection and cancer seek to incorporate cytotoxic T-cell (CTL) epitopes, but defining such epitopes remains a costly and arduous process (4). Understanding the molecular basis of TCR-pMHC recognition will aid the discovery of immunogenic epitopes in infectious and autoimmune diseases.
During thymic development, CD8+ T cells undergo both positive and negative selection to acquire the ability to discriminate antigenic peptides from self peptides (5). Costimulatory signals can enhance this discrimination (6), but a primary event that triggers CD8+ T-cell activation is the noncovalent pMHC–TCR interaction. Proteasomal cleavage patterns and binding affinities of peptides to different MHCs have been studied extensively (7–9). In contrast, the biochemical bases of immunogenic epitopes are less well defined (10). T-cell epitope discovery is complicated by the codominance and polymorphism of MHC alleles, diversity of antigens (both infectious and self antigens), limited mass spectrometry-based confirmation of MHC-bound peptides, and scarcity of experimentally confirmed immunogenic epitopes within the infectious and self proteome (4). As a result, T-cell epitope prediction algorithms have focused on amino acid binding affinity for specific MHC motifs and the protein’s proteasomal cleavage pattern to identify candidate T-cell epitopes (11–14).
Although computational tools have improved over the past decade, they have not been trained to predict immunogenicity. The major limitation when using such prediction algorithms is the presence of a significant number of binders from a given antigen that will never lead to an immune response (15). Thus, immunogenic CTL epitopes fulfill additional criteria that go beyond antigen processing and MHC binding.
Here we sought to identify the biochemical criteria that define immunogenicity within the subset of MHC-I–binding peptides. Using a curated repository of MHC-I epitopes from the Immune Epitope Database (IEDB) (16), we evaluated the biochemical properties of amino acids that discriminate between immunogenic epitopes and nonimmunogenic self peptides. We found a strong bias toward hydrophobicity in amino acid residues of immunogenic CTL epitopes that is highly selective for exposed TCR contact residues. Using these criteria, we trained an artificial neural network (ANN) model to identify immunogenic CTL epitopes from a dataset, and empirically assessed our prediction model for three HIV-1 Gag protein variants in a murine model of immunogenicity. We demonstrate the utility of this ANN model, which has the potential to significantly enhance the efficiency of T-cell epitope discovery.
Results
Amino Acid Use Differs Between Immunogenic and Nonimmunogenic Peptides.
CTLs recognize immunogenic epitopes from a background of poorly immunogenic self peptides. To understand the biochemical basis of differences between these two classes of peptides, we retrieved all known MHC-I–binding peptides reported as T-cell reactive (“immunogenic” hereinafter) and self peptides from MHC ligand elution experiments with no known immunogenicity (“nonimmunogenic” hereinafter) from the IEDB. Any eluted peptide that was immunogenic (either pathogen-derived or self antigen-derived) was excluded, to generate two mutually exclusive datasets that avoid any potential bias. Out of the 34,586 total retrieved peptides from the IEDB, 5,035 8- to 11-mer nonredundant peptides were reported to be immunogenic, and 4,853 were nonredundant nonimmunogenic and were used in further analysis (SI Appendix, Table S1). The frequency distributions of amino acids in 8- to 11-mer immunogenic and nonimmunogenic peptides showed significant variability in amino acid composition (SI Appendix, Fig. S1A).
To identify overrepresentation of certain amino acids in immunogenic epitopes, we computed a probability ratio for each amino acid. We then performed a correlation analysis between the probability ratio of each amino acid and three major biochemical properties—hydrophobicity (Kyte–Doolittle) (17), polarity (Grantham) (18), and side chain bulkiness (Zimmerman) (19)—using independent numeric scales (SI Appendix, Table S2). We found a strong, statistically significant correlation between probability ratios and hydrophobicity values (Spearman ρ = 0.71, P = 4.24 × 10−4; Fig. 1A). Similarly, we also found a negative correlation between probability ratios and polarity of amino acids (Spearman ρ = −0.77, P = 6.97 × 10−5; Fig. 1B), with highly polar amino acids underrepresented in immunogenic epitopes. No significant correlation was observed with amino acid side chain bulkiness (Fig. 1C). Most of the overrepresented and strongly bulky amino acids were strongly hydrophobic as well. Cysteine, a nonpolar hydrophobic amino acid, was an outlier in the immunogenic dataset. Excluding cysteine did not change our results significantly (SI Appendix, Fig. S1D).
Fig. 1.

Probability ratio [P(× I immunogenic)/P(× I nonimmunogenic)] of each amino acid as a function of its corresponding biochemical property. Each probability of each amino acid was computed from the frequency distribution of immunogenic epitopes and nonimmunogenic peptides. Biochemical properties analyzed were (A) hydrophobicity (17), (B) polarity (18), and (C) side-chain bulkiness (19). A probability ratio >1 indicates overrepresentation of the amino acid in the immunogenic dataset. The overrepresented outlier cysteine (C) was omitted for scale. Spearman correlations coefficients (ρ) are shown.
Two potential sources of bias in our analyses were the variation in peptide length of MHC-I peptides and the dominance of HLA-A2 epitopes within existing databases; therefore, we performed the analyses on the 9-mer epitopes (SI Appendix, Fig. S1B) and on HLA class I-restricted peptides excluding HLA-A2 peptides (SI Appendix, Fig. S1C). Within these subsets, the overrepresentation of nonpolar, hydrophobic amino acids in immunogenic epitopes was maintained.
Hydrophobicity Bias in Selective TCR Contact Residues.
We first compared the mean hydrophobicity of each residue between immunogenic and nonimmunogenic peptides using the Kyte–Doolittle numeric hydrophobicity scale. Immunogenic 9-mer epitopes were significantly more hydrophobic than nonimmunogenic 9-mer peptides at each residue (P < 1.6 × 10−5; Fig. 2A and SI Appendix, Table S3). We observed similar results in 10-mer peptides (P < 2 × 10−7 at each residue; SI Appendix, Fig. S2A), and within HLA-A2 excluded 9-mer and 10-mer subsets (SI Appendix, Fig. S2 B and C and Table S3).
Fig. 2.

Hydrophobicity comparison at each residue position between immunogenic and nonimmunogenic MHC-I peptides. Each peptide sequence in the dataset was transformed into a numeric sequence based on amino acid hydrophobicity, and the mean hydrophobicity at each position was calculated. (A) All immunogenic and nonimmunogenic MHC-I 9-mers; every residue had P < 1.6 × 10−5. (B) HLA-A2–restricted immunogenic and nonimmunogenic 9-mers. (C) Murine MHC H-2Db–restricted immunogenic and nonimmunogenic 9-mers. (D) Murine MHC H-2Kb–restricted immunogenic and nonimmunogenic 8-mers. Down-arrows in B–D indicate anchor residues based on specific MHC motifs. *P < 0.008 in that residue position. P values are listed in SI Appendix, Table S3.
Because the immunogenic dataset is biased to pathogen-derived immunogenic epitopes, we performed similar analyses between immunogenic self epitopes and nonimmunogenic self peptides (P < 1 × 10−4 at all residues except P5 and P6; SI Appendix, Fig. S2D). We further compared immunogenic HLA-A2–restricted 9-mer epitopes derived from pathogens with those derived from self antigens and observed no significant difference in hydrophobicity (P > 0.05 at each amino acid residue except P6, P = 0.04; SI Appendix, Fig. S2G), revealing that T cells that escape thymic deletion recognize self peptides with a hydrophobicity profile that is virtually the same as that of pathogen-derived epitopes.
Finally, to evaluate whether potential bias is created by using peptide immunization experiments, we performed the same analysis using immunogenic epitopes identified using whole “organism” as the immunogen (P < 0.01 at all residues except P1 and P5; SI Appendix, Fig. S2E). Thus, our results demonstrate a preference for hydrophobicity in immunogenic epitopes across antigenic sources (self and pathogen) and MHC molecules (HLA-A2 and non–HLA-A2).
The locations of anchor residues and TCR contacts have been mapped for many MHC peptides (20). If the observed bias toward nonpolar hydrophobic amino acids within immunogenic epitopes affects TCR affinity, then we predicted that it would be selective for TCR contact residues. We analyzed the mean hydrophobicity along the peptide for the most well-represented MHC epitopes within the database: HLA-A2 (Fig. 2B), and murine MHC H-2Db and H-2Kb (Fig. 2 C and D). HLA-A2–restricted 9-mer peptides are anchored at residues P2 and P9, with P6 as an auxiliary anchor (7). We observed no statistical difference in hydrophobicity between the anchor residues of immunogenic and nonimmunogenic peptides (P2, P = 0.9; P9, P = 0.08; Fig. 2B). The observed difference in hydrophobicity was at specific TCR contact residues P4, P7, and P8 (P4, P = 6.3 × 10−12; P7, P = 5 × 10−13; P8, P < 2.2 × 10−16). In contrast, the auxiliary anchor P6 was more hydrophobic in nonimmunogenic peptides (P = 3.1 × 10−7). We found similar results for HLA-A2–restricted 10-mer peptides (SI Appendix, Fig. S2F).
To determine whether the difference in hydrophobicity is species-specific, we evaluated the subset of known mouse MHC H-2Kb–restricted 8-mer peptides. Again, we observed a marked increase in relative hydrophobicity for the TCR contact residues P6 and P7 of immunogenic epitopes (P6, P = 7 × 10−5; P7, P = 1.1 × 10−6), but no difference in anchor residues (P5, P = 0.67; P8, P = 0.15) (Fig. 2C). As observed with HLA-A2, the auxiliary anchor residue P3 was more hydrophobic in nonimmunogenic peptides (P = 0.005).
Finally, we analyzed mouse MHC H-2Db–restricted 9-mer peptides and observed that P7 and P8 TCR contact residues were more hydrophobic in immunogenic epitopes (P7, P = 1.1 × 10−4; P8, P = 0.001), with no difference in anchor residue P9 (P = 0.127) (Fig. 2D). One exception was the anchor residue P5, which was more hydrophobic in immunogenic epitopes (P = 4.9 × 10−10). This discrepancy might be related to the presence of other potential anchors at P5 (apart from Asn) within the immunogenic dataset. Thus, we demonstrate that the observed bias toward relative hydrophobic amino acids in immunogenic epitopes is selective for TCR contact residues.
Differential Hydrophobicity Can Predict Immunogenic CTL Epitopes.
Although MHC binding is necessary for antigen presentation, it is not sufficient to stimulate an immune response. We predicted that hydrophobicity could be incorporated into existing binding algorithms to improve the prediction of CTL epitopes. To test this hypothesis, we used the IEDB consensus binding prediction tool to generate peptide predictions for HLA-A2–restricted peptides (9 and 10 mer) for two viral proteins: polyprotein from dengue virus type 1 (DENV1) and tegument protein pp65 from cytomegalovirus (CMV). Using mean hydrophobicity of amino acids in TCR contact residues (all residues except anchors: P2, P6, and P9 or P10), we reranked each predicted peptide with decreasing TCR contact hydrophobicity values (Fig. 3). The rate at which experimentally defined HLA-A2–restricted CTL epitopes (SI Appendix, Table S4) were identified was increased using hydrophobicity-based predictions compared with the IEDB consensus binding predictions (Fig. 3 A and B). As a negative control, we performed reranking of top predictions from the two proteins using the mean hydrophobicity of just anchor residues (Fig. 3 C and D). The rate of prediction of HLA-A2–restricted CTL epitopes was similar to the IEDB consensus binding predictions, confirming that relative hydrophobicity impacts immunogenicity and not HLA binding. These results suggest that using TCR contact hydrophobicity could improve the prediction of immunogenic epitopes.
Fig. 3.

Efficiency of predicting experimentally defined HLA-A0201–restricted immunogenic epitopes using mean hydrophobicity of TCR contact residues (straight lines) compared with IEDB consensus binding tool (IEDB-Bind; dashed lines). Tegument protein pp65 from cytomegalovirus (CMV) and polyprotein from dengue virus type 1 were used for predictions. (A and B) Predicted peptides from the IEDB-Bind were reranked using the mean hydrophobicity of TCR contact residues. (C and D) Predicted peptides from the IEDB-Bind were reranked using the mean hydrophobicity of anchor residues.
Hydrophobicity-Based ANN Prediction Model.
The relative contribution of each amino acid residue to immunogenicity varies among MHC allomorphs and is motif-dependent (Fig. 2 and SI Appendix, Table S3). Furthermore, the immunogenicity of a peptide might result from nonlinear interactions between different TCR contact residues. ANNs are designed to handle such nonlinearity (11, 21); therefore, we developed and trained an ANN-based prediction model of immunogenicity using amino acid hydrophobicity (ANN-Hydro), with the goal of improving existing CTL epitope prediction algorithms for H-2Db and HLA-A2. Each peptide sequence in the H-2Db and HLA-A2 datasets was transformed into a corresponding numeric sequence based on the hydrophobicity value of amino acids, and these served as the training sets for the two ANN-Hydro models (SI Appendix, Fig. S3). An initial assessment of the trained ANN-Hydro model for HLA-A2 assigned a good probability of immunogenicity to 54 of 64 (>80%) experimentally defined HLA-A2–restricted epitopes from three recent studies (15, 22, 23) (P < 0.001, compared with the distribution of probabilities of immunogenicity of 64 randomly generated 9-mer peptides) (SI Appendix, Table S5).
We then developed an epitope discovery strategy incorporating the ANN-Hydro model to predict a previous set of experimentally validated H-2Db and HLA-A2 epitopes from five pathogen antigens and five tumor antigens (SI Appendix, Table S6). We used the IEDB consensus MHC-binding prediction algorithm to obtain a list of predicted peptides for each antigen, each of which was assigned a normalized binding score, SB. Because T-cell epitopes are a subset of predicted peptides that bind to MHC molecules, a normalized score, SI, based on the probability of immunogenicity obtained by ANN-Hydro, was assigned to each peptide (SI Appendix, Fig. S3). We then defined a total score, S, as S = SB⋅SI for the rate of identifying CTL epitopes from the list of predicted H-2Db and HLA-A2 peptides from each antigen; thus, the total score is dependent on the contribution of both scores, reflecting two critical aspects: binding and immunogenicity (SI Appendix, Fig. S3).
Our strategy of reranking by prioritization of high-binding and high-immunogenic peptides over other predicted peptides (SI Appendix, Fig. S3, SI Materials and Methods) scored 42 of the 43 H-2Db and HLA-A2 9-mer epitopes within the top-20 ranked peptides (SI Appendix, Table S6). In contrast, individual prediction algorithms ranked the same epitopes up to rank 133 (SI Appendix, Table S6). Therefore, the ANN-Hydro model can be used in conjunction with IEDB consensus to improve the efficiency of prediction of CTL epitopes.
Prediction Validation by in Vivo Discovery of HIV-1 Gag Epitopes.
To comprehensively evaluate the predictive capacity of our approach for CTL epitope discovery and to correlate immunodominance, we interrogated three HIV-1 Gag variant proteins: Consensus B (ConsB), 96ZM651.8 (ZM96), and 97/CN54 (CN54) (Fig. 4). With no previous knowledge of Gag-specific CTL epitopes, we used our model to generate a list of ranked H-2Db–restricted peptides, of which the top-20 predictions for each interrogated Gag sequence are shown (SI Appendix, Table S7). To validate our predicted epitopes in vivo, we immunized B6 mice independently against each of the three different Gag variants and analyzed the peptide specificity of effector CD8+ T-cell responses using overlapping peptide pools (SI Appendix, Fig. S4). Deconvolution and truncation experiments allowed us to define a unique dominant H-2Db–restricted epitope within each Gag protein (SI9 for ConsB, QL11 for CN54, and RT9 for ZM96), as well as shared subdominant epitopes: Db-restricted RT9, AI9, and YI9 and Kb-restricted VL8 (Fig. 4 F–H and SI Appendix, Fig. S5).
Fig. 4.
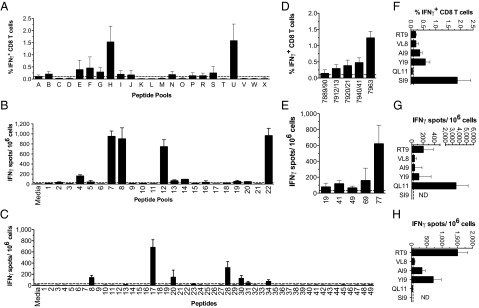
ANN-Hydro model prediction validation by in vivo discovery of HIV-1 Gag epitopes. Predictions for H-2Db epitopes were made for three HIV-1 Gag proteins using the ANN-Hydro model, and then a blinded epitope discovery study was performed in vivo. The top-20 predicted peptides for each protein using the model are listed in SI Appendix, Table S7. (A–C) B6 mice were immunized with AdHu5 vaccines expressing the ConsB, CN54, or ZM96 Gag, and CD8+ T-cell responses determined by intracellular IFN-γ or IFN-γ ELISPOT after ex vivo stimulation with peptide pools of 15-mer peptides (overlapping by 11 mer) spanning the entire Gag sequence (ConsB or CN54, A and B) or with a complete set of overlapping 20-mer peptides spanning ZM96 (C). (D–H) Positive responses to pools were deconvoluted by stimulation with individual 15-mer peptides from the positive pools (ConsB or CN54, D and E). Minimal epitopes were identified by stimulation with truncated peptides and are shown (F–H).
A comparison of empirically defined epitopes with predictions made using ANN-Hydro revealed that H-2Db–restricted 9-mer CTL epitopes for HIV-1 CN54 Gag and ZM96 Gag correlated with ANN-Hydro model epitope sequences predicted within the top-15 ranked peptides and for ConsB Gag within the top-11 ranked peptides (SI Appendix, Table S6). In striking contrast, prediction of the identified Gag epitopes by individual prediction algorithms was more varied, with predictions up to rank number 46, depending on the binding or processing algorithm used. Although the IEDB consensus binding and NetMHCpan algorithms predicted the identified Gag epitopes within the top-six ranked peptides, the performance of these algorithms (unlike the ANN-Hydro model) was highly variable depending on the antigen selected (variance range of 66.72–220.27; SI Appendix, Table S6).
In sum, the ANN-Hydro model predicted 52 out of 53 experimentally validated H-2Db and HLA-A2 9-mer epitopes from 13 different antigens within the top-20 ranked peptides (Fig. 5A), corresponding to a 98% success rate in identifying immunogenic epitopes. Moreover, this predictive improvement was reflected in lower variability of epitope identification, a variance of 37.72 using ANN-Hydro as opposed to 66.72 by IEDB alone (P < 0.05, F-test).
Fig. 5.

Incorporating ANN-Hydro in the IEDB-consensus binding tool improves epitope prediction. (A) Ranked epitopes are shown by scatterplots for 26 H-2Db CTL epitopes from eight well-described antigens (LCMV-GP, LCMV-NP, AdV.E1B, Flu-NP, Flu-NA, and HIV-1 Gag variants ConsB, ZM96, and CN54) and for 27 HLA-A2 CTL epitopes from five tumor antigens (Melan-A, Wt-1, gp100, TRAG-3, and p53). The following prediction algorithms were used: IEDB-Bind, IEDB consensus binding tool; NetMHC-Bind, NetMHCpan binding tool; SYFPEITHI, SYFPEITHI epitope prediction tool; IEDB-Prot, IEDB-recommended processing prediction; and ANN-Prot, IEDB processing predictions using ANN. Epitopes and their corresponding predicted ranks by prediction algorithms are shown in SI Appendix, Table S6. (B–D) Epitope immunodominance as a function of probability of immunogenicity for LCMV-GP, Flu-NP, and Gag-ZM96. (E–G) Epitope immunodominance as a function of predicted MHC binding (IEDB consensus) for LCMV-GP, Flu-NP, and Gag-ZM96. Immunodominance was determined from percentage-specific lysis of target cells ex vivo. (B and E) 9-mer versions of SGV11 and CSA10 were used (25). (C and F) Percent survival of peptide-primed mice on lethal challenge of virus (24). (D and G) IFN-γ spots per million cells on ex vivo peptide stimulation postvaccination with antigen (this study).
Prediction of Immunodominant Epitopes.
The probabilities of immunogenicity assigned by ANN-Hydro were interrogated with respect to epitope immunodominance using three antigens with a clear vertical epitope hierarchy, as identified by ex vivo experimental data (24, 25). The epitope hierarchy defined experimentally in LCMV-GP, Flu-NP, and ZM96 Gag showed robust correlation with the probabilities of immunogenicity assigned by ANN-Hydro (r > 0.94, P < 0.05; Fig. 5 B–D). In contrast, predicted MHC binding assigned by IEDB consensus showed no correlation with epitope immunodominance in LCMV-GP and ZM96-Gag (Fig. 5 E and G). Epitope immunodominance in Flu-NP correlated with both ANN-Hydro’s predicted probability and predicted MHC binding (Fig. 5 C and F). As a further correlate, 7 of 13 epitopes predicted in lower rankings by ANN-Hydro along with the IEDB consensus were modest immunogens derived from LCMV-GP, LCMV-NP, ZM96, CN54, and consensus Gag (SI Appendix, Table S6). Therefore, efficient pMHC-TCR affinity may contribute toward epitope immunodominance. Epitope predictions from ANN-Hydro were consistently less variable, and improved the prediction of immunodominant CTL epitopes.
Discussion
At present, there is no consensus regarding the molecular mechanisms by which CD8+ T cells discriminate immunogenic antigens within the background of poorly immunogenic self peptides. Understanding this discrimination has implications for rational vaccine design and the identification of antigenic targets of malignant and autoimmune diseases. Although several theories have been proposed to explain the concept of self/nonself discrimination (26), to our knowledge the present study is the first attempt to provide a biochemical explanation for this fundamental phenomenon. We show that relative amino acid hydrophobicity within immunogenic epitopes reveals an antigenic pattern that can be recognized by TCRs. We leveraged these findings to design an immunogenicity model, which was trained and validated using experimentally defined epitopes. ANN-Hydro consistently reduced variable standard prediction outputs across multiple antigens, demonstrating an important step forward in reducing the empirical element of T-cell epitope testing.
The majority of antigens within the immunogenic dataset used in this study are derived from intracellular pathogens, such as viruses, which have been shown to favor a lower G+C genomic content, as reflected in their amino acid use (27). Strongly hydrophobic amino acids (e.g., L, I, V, F, M) are characterized by low G+C codons, whereas hydrophilic amino acids are not (28). This suggests the possibility that pathogens generally have a greater use of hydrophobic amino acids that could be exploited for TCR recognition. A second possibility is that antigen presentation inherently favors hydrophobic regions within a protein. A recent study demonstrated that exposing hydrophobic domains significantly enhances the rate of proteasomal degradation and MHC presentation (29). Moreover, immunogenic CTL epitopes are also positionally biased toward the center of their source antigens (30), consistent with the fact that cytosolic proteins with a central hydrophobic core are the major substrates of proteasomal degradation. Thus, protein hydrophobicity can enhance both antigen presentation and immunogenicity, perhaps an evolutionary adaptation of hydrophobicity driven by damage-associated molecular patterns (31).
TCRs are estimated to recognize on average approximately five nonanchor residues of a presented peptide because of the angle of peptide contact (3, 32). For three pMHC allomorphs analyzed by hydrophobicity in this study, only four or five positions on the peptide were significantly different between immunogenic and nonimmunogenic peptides (Fig. 2), similar to published pMHC-TCR structures (20). This hydrophobicity difference is relative, not absolute. Certain amino acid positions in the peptide may be hydrophilic (e.g., P4 in HLA-A2 9-mers; Fig. 2B); however, even in such inherently hydrophilic residues in the peptide, immunogenic epitopes are less hydrophilic (more hydrophobic). Covering exposed hydrophobic residues on the peptide by a TCR may be a thermodynamically favorable process, facilitating the pMHC–TCR interaction, as noted in retrospect by a recent study (33). TCR engagement of pMHC complexes may be enhanced by water exclusion from the immunologic synapse or by increased Kon rates of the TCR-pMHC complex by relatively hydrophobic amino acids.
In the absence of a good understanding of the biochemical composition of peptide ligands that result in T-cell activation, current strategies for epitope discovery either rely on the unbiased synthesis of a large number of overlapping peptides or use MHC-binding/antigen-processing algorithms to select candidate peptides. Whereas the former is an expensive and laborious process, the latter results in a large number of false-positive peptides that are not immunogenic. Advances in the development of combinatorial technologies have allowed the rapid identification and characterization of antigen-specific T cells (34); however, even such novel technologies rely on binding predictions to create lists of candidate peptides that require extensive empirical validation. For instance, 77 candidate good binders for HLA-A2 from the rotavirus proteome were chosen for recombinant pMHC tetramer production based on their MHC-binding capability, but only six (four being 9-mer epitopes) were confirmed to be immunogenic epitopes (15). Therefore, T-cell antigen discovery studies need strategies to improve the efficiency of epitope prediction.
ANN-Hydro assigned high probabilities of immunogenicity to 80% of the HLA-A2 9-mer epitopes described in the three proteome-wide studies. Of note, three of the four rotavirus 9-mer epitopes from the dataset scored a probability of immunogenicity >0.8 (SI Appendix, Table S5). In the HIV-1 Gag study, more than 364 overlapping peptides were tested in vivo from the Gag variants (length, 500 aa) for epitope discovery. Using the ANN-Hydro model combined with SB scores narrowed the validation discovery process down to 11–15 peptides per Gag protein to be tested. Similarly, applying ANN-Hydro also improved predictions of immunogenic H-2Db and HLA-A2 epitopes from 10 independent antigens compared with individual prediction algorithms. Thus, models such as ANN-Hydro add an extra dimension (immunogenicity) to MHC-binding for CTL epitope prediction and could be used to significantly reduce the variability associated with standard prediction algorithms, as well as the time and cost of experimental validation (Fig. 5 and SI Appendix, Table S6). With the advent of tumor exome sequencing in immune therapy settings, we anticipate that immunogenicity models such as ANN-Hydro will be critical in identifying immunogenic neoantigens for tumor immune therapies (35, 36).
The ANN-Hydro model differs from existing MHC-binding/antigen-processing prediction algorithms in two respects. First, ANN-Hydro was trained on a relative hydrophobicity scale, which helps the model discover complex numerical relationships between different amino acid residues. Second, the dataset used for training was immunogenic epitopes and nonimmunogenic self peptides, which do not differ in binding motifs but differ only in immunogenicity. Whereas some high-binding epitopes (e.g., SI9 from ConsB) are readily predicted by all algorithms, other epitopes (e.g., the immunodominant dominant RT9 from ZM96, LL9 from LCMV-GP) are predicted at variable rankings by different algorithms (SI Appendix, Table S6). In comparison, ANN-Hydro rescued these epitopes by virtue of their probability of immunogenicity.
Although ANN-Hydro marks a step forward in efficiently predicting 9-mer epitopes, it is currently limited in terms of predicting longer or shorter epitopes, as exemplified by the 11-mer epitope (QL11) deduced by epitope mapping from the CN54 Gag protein. To improve longer or shorter epitope predictions, larger representative datasets are needed for training. Nonetheless, the model predicted a 9-mer version of this epitope ranked at 35 and 44, which is consistent with the presentation of nested-length peptides (37). A second limitation of the current model is its applicability to predict epitopes for other HLA class I alleles. In theory, the ANN-Hydro model could be applied to predict CTL epitopes for any MHC class I allele, but large representative datasets are needed to train the model for representative MHC allomorphs. We anticipate that advances in mass spectrometry-based MHC peptide discovery will result in more extensive training databases for predicting longer and shorter epitopes from a broader selection of HLA class I molecules (37, 38).
Although immunogenicity models have been developed by others for predicting CTL epitopes (39, 40), they considered only the impact of pMHC stability and positional significance along the peptide for immunogenicity. In contrast, a crucial feature of our approach is the use of ligand-eluted nonimmunogenic self peptides as the comparator set. Because binding and antigen processing are required for all epitopes, we built on existing algorithms for immunogenic pMHC predictions. “Layering” the immunogenicity model on top of existing prediction algorithms enabled us to predict epitopes with increased effectiveness over stand-alone predictions. Importantly, the empirical evaluation of our immunogenicity model and epitope prediction approach without a priori knowledge of the immunodominant HIV-1 Gag epitopes in vivo provides strong support for these results. In summary, integrating amino acid hydrophobicity into pMHC prediction algorithms should significantly enhance the success of epitope discovery. The biological mechanism underlying TCR preferences for nonpolar hydrophobic residues remains to be evaluated.
Materials and Methods
Full details on methods and construction of datasets are provided in SI Appendix, SI Materials and Methods.
Construction of Datasets.
All MHC-I peptides used in this study and design of the ANN-Hydro prediction model were retrieved from IEDB (16) (www.iedb.org). Epitopes with a positive T-cell response represent the immunogenic epitope group. The nonimmunogenic self-peptide group represents cell surface ligand-eluted MHC-I self peptides that have been antigenically processed and MHC-bound. Additional curation and exclusion criteria resulted in a final dataset with 5,035 8- to 11-mer immunogenic epitopes and 4,853 8- to 11-mer nonimmunogenic peptides (SI Appendix, Table S1). Further details are provided in SI Appendix, SI Materials and Methods.
Amino Acid Scales.
These were derived from Expasy’s ProtScale (web.expasy.org/protscale/) (41), specifically the Hydrophobicity (Kyte and Doolittle) (17), Polarity (Grantham) (18), and Bulkiness (Zimmerman) (19) scales. The scales are relative; that is, negative to positive values in the hydrophobicity scale correspond to a relative hydrophobicity increase between amino acids (SI Appendix, Table S2).
Position-Based Hydrophobicity Analysis.
Our datasets of immunogenic and nonimmunogenic peptides were transformed into numeric arrays using R statistical software (42). Separate numeric arrays were generated for immunogenic and nonimmunogenic 8, 9, and 10 mers. Mean hydrophobicity of immunogenic and nonimmunogenic peptides at each position was calculated and compared residue-by-residue through Wilcoxon rank-sum tests to quantify statistical significance.
Hydrophobicity-Based ANN Prediction Model (ANN-Hydro).
The R neuralnet package was used to design and train the two ANN-Hydro models on H-2Db–and HLA-A2–restricted 9-mer peptides known to be immunogenic (n = 204 and n = 374, respectively) or nonimmunogenic (n = 232 and n = 201, respectively). Each peptide sequence in the respective H-2Db and HLA-A2 datasets was transformed into a corresponding numeric sequence based on amino acid hydrophobicity using R statistical software. A three-layer, fully connected, feed-forward ANN was composed of nine input neurons, one hidden layer with three neurons, and one output variable (SI Appendix, Fig. S3).
Application of ANN-Hydro.
For each H-2Db– and HLA-A2–restricted epitope prediction, we used IEDB consensus to generate a list of epitope predictions. Each peptide was assigned a normalized binding score, SB, and a subset of these predicted peptides was then selected by defining an SB threshold of 0.1 for antigen length > 100 amino acids and an SB threshold of 0.2 for antigen length ≤ 100 amino acids. Independently, probabilities of immunogenicity were obtained by applying ANN-Hydro to this subset of binding predictions. Normalized scores, SI, were then assigned based on the probabilities of immunogenicity (SI Appendix, Fig. S3). The list of predicted peptides was ranked based on a total score, S = SB⋅SI, ranging from lowest to highest score. The lower the total score of a predicted peptide, the higher its probability of being an immunogenic epitope. Details are provided in SI Appendix, SI Materials and Methods.
Vaccines.
Recombinant adenovirus type 5 (rAdHu5) vectors encoding codon optimized HIV-1 Gag from ConsB, strain 96ZM651.8 (ZM96) and strain 97CN54 (CN54) (43), are described in SI Appendix, SI Materials and Methods.
Immunization of Mice.
C57BL/6 mice were immunized with 109 virus particles. All animal studies were conducted in accordance with UK Home Office regulations and the King’s College London Ethics Committee.
Peptides.
The 15-mer peptides spanning HIV-1 CN54 Gag and a 20-mer set of peptides spanning HIV-1 ZM96 were provided by the UK Centre for AIDS Reagents. The 15-mer peptides spanning HIV-1 ConsB Gag were provided by the National Institutes of Health’s AIDS Reagent Reference Program. Truncated HIV-1 Gag peptides were purchased from Proimmune.
T-Cell Epitope Mapping.
Spleen cells were restimulated either with media alone or with peptides, either in pools or individually (each at 1 μM final concentration), and IFN-γ production was detected by intracellular cytokine staining or by ELISPOT assay as described previously (43). ConsB and CN54 Gag epitopes were deconvoluted to individual 15 mers from peptide pools, and truncated versions of the 15-mer peptides were synthesized and tested. For ZM96 Gag, 49 individual 20-mer peptides were tested. Reactive peptide sequences were confirmed against the corresponding 15-mer peptide to the reactive sequence, and 9-mer peptides were synthesized and tested.
Supplementary Material
Acknowledgments
We thank J. LaBaer and J. E. Taylor for their helpful comments, J. Patel for assisting with the annotation of the datasets, B. Hahn for providing the ZM96gag plasmid DNA obtained through the Center for Aids Research, D. Garber for providing the ConsB gene, G. Nabel for providing AdHu5, and the National Institutes of Health’s AIDS Reference and Reagent Repository program and the Center for Aids Research for providing peptides. This work was supported in part by institutional funds from Arizona State University (to D.C., S.K., J.N.B., and K.S.A.), National Institutes of Health Grants CA33084 and CA18029 (to P.D.G.), and the Bill and Melinda Gates Foundation (to P.D.G., J.N.B., and L.S.K.). Equipment was made available by the National Institutes of Health’s Biomedical Research Centre based at Guy's and St. Thomas' National Health Service Foundation Trust, King's College London.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1500973112/-/DCSupplemental.
References
- 1.Grakoui A, et al. The immunological synapse: A molecular machine controlling T cell activation. Science. 1999;285(5425):221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 2.Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol. 2013;31:443–473. doi: 10.1146/annurev-immunol-032712-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hennecke J, Wiley DC. T cell receptor–MHC interactions up close. Cell. 2001;104(1):1–4. doi: 10.1016/s0092-8674(01)00185-4. [DOI] [PubMed] [Google Scholar]
- 4.Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discov. 2007;6(5):404–414. doi: 10.1038/nrd2224. [DOI] [PubMed] [Google Scholar]
- 5.Hogquist K, et al. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76(1):17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 6.Medzhitov R, Janeway CA., Jr Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296(5566):298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 7.Falk K, Rötzschke O, Stevanović S, Jung G, Rammensee H-G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351(6324):290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- 8.Rammensee H, Bachmann J, Emmerich NPN, Bachor OA, Stevanović S. SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics. 1999;50(3-4):213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 9.Kubo RT, et al. Definition of specific peptide motifs for four major HLA-A alleles. J Immunol. 1994;152(8):3913–3924. [PubMed] [Google Scholar]
- 10.van der Merwe PA, Dushek O. Mechanisms for T cell receptor triggering. Nat Rev Immunol. 2011;11(1):47–55. doi: 10.1038/nri2887. [DOI] [PubMed] [Google Scholar]
- 11.Honeyman MC, Brusic V, Stone NL, Harrison LC. Neural network-based prediction of candidate T-cell epitopes. Nat Biotechnol. 1998;16(10):966–969. doi: 10.1038/nbt1098-966. [DOI] [PubMed] [Google Scholar]
- 12.Moutaftsi M, et al. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus. Nat Biotechnol. 2006;24(7):817–819. doi: 10.1038/nbt1215. [DOI] [PubMed] [Google Scholar]
- 13.Nielsen M, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS ONE. 2007;2(8):e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tenzer S, et al. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell Mol Life Sci. 2005;62(9):1025–1037. doi: 10.1007/s00018-005-4528-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newell EW, et al. Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Nat Biotechnol. 2013;31(7):623–629. doi: 10.1038/nbt.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vita R, et al. The immune epitope database 2.0. Nucleic Acids Res. 2010;38(Database issue):D854–D862. doi: 10.1093/nar/gkp1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 18.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185(4154):862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 19.Zimmerman JM, Eliezer N, Simha R. The characterization of amino acid sequences in proteins by statistical methods. J Theor Biol. 1968;21(2):170–201. doi: 10.1016/0022-5193(68)90069-6. [DOI] [PubMed] [Google Scholar]
- 20.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 21.Bishop CM. Pattern Recognition and Machine Learning. Springer; New York: 2006. [Google Scholar]
- 22.Assarsson E, et al. Immunomic analysis of the repertoire of T-cell specificities for influenza A virus in humans. J Virol. 2008;82(24):12241–12251. doi: 10.1128/JVI.01563-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weiskopf D, et al. Insights into HLA-restricted T cell responses in a novel mouse model of dengue virus infection point toward new implications for vaccine design. J Immunol. 2011;187(8):4268–4279. doi: 10.4049/jimmunol.1101970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oukka M, et al. Protection against lethal viral infection by vaccination with nonimmunodominant peptides. J Immunol. 1996;157(7):3039–3045. [PubMed] [Google Scholar]
- 25.van der Most RG, et al. Identification of Db- and Kb-restricted subdominant cytotoxic T-cell responses in lymphocytic choriomeningitis virus-infected mice. Virology. 1998;240(1):158–167. doi: 10.1006/viro.1997.8934. [DOI] [PubMed] [Google Scholar]
- 26.Pradeu T, Carosella ED. On the definition of a criterion of immunogenicity. Proc Natl Acad Sci USA. 2006;103(47):17858–17861. doi: 10.1073/pnas.0608683103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calis JJ, Sanchez-Perez GF, Keşmir C. MHC class I molecules exploit the low G+C content of pathogen genomes for enhanced presentation. Eur J Immunol. 2010;40(10):2699–2709. doi: 10.1002/eji.201040339. [DOI] [PubMed] [Google Scholar]
- 28.Khrustalev VV, Barkovsky EV. Percent of highly immunogenic amino acid residues forming B-cell epitopes is higher in homologous proteins encoded by GC-rich genes. J Theor Biol. 2011;282(1):71–79. doi: 10.1016/j.jtbi.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 29.Huang L, Kuhls MC, Eisenlohr LC. Hydrophobicity as a driver of MHC class I antigen processing. EMBO J. 2011;30(8):1634–1644. doi: 10.1038/emboj.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim Y, Yewdell JW, Sette A, Peters B. Positional bias of MHC class I restricted T-cell epitopes in viral antigens is likely due to a bias in conservation. PLOS Comput Biol. 2013;9(1):e1002884. doi: 10.1371/journal.pcbi.1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seong SY, Matzinger P. Hydrophobicity: An ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4(6):469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 32.Burroughs NJ, de Boer RJ, Keşmir C. Discriminating self from nonself with short peptides from large proteomes. Immunogenetics. 2004;56(5):311–320. doi: 10.1007/s00251-004-0691-0. [DOI] [PubMed] [Google Scholar]
- 33.Birnbaum ME, et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell. 2014;157(5):1073–1087. doi: 10.1016/j.cell.2014.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newell EW, Davis MM. Beyond model antigens: High-dimensional methods for the analysis of antigen-specific T cells. Nat Biotechnol. 2014;32(2):149–157. doi: 10.1038/nbt.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snyder A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yadav M, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–576. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 37.Riemer AB, et al. A conserved E7-derived cytotoxic T lymphocyte epitope expressed on human papillomavirus 16-transformed HLA-A2+ epithelial cancers. J Biol Chem. 2010;285(38):29608–29622. doi: 10.1074/jbc.M110.126722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan CT, Croft NP, Dudek NL, Williamson NA, Purcell AW. Direct quantitation of MHC-bound peptide epitopes by selected reaction monitoring. Proteomics. 2011;11(11):2336–2340. doi: 10.1002/pmic.201000531. [DOI] [PubMed] [Google Scholar]
- 39.Calis JJ, et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLOS Comput Biol. 2013;9(10):e1003266. doi: 10.1371/journal.pcbi.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harndahl M, et al. Peptide-MHC class I stability is a better predictor than peptide affinity of CTL immunogenicity. Eur J Immunol. 2012;42(6):1405–1416. doi: 10.1002/eji.201141774. [DOI] [PubMed] [Google Scholar]
- 41.Gasteiger E, et al. Protein identification and analysis tools on the ExPASy server. 2005 doi: 10.1385/1-59259-584-7:531. The Proteomics Protocols Handbook, ed Walker JM (Humana, Totowa, NJ), 1st Ed, pp 571–607. [DOI] [PubMed] [Google Scholar]
- 42.R Development Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2008. [Google Scholar]
- 43.Bachy V, et al. Langerin negative dendritic cells promote potent CD8+ T-cell priming by skin delivery of live adenovirus vaccine microneedle arrays. Proc Natl Acad Sci USA. 2013;110(8):3041–3046. doi: 10.1073/pnas.1214449110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.