

Significance
β-Adrenergic receptors (β-ARs) are hormone and neurotransmitter receptors. The data we present in this paper challenge the assumption that memory reconsolidation is governed by the traditional β-AR/G protein signaling pathway. We found that memory reconsolidation is mediated by a β-arrestin–dependent β-adrenergic signaling pathway. Our experiments demonstrate that upon memory retrieval, a β1-AR/β-arrestin2/ERK pathway is activated in distinct brain areas, stimulating de novo protein synthesis and inducing postretrieval memory restabilization. Moreover, memory reconsolidation can be disrupted by propranolol, but not biased β-blockers such as carvedilol and alprenolol. Our study thus demonstrates that memory reconsolidation is mediated by a β-arrestin–biased β-adrenergic signaling pathway and reveals the therapeutic potential for β-arrestin–biased ligands in the treatment of memory-related disorders.
Keywords: β-arrestin2, β-adrenergic receptor, memory reconsolidation, biased receptor signaling
Abstract
A long-standing hypothesis posits that a G protein-coupled signaling pathway mediates β-adrenergic nervous system functions, including learning and memory. Here we report that memory retrieval (reactivation) induces the activation of β1-adrenergic β-arrestin signaling in the brain, which stimulates ERK signaling and protein synthesis, leading to postreactivation memory restabilization. β-Arrestin2-deficient mice exhibit impaired memory reconsolidation in object recognition, Morris water maze, and cocaine-conditioned place preference paradigms. Postreactivation blockade of both brain β-adrenergic Gs protein- and β-arrestin–dependent pathways disrupts memory reconsolidation. Unexpectedly, selective blockade of the Gs/cAMP/PKA signaling but not the β-arrestin/ERK signaling by the biased β-adrenergic ligands does not inhibit reconsolidation. Moreover, the expression of β-arrestin2 in the entorhinal cortex of β-arrestin 2–deficient mice rescues β1-adrenergic ERK signaling and reconsolidation in a G protein pathway-independent manner. We demonstrate that β-arrestin–biased signaling regulates memory reconsolidation and reveal the potential for β-arrestin–biased ligands in the treatment of memory-related disorders.
Alongside classical G protein pathways, activation of G protein-coupled receptors (GPCRs) stimulates β-arrestin–dependent signaling, leading to ERK phosphorylation and other downstream events (1, 2). Biased agonists, which induce functionally selective or biased receptor states and, thus, selectively activate one of the signaling pathways, have recently been identified for several GPCRs (3). Biased receptor agonism offers theoretical guidance for the discovery of a new generation of GPCR-targeted drugs with greater efficacy but fewer adverse effects. However, the lack of knowledge about the signaling pathways specifically eliciting a beneficial effect is a major obstacle in the understanding of disease mechanisms and the development of biased drugs targeting most GPCRs, especially those expressed in the central nervous system (CNS) with psychiatric importance.
Besides their important roles in the cardiovascular and pulmonary systems, β-adrenergic receptors (β-ARs) are critically involved in CNS functions such as arousal, cognition, and stress-related behaviors (4, 5). β-Adrenergic neuronal signaling is important for neuroplasticity, including long-term potentiation (6) and memory formation (7). Accumulating cell biological evidence suggests that β-ARs also signal via G protein-independent, β-arrestin–dependent pathways (8–10). However, functions of β-AR in the CNS have been primarily ascribed to their classical role of stimulating Gs protein. The differential neurophysiological consequences for the G protein- and β-arrestin–dependent pathways, if any, have not been delineated.
A longstanding hypothesis posits that a β-AR/Gs/protein kinase A (PKA) signaling pathway mediates memory reconsolidation (11–13), a process that strengthens, updates, or erases a previously acquired memory after recall (memory reactivation). This hypothesis is largely based on observations that β-ARs and molecules in the classical GPCR signaling pathway—such as cAMP (cAMP), PKA, and cAMP response element-binding protein (CREB)—are required for reconsolidation, which was determined by using receptor antagonists, kinase inhibitors, or gene knockout mice (11, 14, 15). Most of these molecules are also required for basal neural activity or plasticity, and there has been no direct evidence demonstrating that the function of β-ARs in reconsolidation is mediated by G protein/PKA or other signaling pathway (12). In the current study, we tested the potential involvement of G protein/cAMP/PKA-dependent pathway versus β-arrestin–dependent signaling in memory reconsolidation by using object recognition paradigm.
Results
Reconsolidation of Object Recognition Memory Is Mediated by a Gs Protein-Independent β1-AR Signaling Pathway.
Mice tend to explore a novel object more than the familiar one, and this preference reflects the use of recognition memory (16). In the reconsolidation of object recognition memory (ORM) test, mice were first trained to recognize object A and object B (Fig. S1A), and 24 h after reexposure to the two objects to retrieve/reactivate ORM acquired in the training session, they were subjected to memory retention (reconsolidation) test. During the 5-min memory test, mice were allowed to explore a novel object (object C) and a familiar object (object A). The time spent exploring each object was recorded (Fig. S1 A–G) and the animal’s preference for object C over object A was designated as preference index and compared with those for object B over object A determined during memory reactivation process. We first examined the effect of antagonist treatment given immediately (within 2 min) after memory reactivation on ORM reconsolidation (Fig. 1A). Two-way repeated measures (RM) ANOVA indicates a drug treatment by session test interaction [Ftreatment (4, 93) = 15.082, P < 0.001, Fsession (1, 93) = 80.298, P < 0.001, Ftreatment × session (4, 93) = 13.051, P < 0.001, two-way RM ANOVA]. During the memory retention test, C57BL/6 mice treated with vehicle immediately after reexposed to objects A and B (memory reactivation) exhibited a preferential exploration for object C versus object A, indicating a normal object recognition memory, whereas mice i.p. administrated propranolol (a nonselective blocker of β-AR) or betaxolol (a selective β1-AR antagonist) after memory reactivation did not (Bonferroni’s post hoc comparison, Fig. 1A and Fig. S1 B, H, and I). Moreover, ORM reconsolidation could not be blocked by i.p. administration of nadolol, a blood–brain barrier-impermeable β-blocker (Fig. S1M) or β2-AR–selective antagonist ICI 118, 551 (Fig. 1A and Fig. S1J). These data suggest the critical involvement of brain β1 adrenergic signaling in ORM reconsolidation.
Fig. 1.
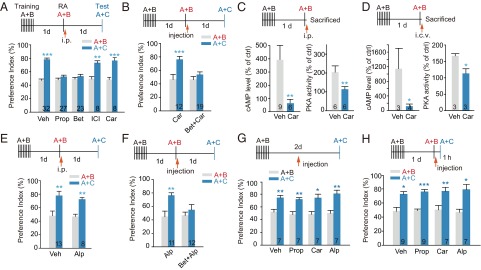
ORM reconsolidation is mediated by a Gs protein-independent β1-AR signaling pathway. Mice trained with object A and object B were reexposed to both objects (A+B) for memory reactivation (RA) 24 h later. Drug injection was given within 2 min after memory reactivation. Memory retention was tested by exposure to object A and object C (A+C). Values in the bar indicate number of mice per group. (A and E) Postreactivation i.p. injection of propranolol (Prop, 10 mg/kg) or betaxolol (Bet; 1.0 mg/kg) inhibited reconsolidation, whereas ICI 118,551 (ICI; 10 mg/kg), carvedilol (Car; 3.0 mg/kg), alprenolol (Alp; 10 mg/kg), or vehicle (Veh; 4.0 mL/kg) did not. **P < 0.01, ***P < 0.001 vs. RA (A+B) with the same drug treatment. (B and F) Administration of Bet (1.0 mg/kg, i.p.) before Car (10 µg i.c.v.) or Alp (10 µg, i.c.v.) decreased preference index, whereas injection of Car or Alp alone did not. **P < 0.01, ***P < 0.001 vs. (A+B) with same drug treatment. (C and D) Injection of Car (3.0 mg/kg, i.p. or 10.0 µg, i.c.v., within 2 min after memory reactivation) decreased cAMP level and PKA activity in the Enc as determined 5 min after RA. Data are expressed as percentage of basal level determined before reactivation. *P < 0.05, **P < 0.01 vs. Veh, t test. (G) ORM retention was tested 2 d after training. β-Blockers were given 1 d after training without memory reactivation. *P < 0.05, **P < 0.01 vs. Training (A+B) with same drug treatment. (H) ORM retention was tested 1 h after memory reactivation. *P < 0.05, **P < 0.01, ***P < 0.001 vs. RA (A+B) within same treatment.
Unexpectedly, in contrast to the strong inhibitory effect of propranolol and betaxolol, postreactivation i.p. administration of the biased β-AR ligand carvedilol, an antagonist of the G protein pathway and a weak agonist of β-arrestin–dependent ERK signaling (8, 9), failed to block ORM reconsolidation (Fig. 1A and Fig. S1K). Intracerebroventricular (i.c.v.) injection of carvedilol immediately after memory reactivation also failed to inhibit ORM reconsolidation, whereas the combined pretreatment of betaxolol and carvedilol impaired ORM reconsolidation [Fig. 1B, Ftreatment (1, 29) = 3.691, P = 0.065, Fsession (1, 29) = 32.048, P < 0.001, Ftreatment × session (1, 29) = 13.220, P = 0.001, two-way RM ANOVA]. The analysis of total time spent exploring each object confirmed the above results (Fig. S1 B and C). These data argue that the β-AR/β-arrestin signaling, but not the β-AR/Gs-protein signaling, is required for ORM reconsolidation.
To confirm that the Gs/cAMP/PKA pathway in the brain was selectively blocked by carvedilol administered via i.p. and i.c.v. injection during ORM reconsolidation, the level of cAMP and the activation of PKA and ERKs in the entorhinal cortex (Enc), a brain region critically involved in ORM, were determined. Upon memory reactivation, cAMP level in the Enc of C57BL/6 mice was increased and reached peak value at ∼5 min after memory reactivation (Fig. S2A). Administration of carvedilol via i.p. or i.c.v. abolished memory reactivation-induced cAMP accumulation and PKA activation (Fig. 1 C and D and Fig. S2 A and B), but stimulated β1-AR–mediated ERK activation (Fig. S2 C and D). Moreover, postmemory reactivation (i.p. or i.c.v.) administration of another biased β-AR ligand alprenolol, which also selectively antagonizes Gs signaling and stimulates the β-arrestin signaling, did not inhibit memory reconsolidation either [Fig. 1E, Ftreatment × session (1, 19) = 0.225, P = 0.650; Fig. S1D; Fig. 1F, Ftreatment (1, 21) = 0.416, P = 0.526, Fsession (1, 21) = 14.413, P = 0.001, Ftreatment × session (1, 21) = 4.767, P = 0.041; Fig. S1 E and L, two-way RM ANOVA]. The preference index for the novel object was not altered when propranolol, carvedilol, or alprenolol was administered at the corresponding time point but without memory reactivation [Fig. 1G, Ftreatment × session (3, 24) = 0.537, P = 0.663, two-way RM ANOVA; Fig. S1F] or determined 1 h after memory reactivation [Fig. 1H, Ftreatment × session (3, 28) = 0.424, P = 0.738; two-way RM ANOVA; Fig. S1G]. These data suggest that Gs/cAMP/PKA-independent β1-AR signaling mediates ORM reconsolidation.
β-Arrestin2 Is Required for Postreactivation Memory Restabilization.
We next explored the possible involvement of β-arrestin/ERK-dependent signaling. In the long-term memory test after memory reactivation, the performance of wild-type C57BL/6 (Arrb2+/+) and β-arrestin2 knockout (Arrb2−/−) mice was compared. Arrb2+/+ showed significant preference for the novel object in ORM test, whereas Arrb2−/− mice exhibited no preference. ANOVA showed a genotype-by-memory session interaction [Fig. 2A, Fgenotype (1, 28) = 9.209, P = 0.005; Fsession (1, 28) = 12.076, P = 0.002; Fgenotype × session (1, 28) = 18.328, P < 0.001 two-way RM ANOVA]. The attenuation of memory retention by β-arrestin2 ablation was memory reactivation-dependent [Fig. 2B, Fgenotype (1, 27) = 0.244, P = 0.625; Fsession (1, 27) = 29.791, P < 0.001; Fgenotype × session (1, 27) = 0.398, P = 0.534, two-way RM ANOVA] and long-lasting (Fig. S3 A and B), but it was not detected within 3 h of reactivation [Fig. 2C, Fgenotype (1, 16) = 0.509, P = 0.486; Fsession (1, 16) = 125.929, P < 0.001; Fgenotype × session (1, 16) = 0.800, P = 0.384, two-way RM ANOVA; Fig. S3C]. Both Arrb2+/+ and Arrb2−/− mice could form consolidated memory 24 h after training (Fig. S3D). These data suggest that β-arrestin2, like β1-AR, functions in ORM reconsolidation via restabilization of the postreactivation long-term memory. Moreover, the postreactivation treatment of carvedilol did not restore the impaired ORM reconsolidation in Arrb2−/− mice [Fig. 2D, Fgenotype × session (1, 12) = 0.153, P = 0.702, two-way RM ANOVA]. The analysis of total time spent exploring each object confirmed the above results (Fig. S3 H–O). Arrb2−/− mice showed no change in locomotor activity in the open field task, whereas the treatment of propranolol did not inhibit locomotion of Arrb2+/+ or Arrb2−/− mice (Table S1). No impairment of ORM reconsolidation was found in β-arrestin1 knockout (Arrb1−/−) mice, suggesting β-arrestin1 is not critically involved in ORM reconsolidation (Fig. S3 E–G).
Fig. 2.

β-Arrestin2 is required for postreactivation restabilization of ORM and spatial memory. Arrb2+/+ and Arrb2−/− mice were tested for ORM retention 48 h after training with (A) or without memory reactivation (B), or tested 1 h after memory reactivation (C). **P < 0.01, ***P < 0.001 vs. (A+B) within the same genotype. (D) Reconsolidation of ORM after postreactivation carvedilol treatment in Arrb2−/− mice (3.0 mg/kg, i.p.). (E and F) Reconsolidation of spatial memory. Mice were trained to find the hidden platform in the Morris water maze; two probe trials were performed sequentially with the platform removed, and the percentage of time spent in each quadrant was calculated. Probe Test 1 (memory reactivation) was carried out 24 h after the last training trial. Probe Test 2 was performed 24 h (E) or 1 h (F) after Probe Test 1. Quadrants: L, left; O, opposite; R, right; T, target. n = 6–10 per group; *P < 0.05 vs. O, L, and R in each probe test, three-way ANOVA.
The role of β-arrestin2 in reconsolidation of spatial memory was tested in the Morris water maze task. Arrb2−/− mice performed comparably to Arrb2+/+ mice in cued or spatial training (Fig. S4 A and B). Two probe trials were sequentially carried out after mice learned to find the hidden platform (Fig. 2 E and F). To avoid possible memory extinction, a short probe trial of 60 s was used as memory reactivation session, which has been shown by other groups to cause no extinction (17–19). The first probe test was carried out 1 d after spatial training, and both Arrb2−/− mice and wild-type littermates demonstrated a similar preference for the target quadrant. The results of the second probe test (memory retention test) revealed that Arrb2−/−, but not Arrb2+/+ mice, forgot the location of the platform 1 d after the first probe test [Fig. 2E and Fig. S4C, Ftarget × genotype (3, 104) = 5.038, P = 0.003], although both genotypes retained this information 1 h after the first probe trial [Fig. 2F, Ftarget × genotype (3, 88) = 2.189, P = 0.095]. No extinction in Probe Test 2 was detected in the wild-type littermates; however, significant decrease of preference for the target quadrant in Arrb2−/− mice was found in the second probe test, indicating that β-arrestin2 is required for the reconsolidation of hippocampus-dependent spatial memory.
Memory Reactivation Triggers β-Arrestin2–Mediated β-AR-ERK Translational Signaling.
The activation of ERK cascade, a major target of β-arrestin–dependent signaling, was tested. Reactivation of ORM induced a time-dependent increase of ERK phosphorylation in the Enc of Arrb2+/+ mice (Fig. S5A), which could be further enhanced by carvedilol treatment [Fig. 3A and Fig. S5B, Ftreatment × session (5, 86) = 7.935, P < 0.001, two-way ANOVA]. The peak of ERK activation was detected 15 min after memory reactivation in the Enc, but not the ventral hippocampus or cerebellum in Arrb2+/+ mice (Fig. S5 A and C). Postreactivation inhibition of ERK activation in the brain by U0126 blocked ORM reconsolidation (Fig. S5D). The increase in phosphorylation of ERK in Enc neurons induced by memory reactivation could be abolished by postreactivation administration of propranolol or betaxolol, or ablation of β-arrestin2 [Fig. 3B and Fig. S5E, Fgenotype × treatment × session (1, 55) = 5.726, P = 0.020; Fig. 3 C and D, Fgenotype × treatment × session (1, 36) = 5.230, P = 0.028, three-way ANOVA; Fig. S5F]. The i.c.v. injection of isoproterenol increased pERK level in the Enc of Arrb2+/+, but not Arrb2−/− mice, and this increase could be suppressed by pretreatment of betaxolol (Fig. S5G).
Fig. 3.
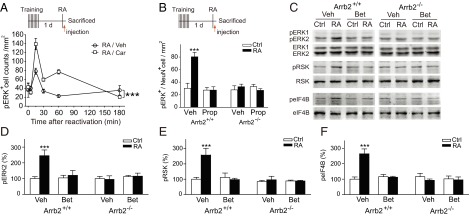
Memory reactivation induces β-arrestin2–mediated β-AR-ERK translational signaling in the Enc. (A) Postreactivation immediate injection of Car (3.0 mg/kg, i.p.) enhanced reactivation-stimulated ERK activation in the Enc. n = 4–11. ***P < 0.001. (B–F) Arrb2+/+ and Arrb2−/− mice were treated with β-blocker or vehicle immediately after ORM reactivation, and brain samples were collected 15 min later for analysis of phosphorylation status. (B) Postreactivation injection of Prop (10 mg/kg, i.p.) and ablation of β-arrestin2 inhibited reactivation-induced increase in phosphorylated ERK (pERK)/NeuN double-positive cell counts in the Enc (n = 5–7 mice per group). ***P < 0.001 vs. no RA control (Ctrl). (C–F) Postreactivation injection of Bet (1.0 mg/kg, i.p) and ablation of β-arrestin2 inhibited reactivation-induced increase of phosphorylation of ERK (pERK, n = 5–6 per group), phosphorylation of ribosomal S6 kinase (pRSK, n = 5–6 per group), and phosphorylation of eukaryotic translation initiation factor 4B (peIF4B, n = 5–6 per group) in the Enc. ***P < 0.001 vs. Ctrl.
Previous studies have shown that reconsolidation of spatial and fear memories requires postreactivation neurotransmission and de novo protein synthesis (20, 21), and that the binding of β-arrestin–biased ligand with AT1R stimulates ERK-dependent protein translation in HEK293 cells (22, 23). As shown in Fig. 3 C, E, and F, increased phosphorylation of 90-kDa ribosomal S6 kinase (p90-RSK) and eukaryotic translation initiation factor 4B (eIF4B), downstream targets of ERKs and key regulators of protein synthesis, was observed in the Enc of Arrb2+/+ mice after ORM reactivation [Fig. 3E, Fgenotype × treatment × session (1, 36) = 11.109, P = 0.003; Fig. 3F, Fgenotype × treatment × session (1, 36) = 12.55, P = 0.001, three-way ANOVA]. In contrast, no increase in phosphorylation of ERK1/2, p90-RSK, or eIF4B was observed in Arrb2−/− mice, or mice treated with betaxolol after reactivation (Fig. 3 C–F). These data suggest that memory recall stimulates β1-AR/β-arrestin/ERK–mediated de novo protein synthesis, which could positively regulate memory reconsolidation.
β1-AR/β-Arrestin2/ERK Signaling Mediates Memory Reconsolidation.
The physiological consequence of β-arrestin–biased β1-AR signaling was investigated. Local viral expression of β-arrestin2/GFP, but not the control protein β-galactosidase/GFP, in the Enc of Arrb2−/− mice rescued the ORM reconsolidation phenotype [Fig. 4A, Fgenotype (1, 64) = 5.186, P = 0.026; Fviral construct (1, 64) = 2.126, P = 0.150; Fsession (1, 64) = 55.898, P < 0.001; Fgenotype × viral construct × session (1, 64) = 12.642, P < 0.001, three-way ANOVA]. The number of β-arrestin2/GFP-expressing neurons in the Enc of these mice was positively correlated with ORM reconsolidation (Fig. 4B and Fig. S6A). Expressing β-arrestin2 in the Enc of Arrb2−/− mice restored β-AR–mediated ERK activation in infected neurons [Fig. 4C and Fig. S6B, Fviral expression × treatment (1, 20) = 5.106, P = 0.035 for pERK; Fviral expression × treatment (1, 20) = 12.494, P = 0.002 for pERK/GFP, two-way ANOVA]. The restored ORM reconsolidation by infection of AAV-Arr2 in the Enc of Arrb2−/− mice could be interrupted by postmemory reactivation treatment of betaxolol [Fig. 4D, Fviral construct (1, 66) = 5.125, P = 0.027; Ftreatment (1, 66) = 6.300, P = 0.014; Fviral construct × session × treatment (1, 66) = 7.993, P = 0.006, three-way ANOVA], just as what was observed with the AAV-Gal injected wild-type control mice (Fig. S6C). Similar to the results obtained with the wild-type C57BL/6 mice (Fig. 1 B and F), betaxolol plus carvedilol, but not carvedilol treatment alone showed a significant inhibition on memory reconsolidation in Arrb2−/− mice infected with AAV-Arr2 (Fig. S6D). These results indicate that the β-arrestin/ERK-biased β1-AR signaling, but not the conventional β-adrenergic Gs/cAMP/PKA signaling in the Enc mediates reconsolidation of ORM.
Fig. 4.
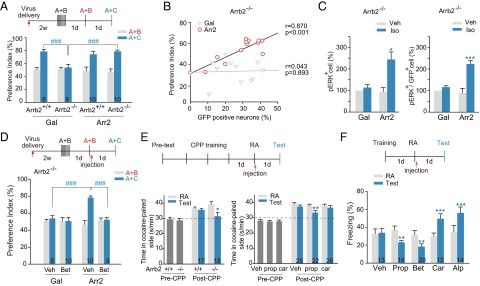
β1-AR/β-arrestin2/ERK signaling mediates memory reconsolidation. (A) Reconsolidation of ORM in Arrb2+/+ and Arrb2−/− mice infected with AAV-encoding β-galactosidase (Gal) or β-arrestin2 (Arr2) in the Enc. ###P < 0.001 vs. indicated groups. (B) Plot of preference index for object C against percentage of GFP-positive neurons in the Enc of Arrb2−/− mice infected with Gal or Arr2. Each symbol represents data obtained from a single animal. (C) Levels of pERK- and pERK/GFP-positive cells in the Enc of Arrb2−/− mice locally infected with virus 15 min after i.c.v. injection of vehicle or isoproterenol (Iso, 10 μg). n = 4–9. *P < 0.05, ***P < 0.001 vs. Veh. (D) Reconsolidation of ORM. Arrb2−/− mice expressing Gal or Arr2 in the Enc were given Bet (1.0 mg/kg, i.p.) or Veh injection after memory reactivation. ###P < 0.001 vs. indicated groups. (E) Reconsolidation of cocaine CPP memory. Time (s/min) spent in drug-paired side before and after 3 d of place preference conditioning of cocaine (10 mg/kg) was presented. (Left) Arrb2+/+ and Arrb2−/− mice were tested. (Right) Wild-type mice were injected with Prop (10 mg/kg, i.p.) or Car (10 μg, i.c.v.) after memory reactivation. **P < 0.01, ***P < 0.001 vs. RA. (F) Reconsolidation of contextual fear memory. Postreactivation treatment with Prop (10 mg/kg, i.p.) or Bet (1.0 mg/kg, i.p.) inhibited freezing, whereas Car (10 µg, i.c.v.) or Alp (10 µg, i.c.v.) increased cue-induced freezing as determined 24 h after. **P < 0.01, ***P < 0.001 vs. RA.
Combining memory reactivation with the pharmacological disruption of reconsolidation was recently proposed as a strategy to treat drug addiction and fear-related disorders, including posttraumatic stress disorder; however, the distinct receptor-mediated signaling pathway responsible for reconsolidation has not been identified. We hypothesized that β-arrestin–biased, and not Gs protein-mediated signaling, also underlies reconsolidation of drug-conditioned place preference (CPP) and conditioned fear memories, and possibly other types of memories. Consistent with the results of ORM, Arrb2−/− mice showed reduced preference for the drug-paired chamber 1 d after reactivation of a cocaine-associated memory [Fig. 4E, Left, Fgenotype × session (1, 33) = 6.028, P = 0.020, two-way RM ANOVA]. The reconsolidation of cocaine-related memory was disrupted by postreactivation injection of propranolol, but not carvedilol [Fig. 4E, Right, Ftreatment × session (2, 70) = 6.820, P = 0.003, two-way RM ANOVA]. The involvement of β-arrestin–biased signaling was also shown in conditioned fear memory model. During training phase, the freezing levels before and right after footshock were not different between Arrb2+/+ and Arrb2−/− mice (Fig. S6E). In the memory retention test, propranolol and betaxolol inhibited, whereas carvedilol and alprenolol enhanced, freezing behavior when memory retention was tested 1 d after reexposure to the context, in which a one-trial fear training was given [Fig. 4F, Ftreatment × session (4, 68) = 17.249, P < 0.001, two-way RM ANOVA].
Discussion
Consolidated memories are transiently destabilized upon reactivation, and subsequently restabilized through reconsolidation, an active, de novo protein synthesis-dependent process (21, 24, 25). Studies have shown that blocking β-adrenergic transmission by propranolol impairs memory reconsolidation in auditory fear conditioning, spatial radial maze, and cocaine-induced CPP (12, 26–30); inhibition of basal and stimulated activities of certain components of G protein-coupled pathways associated with neuroplasticity, such as PKA, ERK, and CREB, disrupts reconsolidation of fear and object recognition memories (31–34). In cocaine-associated reward memory task, bilateral intraamygdalar infusions of the PKA inhibitor Rp-cAMPS following light/tone stimulus reactivation decreases subsequent cue-induced reward memory reconsolidation (35). Activation of amygdalar PKA is sufficient to enhance memory only when it is retrieved; in contrast, PKA inhibition impaired reconsolidation (11). Although it has also been reported that memory reconsolidation requires protein synthesis but not PKA activation 24 h after training (36), the classical G protein/cAMP/PKA signaling pathway has been proposed to mediate the function of β-ARs in memory (6), and the signaling pathways leading to protein synthesis and memory restabilization are not clear. In this study, we showed that upon reactivation of a particular memory trace, β1-AR/β-arrestin2/ERK signaling and downstream effectors involved in protein translation, such as p90RSK and eIF4B (9, 37), are activated in a distinct brain area. We found that this pathway, but not the conventional Gs protein-coupled PKA pathway, mediates memory reconsolidation, revealing an unexpected role of β-arrestin–biased signaling in brain physiology. Our data suggest that memory reactivation-triggered β1-AR/β-arrestin2/ERK signaling positively regulates postreactivation protein synthesis and memory restabilization. Our data showed that in parallel to β-arrestin-dependent ERK activation, reactivation of ORM induced an increase of cAMP production and PKA activation in the Enc; however, selective blockade of β-AR–mediated Gs/PKA signaling by G protein-biased β-AR antagonist failed to inhibit ORM reconsolidation. The physiological consequence of memory reactivation-stimulated neuronal β-AR/G protein signaling remains to be investigated.
The signaling pathways that mediate or regulate memory reconsolidation are therefore potential pharmacological targets for memory enhancement or erasure. It has been reported earlier that in HEK293 cells stably expressing β-AR, carvedilol and alprenolol can stimulate ERK1/2, but have inverse efficacy for Gs-dependent adenylyl cyclase activation (8, 9). Consistently, our results showed that carvedilol enhanced memory reactivation-induced ERK activation and inhibited cAMP accumulation and PKA activation in the brain. Our results suggest that β-arrestin–dependent adrenergic signaling regulates postreactivation memory retention, which implicates that agents up-regulating this pathway may improve memory.
In therapeutic contexts, disrupting the process of reconsolidation could change or erase pathophysiological memories (28, 30, 38). Blockade of β-AR signaling disrupts reconsolidation of memory for learned behaviors. In human and clinical studies, administration of propranolol before memory reactivation suppresses the behavioral expression of the fear memory (39), and postreactivation treatment reduces symptoms of posttraumatic stress disorder (40, 41). In animal studies, propranolol has been shown to inhibit reconsolidation of cocaine- and morphine-conditioned place preference (27, 42). Postreactivation propranolol administration also attenuates reconsolidation of memories for craving and cue reactivity in cocaine addicts and abstinent heroin addicts (31, 43). Previous studies have shown that Arrb2−/− mice respond well to cocaine in CPP (44), but not to amphetamine-induced locomotor activity, compared with wild-type mice (45). In this study, we show that Arrb2−/− mice could acquire cocaine CPP, but exhibit impaired reconsolidation of CPP. Moreover, propranolol, but not carvedilol inhibits reconsolidation of CPP and conditioned fear memory. The proposal that the function of β-AR in reconsolidation is regulated by β-arrestin/ERK signaling, not the conventional Gs protein/PKA pathway, suggests that β-AR/β-arrestin pathway may be an effective target of β-blockers in pharmacological intervention of memory reconsolidation. To our knowledge, β-arrestin–biased β-AR antagonist has not been reported. Our results highlight a need to develop novel β-blockers with specific β-arrestin–biased antagonism, which may produce fewer side effects than antagonists against both the G protein- and β-arrestin–dependent pathway for the treatment of posttraumatic stress disorder and drug addiction.
In addition, β-blockers are routinely used to treat hypertension and heart failure. Consistent with the report that β-blockers can aggravate memory loss in cognitively impaired elderly patients and cause clinically significant side effects (46), our data suggest that chronic use of β-blockers that antagonize both G protein and β-arrestin signaling and are capable of penetrating the blood–brain barrier may have secondary effects on cognitive processes.
Materials and Methods
Object Recognition Memory Task.
Mice were submitted to a 30-min familiarization session daily in the empty arena for 3 d. In the extensive training session, mice were exposed to Object A and Object B for 4 blocks of two 5-min trials, with 60-min interval between blocks and 15-min interval between trials. In the memory reactivation session, mice were reexposed to Object A and Object B for 5 min 24 h after training to reactivate the memory trace. To test reconsolidation of memory for objects A and B, a 5-min memory retention test was carried out 1 h, 3 h, 24 h, or 6 d after reactivation by presenting mice with a duplicate of Object A and a novel object (Object C) in the same location of Object B.
Additional Methods.
The memory tasks, cannula implantation and drug delivery, Western blotting, cAMP assay, PKA activity assay, immunohistochemistry, viral constructs, and microinjection methods are described in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Dr. R. J. Lefkowitz (Duke University Medical Center) for Arrb1−/− and Arrb2−/− mice. This research was supported by Ministry of Science and Technology Grants 2014CB942801 (to Lan Ma) and 2013CB835102 (to X.L.), and National Natural Science Foundation of China Grants 31371136 (to X.L.) and 91232307, 31430033, and 31421091 (to Lan Ma).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1421758112/-/DCSupplemental.
References
- 1.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: Biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9(5):373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci. 2011;36(9):457–469. doi: 10.1016/j.tibs.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol Med. 2011;17(3):126–139. doi: 10.1016/j.molmed.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kopin IJ. Avenues of investigation for the role of catecholamines in anxiety. Psychopathology. 1984;17(Suppl 1):83–97. doi: 10.1159/000284081. [DOI] [PubMed] [Google Scholar]
- 5.McCormick DA, Pape HC, Williamson A. Actions of norepinephrine in the cerebral cortex and thalamus: implications for function of the central noradrenergic system. Prog Brain Res. 1991;88:293–305. doi: 10.1016/s0079-6123(08)63817-0. [DOI] [PubMed] [Google Scholar]
- 6.O’Dell TJ, Connor SA, Gelinas JN, Nguyen PV. Viagra for your synapses: Enhancement of hippocampal long-term potentiation by activation of beta-adrenergic receptors. Cell Signal. 2010;22(5):728–736. doi: 10.1016/j.cellsig.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cahill L, Prins B, Weber M, McGaugh JL. Beta-adrenergic activation and memory for emotional events. Nature. 1994;371(6499):702–704. doi: 10.1038/371702a0. [DOI] [PubMed] [Google Scholar]
- 8.Wisler JW, et al. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci USA. 2007;104(42):16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim IM, et al. Beta-blockers alprenolol and carvedilol stimulate beta-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci USA. 2008;105(38):14555–14560. doi: 10.1073/pnas.0804745105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel PA, Tilley DG, Rockman HA. Beta-arrestin-mediated signaling in the heart. Circ J. 2008;72(11):1725–1729. doi: 10.1253/circj.cj-08-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tronson NC, Wiseman SL, Olausson P, Taylor JR. Bidirectional behavioral plasticity of memory reconsolidation depends on amygdalar protein kinase A. Nat Neurosci. 2006;9(2):167–169. doi: 10.1038/nn1628. [DOI] [PubMed] [Google Scholar]
- 12.Johansen JP, Cain CK, Ostroff LE, LeDoux JE. Molecular mechanisms of fear learning and memory. Cell. 2011;147(3):509–524. doi: 10.1016/j.cell.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stern SA, Alberini CM. Mechanisms of memory enhancement. Wiley Interdiscip Rev Syst Biol Med. 2013;5(1):37–53. doi: 10.1002/wsbm.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Besnard A, Caboche J, Laroche S. Recall and reconsolidation of contextual fear memory: differential control by ERK and Zif268 expression dosage. PLoS ONE. 2013;8(8):e72006. doi: 10.1371/journal.pone.0072006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tronson NC, et al. Distinctive roles for amygdalar CREB in reconsolidation and extinction of fear memory. Learn Mem. 2012;19(5):178–181. doi: 10.1101/lm.025783.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Antunes M, Biala G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn Process. 2012;13(2):93–110. doi: 10.1007/s10339-011-0430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki A, et al. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24(20):4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Artinian J, De Jaeger X, Fellini L, de Saint Blanquat P, Roullet P. Reactivation with a simple exposure to the experimental environment is sufficient to induce reconsolidation requiring protein synthesis in the hippocampal CA3 region in mice. Hippocampus. 2007;17(3):181–191. doi: 10.1002/hipo.20256. [DOI] [PubMed] [Google Scholar]
- 19.Artinian J, et al. Protein degradation, as with protein synthesis, is required during not only long-term spatial memory consolidation but also reconsolidation. Eur J Neurosci. 2008;27(11):3009–3019. doi: 10.1111/j.1460-9568.2008.06262.x. [DOI] [PubMed] [Google Scholar]
- 20.Morris RG, et al. Memory reconsolidation: sensitivity of spatial memory to inhibition of protein synthesis in dorsal hippocampus during encoding and retrieval. Neuron. 2006;50(3):479–489. doi: 10.1016/j.neuron.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 21.Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406(6797):722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- 22.Hansen JL, et al. Lack of evidence for AT1R/B2R heterodimerization in COS-7, HEK293, and NIH3T3 cells: How common is the AT1R/B2R heterodimer? J Biol Chem. 2009;284(3):1831–1839. doi: 10.1074/jbc.M804607200. [DOI] [PubMed] [Google Scholar]
- 23.Doyère V, Debiec J, Monfils MH, Schafe GE, LeDoux JE. Synapse-specific reconsolidation of distinct fear memories in the lateral amygdala. Nat Neurosci. 2007;10(4):414–416. doi: 10.1038/nn1871. [DOI] [PubMed] [Google Scholar]
- 24.Tronson NC, Taylor JR. Molecular mechanisms of memory reconsolidation. Nat Rev Neurosci. 2007;8(4):262–275. doi: 10.1038/nrn2090. [DOI] [PubMed] [Google Scholar]
- 25.Chen X, et al. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci. 2005;8(7):925–931. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- 26.Bernardi RE, Lattal KM, Berger SP. Postretrieval propranolol disrupts a cocaine conditioned place preference. Neuroreport. 2006;17(13):1443–1447. doi: 10.1097/01.wnr.0000233098.20655.26. [DOI] [PubMed] [Google Scholar]
- 27.Diergaarde L, Schoffelmeer AN, De Vries TJ. Beta-adrenoceptor mediated inhibition of long-term reward-related memory reconsolidation. Behav Brain Res. 2006;170(2):333–336. doi: 10.1016/j.bbr.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 28.Przybyslawski J, Roullet P, Sara SJ. Attenuation of emotional and nonemotional memories after their reactivation: role of beta adrenergic receptors. J Neurosci. 1999;19(15):6623–6628. doi: 10.1523/JNEUROSCI.19-15-06623.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Debiec J, Ledoux JE. Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience. 2004;129(2):267–272. doi: 10.1016/j.neuroscience.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 30.Gazarini L, Stern CA, Carobrez AP, Bertoglio LJ. Enhanced noradrenergic activity potentiates fear memory consolidation and reconsolidation by differentially recruiting α1- and β-adrenergic receptors. Learn Mem. 2013;20(4):210–219. doi: 10.1101/lm.030007.112. [DOI] [PubMed] [Google Scholar]
- 31.Zhao LY, et al. Effects of β-adrenergic receptor blockade on drug-related memory reconsolidation in abstinent heroin addicts. Drug Alcohol Depend. 2011;118(2-3):224–229. doi: 10.1016/j.drugalcdep.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 32.Duvarci S, Nader K, LeDoux JE. Activation of extracellular signal-regulated kinase- mitogen-activated protein kinase cascade in the amygdala is required for memory reconsolidation of auditory fear conditioning. Eur J Neurosci. 2005;21(1):283–289. doi: 10.1111/j.1460-9568.2004.03824.x. [DOI] [PubMed] [Google Scholar]
- 33.Kelly A, Laroche S, Davis S. Activation of mitogen-activated protein kinase/extracellular signal-regulated kinase in hippocampal circuitry is required for consolidation and reconsolidation of recognition memory. J Neurosci. 2003;23(12):5354–5360. doi: 10.1523/JNEUROSCI.23-12-05354.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller CA, Marshall JF. Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron. 2005;47(6):873–884. doi: 10.1016/j.neuron.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez H, Quinn JJ, Torregrossa MM, Taylor JR. Reconsolidation of a cocaine-associated stimulus requires amygdalar protein kinase A. J Neurosci. 2010;30(12):4401–4407. doi: 10.1523/JNEUROSCI.3149-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kemenes G, Kemenes I, Michel M, Papp A, Müller U. Phase-dependent molecular requirements for memory reconsolidation: Differential roles for protein synthesis and protein kinase A activity. J Neurosci. 2006;26(23):6298–6302. doi: 10.1523/JNEUROSCI.0890-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tilley DG, Kim IM, Patel PA, Violin JD, Rockman HA. beta-Arrestin mediates beta1-adrenergic receptor-epidermal growth factor receptor interaction and downstream signaling. J Biol Chem. 2009;284(30):20375–20386. doi: 10.1074/jbc.M109.005793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sara SJ. Retrieval and reconsolidation: Toward a neurobiology of remembering. Learn Mem. 2000;7(2):73–84. doi: 10.1101/lm.7.2.73. [DOI] [PubMed] [Google Scholar]
- 39.Kindt M, Soeter M, Vervliet B. Beyond extinction: Erasing human fear responses and preventing the return of fear. Nat Neurosci. 2009;12(3):256–258. doi: 10.1038/nn.2271. [DOI] [PubMed] [Google Scholar]
- 40.Pitman RK, et al. Pilot study of secondary prevention of posttraumatic stress disorder with propranolol. Biol Psychiatry. 2002;51(2):189–192. doi: 10.1016/s0006-3223(01)01279-3. [DOI] [PubMed] [Google Scholar]
- 41.Donovan E. Propranolol use in the prevention and treatment of posttraumatic stress disorder in military veterans: Forgetting therapy revisited. Perspect Biol Med. 2010;53(1):61–74. doi: 10.1353/pbm.0.0140. [DOI] [PubMed] [Google Scholar]
- 42.Robinson MJ, Franklin KB. Central but not peripheral beta-adrenergic antagonism blocks reconsolidation for a morphine place preference. Behav Brain Res. 2007;182(1):129–134. doi: 10.1016/j.bbr.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 43.Saladin ME, et al. A double blind, placebo-controlled study of the effects of post-retrieval propranolol on reconsolidation of memory for craving and cue reactivity in cocaine dependent humans. Psychopharmacology (Berl) 2013;226(4):721–737. doi: 10.1007/s00213-013-3039-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bohn LM, et al. Enhanced rewarding properties of morphine, but not cocaine, in beta(arrestin)-2 knock-out mice. J Neurosci. 2003;23(32):10265–10273. doi: 10.1523/JNEUROSCI.23-32-10265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beaulieu JM, et al. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122(2):261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 46.Gliebus G, Lippa CF. The influence of beta-blockers on delayed memory function in people with cognitive impairment. Am J Alzheimers Dis Other Demen. 2007;22(1):57–61. doi: 10.1177/1533317506295889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.