

Significance
The proteasome of Mycobacterium tuberculosis is required to cause lethal infections and is thus a potential drug target. Bacterial proteasomes degrade proteins modified by pupylation, but evidence suggests that the M. tuberculosis proteasome possesses additional functions. In this work, we describe a degradation pathway in M. tuberculosis controlled by a previously unidentified proteasomal cofactor, Rv3780. Rv3780 enhanced the ATP-independent proteasomal degradation of peptides and proteins and was required to maintain levels of a unique set of putative proteasome substrates. Importantly, an Rv3780 mutant was attenuated for growth in mice. To our knowledge, these studies show for the first time that an ATP-independent proteasomal-degradation pathway plays a role in the physiology of an important human pathogen.
Keywords: mycobacterium, tuberculosis, proteasome, activator, degradation
Abstract
Mycobacterium tuberculosis encodes a proteasome that is highly similar to eukaryotic proteasomes and is required to cause lethal infections in animals. The only pathway known to target proteins for proteasomal degradation in bacteria is pupylation, which is functionally analogous to eukaryotic ubiquitylation. However, evidence suggests that the M. tuberculosis proteasome contributes to pupylation-independent pathways as well. To identify new proteasome cofactors that might contribute to such pathways, we isolated proteins that bound to proteasomes overproduced in M. tuberculosis and found a previously uncharacterized protein, Rv3780, which formed rings and capped M. tuberculosis proteasome core particles. Rv3780 enhanced peptide and protein degradation by proteasomes in an adenosine triphosphate (ATP)-independent manner. We identified putative Rv3780-dependent proteasome substrates and found that Rv3780 promoted robust degradation of the heat shock protein repressor, HspR. Importantly, an M. tuberculosis Rv3780 mutant had a general growth defect, was sensitive to heat stress, and was attenuated for growth in mice. Collectively, these data demonstrate that ATP-independent proteasome activators are not confined to eukaryotes and can contribute to the virulence of one the world's most devastating pathogens.
Proteasomes are compartmentalized proteases essential for the viability of all eukaryotes and archaea. The proteolytically active component of the proteasome is the 20S core particle (CP), a ∼700-kDa complex composed of four heptameric rings stacked axially to form a barrel (reviewed in refs. 1 and 2). The active site of the 20S CP is buried deep within its core, rendering it inaccessible to folded proteins (3, 4). Further protection of the active site is afforded by “gating” amino acids that block the entrance to the proteasome core. Thus, proteasomal degradation is critically dependent on proteasome activators, which bind to either end of the 20S CP to reposition its gating residues and allow access to doomed substrates (reviewed in ref. 5).
The best-characterized proteasome activator is the eukaryotic 19S regulatory particle (RP). The 19S RP is a ∼700-kDa heteromeric complex that binds either or both ends of the 20S CP to form the 26S proteasome, which is responsible for degrading proteins in an adenosine triphosphate (ATP)-dependent manner (6–8). The unfolding activities of the 19S RP come from its base, a heterohexameric ring of ATPases associated with diverse cellular activities (AAA ATPases) that directly bind to the 20S CP (reviewed in ref. 2). Ubiquitylation, which is the posttranslational modification of doomed proteins with the small protein ubiquitin, is the major signal that targets proteins for degradation by the 26S proteasome. Because ubiquitylation is an essential regulator of virtually every cellular pathway in eukaryotes, a considerable amount of work has focused on the biochemistry and biology of the 26S proteasome, leading to the identification of many substrates and the reconstitution of robust in vitro degradation (reviewed in refs. 2 and 9).
In addition to the 19S RP, there are several cofactors that activate proteasome activity without ATP. These include Blm10/PA200 (10, 11), archaeal PbaB (12), and the 11S activators. The 11S activators are a family of small proteins that form heptameric rings and enhance peptide degradation upon binding to 20S CPs. Simple eukaryotes encode a single 11S activator, PA26 (13), whereas higher eukaryotes encode three 11S activator isoforms: PA28-α, -β, and -γ (14–16). An abundance of work has detailed the biochemical characteristics of the 11S activators, which has been crucial in understanding the structural basis of proteasomal gate opening (17–21). However, their precise biological roles remain controversial. A multitude of studies have demonstrated that PA28-α/β alter the peptide products of proteasomal degradation to influence major histocompatibility complex class I antigen presentation (22, 23). However, PA28-α/β–deficient mice are not remarkably defective in antigen presentation or immunity (24). Similarly, only a handful of PA28-γ–dependent protein substrates have been described, and few studies have demonstrated a significant physiologic role for PA28-γ in mammals (25–29). As a result, our understanding of the biological relevance of eukaryotic ATP-independent activators has lagged behind that of the 19S RP.
Although the bulk of research into proteasomes has been in eukaryotes, they are also present in bacteria of the orders Nitrospirales and Actinomycetales, which includes the human pathogen Mycobacterium tuberculosis. M. tuberculosis infects one-third of the world’s population, killing >1.5 million people each year (www.who.int/mediacentre/factsheets/fs104/en/). There is an urgent need for the identification of novel antitubercular drug targets because resistance to currently available drugs is on the rise. One potential target is the prokaryotic ubiquitin-like protein (Pup)-proteasome system (PPS), a pathway used by M. tuberculosis and other proteasome-bearing bacteria to target proteins for degradation. The PPS centers on the small protein modifier Pup (30). The C terminus of Pup is covalently attached to a lysine of a target protein by the Pup ligase proteasome accessory factor A (PafA) (31) and is then bound by the homohexameric AAA ATPase mycobacterial proteasome ATPase (Mpa), which resembles the Rpt ATPases found in the bases of eukaryotic 19S RPs (32–35). Mpa unfolds pupylated proteins to be degraded by the M. tuberculosis 20S CP, which is structurally similar to the eukaryotic 20S CP but is composed of just two unique subunits, PrcA and PrcB (36, 37). Because the PPS is chemically distinct from the eukaryotic ubiquitin–proteasome pathway, and because proteasomal degradation of pupylated proteins is required for full M. tuberculosis virulence (32, 38–40), the PPS represents an attractive therapeutic target for tuberculosis.
To date, pupylation is the only pathway known to target proteins for proteasomal degradation in bacteria. However, several studies suggest that the 20S CP may have pupylation-independent functions, because chemical inhibition of the 20S CP or genetic deletion of the 20S CP genes, but not mpa or genes involved in pupylation, produces a severe growth defect in liquid culture and on solid medium (38–41). Furthermore, although mpa and pupylation mutants are severely attenuated for growth in mice, they are able to persist for the natural lifespan of infected animals; in contrast, 20S CP mutants fail to persist in mice (40, 41). It is therefore likely that the M. tuberculosis proteasome has Pup-independent functions.
In an effort to identify proteasomal cofactors in M. tuberculosis that might contribute to new degradation pathways, we used a catalytically inactive proteasome trap to stabilize normally transient interactions of the M. tuberculosis 20S CP. The proteasome trap interacted with Rv3780, a previously uncharacterized protein. Rv3780 formed oligomeric rings and bound to M. tuberculosis 20S CPs. This interaction required a conserved C-terminal motif in Rv3780, reminiscent of what has been described for eukaryotic proteasome activators. We show that Rv3780 promoted the robust ATP-independent proteasomal degradation of short peptides, an unfolded protein, and a native M. tuberculosis protein in vitro. Importantly, Rv3780 contributed to M. tuberculosis resistance to heat stress and virulence in a mouse infection model.
Results
Identification of a Proteasome Binding Protein in M. tuberculosis.
Previous studies using the M. tuberculosis 20S CP have indicated that interactions with its cofactors are relatively weak (35). We hypothesized that a catalytically inactive 20S CP (“proteasome trap,” 20ST1A CP) may stabilize these interactions by capturing the proteasome in an intermediate degradation state, still bound to cofactors and substrates, as has been shown for a catalytically inactive Escherichia coli ClpP trap (42). To identify new putative substrates and cofactors, we overproduced either active (WT) or catalytically inactive (Trap) tandem affinity purification (TAP)-tagged proteasome complexes in M. tuberculosis. We visualized affinity-purified proteins by silver staining, which showed that more proteins copurified with the Trap than with WT proteasomes (Fig. 1A). We also analyzed the copurifying proteins by tandem mass tag labeling and mass spectrometry (TMT-MS), which allows for the quantitative comparison of relative protein levels between samples (43). The protein that most abundantly copurified with the proteasome trap was the proteasomal ATPase Mpa (Fig. 1B, Left, and Table S1), which was significant because no previous study has demonstrated a robust in vitro interaction between Mpa and the 20S CP. Importantly, this result supported the hypothesis that catalytically inactive 20S CPs could capture cofactors that interact weakly with WT 20S CPs.
Fig. 1.

Rv3780 interacts with the M. tuberculosis proteasome. (A) A proteasome trap identifies interacting proteins. A TAP-tagged active (WT) or catalytically inactive (Trap) 20S CP was overproduced in M. tuberculosis and isolated from lysates by Ni-NTA and anti-FLAG affinity purification. Proteins were separated by 12% (wt/vol) SDS/PAGE and visualized by silver staining. Molecular mass markers in kDa. (B) Proteins that were enriched in pull-downs using the proteasome trap. The eluates generated in A were modified by TMT labeling and their relative levels were quantified by MS. The log2 ratio of relative abundance in the Trap pull-down vs. the WT pull-down from WT M. tuberculosis (Left) or an mpa mutant (Right) is plotted against the log10 p-value as determined by t test using three biological replicates. Data points representing the top 10 copurified proteins of highest abundance with the Trap are enlarged. Data points representing Mpa and Rv3780 are in red. (C) Rv3780 requires a conserved penultimate Tyr to copurify with 20S CPs. His6-tagged WT or Y173A Rv3780 was overproduced in M. tuberculosis and isolated from lysates by Ni-NTA affinity purification. Proteins were visualized by immunoblotting for PrcB or Rv3780 using antibodies raised against PrcB–His6 or Rv3780–His6, respectively. PD, pull-down.
When we purified proteins from WT M. tuberculosis, we found 78 proteins enriched more than fivefold with the proteasome trap than with the WT 20S CPs (Fig. 1B, Left, and Table S1). When we purified proteins from an mpa mutant, 27 proteins were enriched more than fivefold with the Trap over the WT 20S CPs (Fig. 1B, Right, and Table S2), 19 of which were enriched in both strains. Importantly, many established pupylated substrates (44, 45) were enriched with the Trap in an Mpa-dependent manner, further suggesting that these represented stalled degradation complexes.
Of the many proteasome-associated proteins that we identified, we chose to further characterize Rv3780 for three reasons. First, Rv3780 was one of the most abundant proteins that copurified with the Trap (Tables S1 and S2). Second, STRING-DB analysis (46) demonstrated that it co-occurs evolutionarily with the bacterial proteasome, suggesting that the two are functionally related. Third, the last four amino acids of Rv3780 are identical to those of Mpa (Fig. 1C).
Previously, we determined that deletion or mutagenesis of the penultimate tyrosine (Tyr; Y) of Mpa abolishes substrate degradation (30, 45). Therefore, we tested if mutagenesis of the equivalent Tyr to alanine (Ala; A) in Rv3780 (Y173A) would abrogate binding to 20S CPs. Nickel-nitrilotriacetic acid (Ni-NTA) resin purification of amino (N)-terminally hexahistidine (His6)-tagged Rv3780 overproduced in M. tuberculosis demonstrated that 20S CPs copurified with WT Rv3780, but not with the Rv3780Y173A mutant (Fig. 1C). Rv3780 is therefore a previously unidentified proteasome-interacting protein that uses a specific C-terminal sequence to bind to 20S CPs. Importantly, this experiment also confirmed that Rv3780 interacts with WT, endogenous 20S CPs.
M. tuberculosis Rv3780 is annotated as a 178-amino acid protein with a predicted molecular mass of 19.5 kDa (Fig. S1). We noted the presence of two or three Rv3780 species in our immunoblots when we used polyclonal antibodies to detect Rv3780 protein from the purifications (Fig. 1C). The pafE gene has three in-frame, closely spaced potential start codons with putative ribosome-binding sites that could result in the synthesis of slightly different proteins (Fig. S2, Left). The use of “start codon 1” would produce a protein with an N-terminal extension of arginine-lysine-arginine (Arg-Lys-Arg; RKR), which might be unstable based on the N-end rule pathway (47). Moreover, some mycobacterial species encode an Rv3780 homolog that only contains start codons 2 and 3 (Fig. S2, Left). Because start codon 2 is an ATG, which is generally a better start codon than GTG, we used this allele for our experiments; all codon numbering in this work refers to this allele, which is 174 amino acids long and has a predicted molecular mass of 18.9 kDa. For in vitro experiments, we produced PafE translated from start codon 2 with an N-terminal, thrombin-cleavable His6 tag and purified it to homogeneity with subsequent rounds of Ni-NTA and size-exclusion chromatography (SEC); this protein is predicted to be 20.4 kDa (Fig. S2, Right).
Rv3780 Forms Oligomeric Rings and Caps M. tuberculosis 20S CPs.
Rv3780 does not have significant similarity to any protein of known function based on a BLAST homology search (48). To gain a better idea of a potential function, we purified Rv3780 protein from E. coli with an N-terminal His6 tag and assessed its structure using negative stain electron microscopy (EM) and computational image classification. Rv3780 formed homo-oligomeric rings as seen in a raw EM micrograph (Fig. 2A), as well as in class averages after reference-free 2D classification (Fig. 2B). Some averaged rings were not perfectly circular but were instead elongated, suggesting that Rv3780 rings might be somewhat flexible or slightly tilted on the carbon film substrate. The approximate diameter of the Rv3780 rings as measured from the class averages was 112.0 ± 2.3 Å. In comparison, the measured average width of the M. tuberculosis open gate proteasome CP (20SOG) under the same imaging condition was 144.2 ± 4.0 Å. SEC with multiangle light scattering (SEC-MALS) analysis indicated that Rv3780 oligomers have a molecular mass of ∼247 kDa, suggesting that an Rv3780 ring consists of 12 subunits (Fig. 2C).
Fig. 2.

Rv3780 forms oligomeric rings and caps 20S CPs. (A) A raw electron micrograph of negatively stained His6–Rv3780 particles expressed and purified as a recombinant protein from E. coli. White circles mark three Rv3780 rings. (B) Shown are 24 representative reference-free 2D class averages of His6–Rv3780 ring structure. (C) Recombinant purified His6–Rv3780 was separated by SEC and analyzed by MALS. (D) Six representative reference-free 2D class averages of presumptive endogenous Rv3780 rings capping one end of TAP-tagged 20ST1A CP purified from an M. tuberculosis mpa mutant. (E) Six representative reference-free 2D class averages of an in vitro reconstituted Rv3780–20S CP complex with both M. tuberculosis His6–Rv3780 and 20ST1A CP–His6 proteins purified from E. coli. (F) Selected reference-free class averages of the singly capped (Upper) and doubly capped (Lower) Rv3780–20SOG CP complex reconstituted in vitro by incubating M. tuberculosis His6-tagged proteins purified from E. coli in the presence of bortezomib. D–F are on the same scale.
We next used EM to assess the characteristics of the 20S CP–Rv3780 complex. We used two approaches: (i) We purified TAP-tagged 20ST1A CPs from an M. tuberculosis mpa mutant; and (ii) we combined purified recombinant Rv3780 with either 20ST1A or 20SOG CPs that lack the seven N-terminal gating residues of PrcA (37) in the presence of the proteasome inhibitor bortezomib. Our rationale for using the open-gate CPs was based on previous data demonstrating that use of the 20SOG CP helped stabilize an interaction with Mpa long enough to visualize Mpa–CP complexes by EM (35). The purifications from an M. tuberculosis mpa mutant revealed ring structures capping the 20S CPs (Fig. 2D), which strongly resembled Rv3780–20ST1A and Rv3780–20SOG CP complexes reconstituted from recombinant purified proteins (Fig. 2 E and F). Importantly, these data indicate that Rv3780 is capable of binding to M. tuberculosis 20S CPs without additional cofactors.
Rv3780 Is an ATP-Independent Proteasome Activator.
Based on our data, Rv3780 is reminiscent of eukaryotic 11S activators, which are known to form oligomeric rings, engage 20S CPs by using C-terminal residues, and open proteasomal gates to enhance peptidase activity (reviewed in ref. 5). To test if Rv3780 is capable of activating proteasomal peptidase activity, we monitored the degradation of fluorogenic peptide substrates by M. tuberculosis 20S CPs in the presence of increasing amounts of purified recombinant Rv3780.
M. tuberculosis 20S CPs are capable of hydrolyzing the tetrapeptide substrate succinyl–Leu-Leu-Val-Tyr-7–amino-4-methylcoumarin (Suc-LLVY-AMC) in the absence of other cofactors (37). Addition of Rv3780 enhanced degradation of Suc-LLVY-AMC a maximum of approximately threefold (Fig. 3A). However, Rv3780 produced a much more pronounced stimulation of proteasome activity on a larger nonapeptide substrate, LF-2. In the absence of Rv3780, LF-2 was hydrolyzed extremely slowly (Fig. 3B, Inset). In contrast, Rv3780 enhanced degradation of LF-2 at least 150-fold compared with 20S CPs alone. As a control, we used an Rv3780 allele that could not interact with 20S CPs (Fig. 1C) and found that Rv3780Y173A could not stimulate degradation of LF-2 (Fig. 3B). These data suggest that the M. tuberculosis 20S CP allows nearly free passage of smaller peptides, but requires activation by a cofactor to accommodate larger substrates, as has been described for archaeal 20S CPs and E. coli ClpXP (49, 50). The addition of ATP did not improve activation, confirming that PafE acts in an ATP-independent manner (Fig. S3).
Fig. 3.

Rv3780 is an ATP-independent proteasome activator. (A) Rv3780 stimulates proteasomal degradation of a tetrapeptide substrate. Recombinant 20S CP–His6 and His6–Rv3780 were purified from E. coli, mixed, and incubated with Suc-LLVY-AMC. Data are shown as fold change in degradation rate compared with the 20S CP–His6 alone. Molar ratios represent His6–Rv3780 rings:20S CPs. (Inset) Absolute degradation rate in relative fluorescence units (RFU) generated per minute for the 20S CP–His6 alone or with a 58.3-fold molar excess of His6–Rv3780. (B) Rv3780 stimulates proteasomal degradation of a nonapeptide substrate. Reactions were performed as in A, except with LF-2 used as a substrate. Rv3780Y173A, mutant Rv3780 that was unable to bind 20S CPs (Fig. 1C). (C) Rv3780 enhances proteasomal degradation of an unfolded protein. Recombinant purified 20S CP–His6 and His6–Rv3780 were mixed with FITC-labeled β-casein at room temperature. Samples were removed at the indicated time points and separated by 12% (wt/vol) SDS/PAGE, and FITC–β-casein was visualized and quantified by in-gel fluorescence.
We next wondered if Rv3780 could promote the degradation of larger unfolded proteins as has been observed for other bacterial barrel-shaped proteases (reviewed in ref. 51). To this end, we assessed the ability of either the 20S CP alone or in complex with Rv3780 to degrade the model unfolded protein β-casein. The addition of Rv3780 greatly enhanced proteolysis of β-casein by M. tuberculosis 20S CPs (Fig. 3C). Based on these results, we now refer to Rv3780 as PafE for proteasome accessory factor E.
Identification of a Conserved C-Terminal Motif Required for the Activity of Mycobacterial Proteasome Cofactors.
In eukaryotes and archaea, many proteasome cofactors are characterized by the presence of a C-terminal “HbYX” motif (hydrophobic–Tyr–any amino acid), which is required to bind the 20S CP and stimulate gate opening (20, 52, 53). As noted earlier, Mpa and PafE each contain a C-terminal GQYL sequence (glycine-glutamine-Tyr-leucine; Gly-Gln-Tyr-Leu), which has a penultimate Tyr but is otherwise distinct from the HbYX motif. The C-terminal GQYL is absolutely conserved among PafE and Mpa homologs in proteasome-bearing bacteria, but is missing from Mpa homologs in Corynebacteria, which do not encode a 20S CP (Fig. 4B). These observations suggested that the GQYL sequence is important for proteasome-related functions. Thus, to determine which residues of the GQYL motif are functionally important, we performed mutational analysis of PafE and Mpa.
Fig. 4.

Identification of a C-terminal motif required for the activity of PafE and Mpa. (A) A BLAST homology search and ClustalW2 alignment (78) demonstrated that the C-terminal GQYL of PafE is conserved. Suc-LLVY-AMC degradation assays were performed as described above, except by using recombinant His6–PafE containing an Ala substitution at each indicated site. Statistical analysis was done by a nonparametric Student’s t test. *P < 0.05; ***P < 0.001; ****P < 0.0001; n.s., not significant. (B) Identification of C-terminal residues required for Mpa function. ClustalW2 alignment demonstrated that the C-terminal GQYL of Mpa is conserved among homologs in proteasome-bearing bacteria. An M. tuberculosis mpa strain was transformed with integrative plasmids encoding Mpa variants with Ala substitutions at the C terminus. Mpa activity was assessed by immunoblotting to monitor accumulation of the Mpa-dependent substrate malonyl CoA–acyl carrier protein transacylase (FabD). Dihydrolipoamide acyltransferase (DlaT) was used as a loading control. (C) PafE C-terminal peptides are sufficient for proteasome activation. LF-2 degradation assays and statistical analyses were performed as described above, except by using peptides corresponding to the C terminus of PafE at either 200 µM or 2 mM as indicated. (D) Mpa does not strongly compete with PafE for proteasome activation. Suc-LLVY-AMC degradation assays were performed as described above. To test for activation by Mpa alone, Mpa was added at a molar excess of 10:1 (Mpa:20S). To test if Mpa could outcompete PafE activation of the 20S CP, Mpa was added at a molar excess of 8.5:1.7:1 (Mpa:20S:PafE).
In vitro degradation assays with recombinant PafE indicated that single Ala mutations of Gly-171, Tyr-173, or Leu-174 disrupted the ability of PafE to promote Suc-LLVY-AMC degradation, whereas Ala substitutions at Thr-170 or Gln-172 had minimal to no effect (Fig. 4A). To determine if these residues were also important in live M. tuberculosis, we attempted to complement an mpa-null mutant with integrating plasmids encoding Mpa GQYL mutant alleles and assessed their ability to degrade the established Pup–proteasome substrate FabD (45). Consistent with our in vitro data using PafE and with our previous work using Mpa mutants (45), we found that Gly-605, Tyr-607, and Leu-608, but not Gln-606, were required for FabD degradation (Fig. 4B). Based on the absolute conservation of the GQYL residues in PafE and Mpa homologs and our mutagenesis analysis, these data strongly suggest that the GQYL motif is required for the activity of proteasomal cofactors in bacteria. Interestingly, in contrast to disruption of the equivalent residue in PafE, replacement of Leu-604 just preceding GQYL in Mpa prevented FabD degradation (Fig. 4B). Consistent with this observation, the residues preceding GQYL are conserved in Mpa homologs but diverge in PafE homologs, suggesting that this region is important for the function of Mpa, but not PafE.
The C termini of eukaryotic and archaeal proteasomal ATPases, which contain HbYX motifs, are sufficient to elicit proteasome activation, whereas the C termini of the 11S activators are not (17, 20). To test if the C terminus of PafE was capable of activation on its own, we tested activity of the 20S CP in the presence of peptides corresponding to the C-terminal four (GQYL), six (GTGQYL), and eight (GHGTGQYL) residues of PafE. All three peptides activated the 20S CP, with the longer peptides acting as slightly stronger activators, indicating that binding of just the GQYL motif is sufficient for activation (Fig. 4C). Because Mpa and PafE each use this motif to bind to the 20S CP, we next tested if Mpa was capable of activating the 20S CP itself. However, we were unable to detect an increase in proteasomal peptidase activity with the addition of Mpa (Fig. 4D). Moreover, the addition of an 8.5-fold molar excess of Mpa to PafE was unable to outcompete PafE activation. Although it is possible that Mpa is still capable of activating proteasomal degradation, these data suggest that the affinity of the 20S CP is likely higher for PafE than it is for Mpa.
PafE Contributes to M. tuberculosis Growth and Virulence.
We next determined if PafE plays a significant role in M. tuberculosis physiology. Our initial efforts to generate a pafE deletion strain were unsuccessful, likely due to polar effects on several downstream genes that are predicted to be essential for growth (54, 55). To circumvent this problem, we generated a merodiploid “parental” strain by integrating the genes downstream of pafE (Rv3781–3783) under control of the operon’s presumed native promoter into the chromosomal L5 attB site (Fig. 5A). We then deleted and disrupted pafE from this parental strain and confirmed disappearance of PafE from cell lysates by immunoblotting (Fig. 5A, Lower). For complementation, we introduced a single copy of pafE including all possible start codons into the chromosome at the Tweety attB site (56) and showed restoration of PafE in lysates.
Fig. 5.

PafE contributes to M. tuberculosis virulence. (A) Strategy to generate a pafE mutant strain of M. tuberculosis. A parental strain was generated by integrating the genes downstream of pafE under control of their putative native promoter at the chromosomal L5 attB site. pafE was then deleted and disrupted with a hygromycin-resistance gene. Complementation of pafE was achieved by integrating a copy of pafE under control of its putative native promoter at the chromosomal Tweety attB site. The presence or absence of PafE in each strain was shown by immunoblotting. (B) A pafE mutant is growth-defective on solid medium. The indicated M. tuberculosis strains were grown to logarithmic phase, diluted to OD580 = 0.08, serially diluted as indicated, spotted onto Middlebrook 7H11 agar, and incubated at 37 °C for ∼2 wk. (C) A pafE mutant is growth-defective in broth culture. OD580 was measured at the indicated time points. Data show the average of three cultures and is representative of two independent experiments. (D and E) A pafE mutant is attenuated in a mouse infection model. C57BL6/J mice were infected with the indicated M. tuberculosis strains, and lungs and/or spleens were harvested at days 1 (n = 6), 21 (n = 8), and 56 (n = 8) after infection. Lungs (D) and spleens (E) were homogenized and plated on Middlebrook 7H11 agar to enumerate CFU. Statistical analysis was done by nonparametric Student’s t test. *P < 0.05; **P < 0.01; ***P < 0.005; n.s., not significant.
The pafE mutant demonstrated a conspicuous growth defect on solid medium (Fig. 5B) and a slight growth defect in broth culture (Fig. 5C), phenotypes that were complemented by reintroducing pafE into the M. tuberculosis chromosome. Importantly, introduction of the pafEY173A allele did not complement the growth defect (Fig. 5 B and C). Consistent with PafE playing a role in M. tuberculosis physiology, the pafE mutant was attenuated for growth in mice (Fig. 5 D and E). Although complementation of the in vivo growth defect was incomplete, we noted that PafE was not fully restored to WT levels (Fig. 5A, Lower), suggesting that WT PafE levels are more critical during infection than under standard laboratory conditions. Collectively, our data show that PafE contributes to the normal growth and virulence of M. tuberculosis.
PafE Promotes in Vitro Degradation of a Heat Shock Repressor and Is Required for Resistance to Heat Stress.
The only known mechanism for targeting proteins to a bacterial proteasome for degradation is pupylation (30). We therefore tested if PafE was required for the degradation of known Pup–proteasome substrates (44, 45). To our initial surprise, immunoblot analysis of an M. tuberculosis pafE mutant showed WT levels of all pupylation substrates tested, in contrast to an mpa mutant where these substrates accumulated (Fig. 6A). Although we cannot rule out the possibility that PafE contributes to the degradation of pupylated substrates under certain conditions or a different subset of pupylated proteins, these data indicate that it is not an essential part of the PPS and may instead contribute to a distinct proteasomal-degradation pathway. Consistent with this hypothesis, genetic co-occurrence analysis using STRING-DB demonstrated that pafE is present in bacteria that encode a 20S CP, but absent from Corynebacteria, which lack 20S CP genes, yet encode Mpa and the pupylation system (Fig. S4A). These observations led us to investigate the possibility that PafE is involved in pupylation-independent proteasomal protein degradation.
Fig. 6.

HspR is a native PafE-dependent proteasome substrate. (A) PafE is not required for the degradation of several pupylated proteins. Total cell lysates were prepared from the indicated M. tuberculosis strains, separated by 10% (wt/vol) SDS/PAGE, and analyzed by immunoblotting for the known pupylated proteasome substrates Mpa, myo-inositol-1-phosphate synthase (Ino1), and FabD. DlaT was used as a loading control. MW, molecular mass markers in kDa. (B) Proteomic identification of putative PafE-dependent proteasome substrates in M. tuberculosis. Protein lysates from parental and pafE strains were prepared and compared by TMT-MS analysis. The log2 ratio of abundance in pafE vs. parental strains was plotted against the log10 P value as determined by t test using three biological replicates. The data point representing HspR is in red. (C) HspR accumulates in pafE and prcBA mutants. Total cell lysates were prepared from the indicated M. tuberculosis strains and separated by 15% (wt/vol) SDS/PAGE, and endogenous HspR was detected by immunoblotting with polyclonal antibodies to HspR–His6. DlaT was used as a loading control. (D) PafE promotes proteasomal degradation of HspR in vitro. Recombinant 20S CP–His6, HspR–His6, and either His6–PafE or –PafEY173A were purified from E. coli, mixed, and incubated at room temperature. Aliquots were removed at the indicated time points, separated by 13% (wt/vol) SDS/PAGE, and HspR–His6 degradation was monitored by Coomassie Brilliant Blue staining. MW, molecular mass markers in kDa; o/n, overnight. (E) Repression of HspR target genes in an M. tuberculosis pafE mutant. The indicated M. tuberculosis strains were grown to OD ∼ 1.0, and RNA was purified and reverse-transcribed into cDNA for quantitative real-time PCR analysis. Gene expression was normalized to dlaT. Statistical analysis was done by nonparametric Student’s t test. *P < 0.05; **P < 0.01. (F) An M. tuberculosis pafE mutant is hypersensitive to heat stress. The indicated M. tuberculosis strains were incubated at 45 °C for 24 h, serially diluted, and inoculated onto Middlebrook 7H11 agar to enumerate surviving bacteria.
To search for PafE-dependent proteasome substrates, we carried out two proteomic studies with M. tuberculosis using TMT-MS analysis. First, we compared the proteomes of parental vs. pafE strains to identify proteins that accumulate in the absence of PafE. Second, we treated a pupylation-deficient pafA strain with the proteasome inhibitor epoxomicin to identify proteins whose levels are maintained by the proteasome in a pupylation-independent manner. Proteins that increased in abundance were considered putative PafE-dependent proteasome substrates (Fig. 6B, Fig. S4B, and Tables S3 and S4). Known pupylated proteins did not increase in abundance in either experiment, indicating that the proteins we identified represented a distinct class of putative proteasome substrates.
The protein that significantly increased the most in abundance in both experiments was heat shock protein repressor (HspR). We raised rabbit polyclonal antibodies to HspR–His6 and examined endogenous HspR in M. tuberculosis cell lysates, finding that HspR levels increased in the pafE and prcBA mutants compared with the parental strain, but not in an mpa mutant (Fig. 6C). HspR denatures when heated briefly at 42 °C, suggesting that it is intrinsically unstable (57, 58). Because we previously found that PafE promoted the degradation of the unfolded protein β-casein, we performed an in vitro degradation assay to determine if PafE could also directly stimulate the proteolysis of HspR. Remarkably, PafE promoted the robust degradation of HspR, which was undetectable after 3 h. In contrast, HspR was stable for >24 h when we used PafEY173A (Fig. 6D). The degradation rate of HspR increased at higher temperatures, further supporting the hypothesis that denaturation of HspR promotes its PafE-mediated degradation (Fig. S4C).
Because eukaryotic ATP-independent proteasome activators open the gates of 20S CPs, we wondered if gate opening might be the mechanism of PafE-dependent activation of HspR degradation. Specifically, does PafE simply open the 20S CP gate, or does PafE actively recruit HspR to the 20S CP? We performed a degradation assay using 20SOG CPs that lack gating residues and therefore should not require PafE for gate opening. We found that the 20SOG CPs degraded HspR in a similar time scale as the PafE-activated WT 20S CPs. The addition of PafE did not increase the rate of proteolysis, suggesting that gate opening is the sole mechanism by which it enhanced HspR degradation (Fig. S4D).
We next tested if gate opening was sufficient to degrade a known pupylated proteasome substrate. We found that 20SOG CPs were unable to degrade the Mpa-dependent proteasome substrate Pup∼PanB (3-methyl-2-oxobutanoate hydroxymethyltransferase) with or without PafE present (Fig. S4E). Together, our data suggest that PafE opens the proteasomal gate to enhance degradation of a specific subset of proteins, most likely those that are partially or completely unfolded.
Degradation of misfolded proteins is an important component of the cellular defense against proteotoxic stress. Under conditions such as heat shock that lead to the accumulation of misfolded and aggregated proteins, bacteria rely on chaperones to disaggregate and refold some proteins, and proteases to degrade others (reviewed in refs. 59 and 60). DnaK and ClpB are chaperones that are critical for the M. tuberculosis heat shock response, and their expression is repressed by the PafE-dependent substrate HspR (61, 62). We therefore hypothesized that the accumulation of HspR would render a pafE mutant hypersensitive to heat. We found that PafE-deficient M. tuberculosis had somewhat reduced expression of HspR target genes (Fig. 6E), suggesting that its heat shock response might be compromised. Indeed, the pafE mutant was more sensitive to killing than the parental strain when incubated at 45 °C (Fig. 6F). As with the animal experiments, complementation was partial, but survival of the mutant was significantly improved.
Discussion
In this study, we describe the discovery and characterization of PafE, a functional homolog of the eukaryotic 11S proteasome activators. Although it shares no sequence homology with its eukaryotic counterparts, PafE nonetheless forms a ringed oligomer, binds 20S CPs by using its C terminus, and enhances proteasomal peptidase activity. Our proteomic studies identified a unique set of putative PafE-dependent substrates, which was completely distinct from known pupylation substrates. The failure to degrade one or more of these substrates appears to have significant biological consequences because an M. tuberculosis pafE mutant has a general growth defect and is somewhat attenuated in mice. Importantly, we found that PafE stimulates the ATP-independent degradation of HspR, representing, to our knowledge, the first demonstration of robust in vitro degradation of an endogenous substrate by a bacterial proteasome, and that the accumulation of HspR in a pafE mutant leads to repression of the heat shock response genes and increased sensitivity to heat stress.
The discovery that PafE promotes proteasomal degradation of both HspR and a model unfolded protein (β-casein) suggests two potential mechanisms for the heat shock sensitivity of a pafE mutant. First, it is possible that the PafE–proteasome system combats proteotoxic stress by degrading misfolded proteins that would otherwise accumulate during heat shock or other stresses that induce misfolding. M. tuberculosis does not encode a homolog of Lon protease, which in many bacteria is induced by heat shock and is critical for the degradation of misfolded proteins (63–67). Thus, M. tuberculosis may rely on the PafE–proteasome system to dispose of small, misfolded proteins that would otherwise be degraded by Lon. Second, the accumulation of HspR in a pafE mutant might dampen induction of the heat shock response. HspR represses expression of the dnaK operon, of which hspR is a member (62), and clpB (61), both of which encode chaperones that are induced by and required to resist killing by heat shock, and in a pafE mutant, expression of both clpB and hspR itself was significantly reduced. Thus, one model for the heat shock sensitivity of a pafE mutant is that upon encountering an elevated temperature, an M. tuberculosis pafE mutant accumulates misfolded proteins and is unable to quickly degrade HspR, resulting in the bacteria being unable to fully induce expression of its heat shock genes. As a result, the bacteria would be unable to cope with proteotoxic stress, leading to cell death.
The identification of a new proteasomal cofactor in M. tuberculosis has also revealed that actinobacterial activators possess a unique C-terminal 20S CP-binding motif. Both Mpa and PafE have a C-terminal GQYL motif. We have demonstrated that for both proteins, the Gly, Tyr, and Leu are all required for function, whereas the Gln is dispensable. This finding was a surprise, because the C-terminal HbYX motif characteristic of eukaryotic and archaeal cofactors relies critically on a residue just before the penultimate Tyr (18, 20, 53, 68). Thus, bacteria and eukaryotes appear to have evolved slightly different mechanisms to achieve proteasome activation. Interestingly, eukaryotic 11S activators do not have HbYX motifs, despite binding to 20S CPs with their C termini. Whereas the HbYX motif is sufficient for ATP-dependent activators to both bind and reposition the proteasomal gate, 11S activators use their C termini to bind but require a separate activation loop to open the proteasomal gate. The finding that a GQYL peptide alone is sufficient to activate the M. tuberculosis 20S CP (Fig. 4C) demonstrated that bacterial proteasome activators more closely resemble proteasomal ATPases in this regard, with binding to the C terminus sufficient to induce activation. Gate opening in eukaryotes relies on the repositioning of a reverse turn that positions the N-terminal gating residues. Proteasomal ATPases reposition the reverse turn by using interactions with the HbYX alone, whereas the 11S activators use an activation loop (17, 19). However, the reverse turn is absent from the M. tuberculosis 20S CP (69); the mechanism of activation for bacteria is therefore likely to be novel from that seen in other organisms.
Because the 20S CP is composed of heptameric rings, it is intriguing to consider how it might interface with an activator of 12-fold symmetry. The interaction between PafE and the 20S CP appears to be considerably weaker than that of eukaryotic activator/20S CP complexes; thus, it is possible that the presence of additional binding sites might be required to improve binding avidity to enhance activation. Alternatively, it is possible that “free” GQYL motifs that are not actively involved in binding the 20S CP might be involved in gate-opening interactions distinct from binding interactions, because GQYL alone appears to be sufficient for gate-opening. Elucidation of the cocrystal structure of PafE–20S CP complexes will be important for establishing precisely how the GQYL motif opens the proteasome gate.
Although a handful of 11S activator-dependent proteasome substrates have been described in eukaryotes, attempts to reconstitute their degradation in vitro have not been robust (26, 28, 29). As a result, we lack a clear understanding of the mechanisms governing ATP-independent proteasomal protein degradation. By contrast, we have now reconstituted robust degradation of a native M. tuberculosis substrate using only three proteins: PrcB, PrcA, and PafE. Further analysis of this simplified system will be extremely useful for mechanistic studies into ATP-independent proteasomal protein degradation in bacteria, which should, in turn, help to guide research on eukaryotic systems.
The identification of the PafE-proteasome pathway indicates that M. tuberculosis has at least two proteasome systems with distinct biological roles (Fig. 7). Importantly, our data have begun to explain the phenotypic differences observed between pupylation/Mpa-deficient and 20S CP-deficient M. tuberculosis strains. Further studies will elucidate details regarding the mechanism and the physiologic roles of ATP-independent proteasomal protein degradation in M. tuberculosis, which may identify new therapeutic targets as well as contribute to a better understanding of ATP-independent proteasome functions in all domains of life.
Fig. 7.
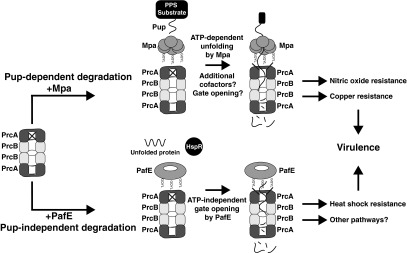
Model of two independent pathways of proteasomal degradation in M. tuberculosis. For Pup-dependent degradation, a substrate protein must first be modified by Pup, which binds to Mpa. Mpa then unfolds the pupylated protein in an ATP-dependent manner, and the 20S CP degrades the unfolded protein. For Pup-independent degradation, substrates are likely to be small, unfolded, or easily misfolded proteins such as HspR. These substrates are degraded by the proteasome upon gate opening, which is induced by PafE.
Experimental Procedures
Bacterial Strains, Growth Conditions, and Primers.
Bacterial strains, plasmids, and primers are listed in Table S5. Plasmid construction and culture conditions are described in detail in SI Experimental Procedures.
Recombinant Protein Purification, Antibodies, and Immunoblotting.
To generate rabbit polyclonal antisera, we produced His6-tagged M. tuberculosis H37Rv PafE, Mpa, and HspR in E. coli and purified proteins under native (for Mpa and PafE) or denaturing (for HspR) conditions as described in the QIAexpressionist manual (Qiagen). A total of 500 μg of purified protein was used to immunize rabbits (Covance). A detailed description of protein purification procedures is included in SI Experimental Procedures. Antibodies to FabD and Ino1 are described elsewhere (30, 44).
To assess protein levels in M. tuberculosis lysates by immunoblotting, cultures were grown to an optical density of absorbance at 580 nm (OD580) ∼1.2, and four OD580 equivalents of bacterial culture were pelleted and resuspended in lysis buffer (10 mM Tris, pH 8.0, 10 mM NaCl, 1 mM EDTA). Cells were lysed by bead beating (BioSpec), mixed with reducing and denaturing sample buffer (70), and boiled for 10 min. Samples were then separated by SDS polyacrylamide gel electrophoreses (SDS/PAGE), transferred to nitrocellulose membranes (GE Amersham), and analyzed by immunoblotting using antibodies as indicated. As a loading control, we stripped the membranes (70) and subsequently incubated the blots with antibodies to dihydrolipoamide acyltransferase (DlaT) (71).
Purification of the Proteasome Trap and Preparation of Total Cell Lysates of M. tuberculosis for TMT-MS.
Design and expression of the proteasome trap construct is described in SI Experimental Procedures. To identify proteins that copurified with the proteasome trap, lysates were prepared from 120 OD580 equivalents of M. tuberculosis culture. Cells were resuspended in low-salt PBS (50 mM NaH2PO4, 100 mM NaCl, pH 8.0) with 10 mM imidazole, lysed by bead beating, filter sterilized, and subjected to affinity purification by using 30 µL of packed, equilibrated Ni-NTA resin (Qiagen). After washing with 20 mM imidazole and eluting with 250 mM imidazole, the eluate was buffer exchanged into low-salt PBS (pH 7.4) and further purified by using 30 µL of packed, equilibrated M2 anti-FLAG affinity gel as described by the manufacturer (Sigma-Aldrich). The final elution of the proteasome and associated proteins was performed by using 60 µL of 3× FLAG peptide at a concentration of 150 ng/µL in low-salt PBS (pH 7.4). Eluted proteins were analyzed by SDS/PAGE and silver staining (72) or prepared for MS analysis by TMT labeling as described in SI Experimental Procedures. For EM analysis of in vivo-purified proteasome complexes, we purified protein with anti-FLAG affinity gel alone.
To prepare lysates from parental vs. pafE mutant M. tuberculosis for TMT labeling, biological triplicate cultures of each strain were grown to OD580 ∼ 1.2, washed with PBS–0.05% Tween-80, resuspended in lysis buffer, lysed by bead beating, and filter-sterilized. To prepare lysates from the M. tuberculosis pafA mutant treated with epoxomicin vs. dimethyl sulfoxide (DMSO), biological duplicate cultures were grown to OD580 ∼ 0.75, and then 50 µM epoxomicin (Boston Biochem) in DMSO or DMSO alone was added. After 4 d, bacterial lysates were prepared as described above. For all samples, protein concentration was assessed by protein assay (Bio-Rad). Equal amounts of protein were precipitated by adding six volumes of cold acetone, incubated at −20 °C for 20 min, and collected by 15 min of centrifugation at 16,000 × g at 4 °C. Protein pellets were air-dried.
Copurification of PafE and 20S CP from M. tuberculosis Lysates.
A total of 40 OD580 equivalents of culture from M. tuberculosis strains overproducing PafE or PafEY173A with an N-terminal His6 tag were lysed in low-salt PBS with 10 mM imidazole by bead beating and filter sterilized. Lysates were incubated with preequilibrated Ni-NTA resin for 3 h, nonspecific interacting proteins were washed off with 20 mM imidazole, and PafE and other bound proteins were eluted by using 250 mM imidazole. The presence of PafE and PrcB was assessed by immunoblotting.
Negative-Stain EM and Single-Particle Image Processing of PafE–20S Complexes.
For analysis of in vivo-purified proteasome complexes, we purified the TAP-tagged proteasome trap from an M. tuberculosis mpa mutant as described above, but used anti-FLAG affinity gel only. For reconstitution of PafE–20S complexes using purified recombinant proteins, PafE and 20S CPs were purified as described in SI Experimental Procedures. The 20SOG CPs were incubated with bortezomib (LC Laboratories) at a molar ratio of 1:8 (20SOG:inhibitor) at 37 °C for 45 min before addition of PafE. Purified PafE was then added to 20SOG CP preincubated with inhibitor or 20ST1A at molar ratio of 1:4 or 1:12 (20S CP:PafE), respectively, at 37 °C for 20 min, followed by an additional 2-h incubation at 20 °C.
EM analysis of PafE and PafE-20S CP complexes is detailed in SI Experimental Procedures.
SEC-MALS.
A total of 200 µg of purified His6–PafE was loaded onto a Shodex KW804 gel permeation chromatography column equilibrated and running with 20 mM Hepes (pH 7.4) and 150 mM NaCl by using a 1515 Isocratic HPLC Pump (Waters). To determine molecular mass, samples were analyzed by refractive index using an Optilab Rex differential refractometer and by MALS using a MiniDawn TREOS light-scattering detector (Wyatt).
Peptide and Protein Degradation Assays.
Suc-LLVY-AMC was from Sigma-Aldrich. Peptides corresponding to the PafE C terminus were synthesized by Peptide 2.0 Inc. with a purity of 95%. The syntheses of LF-2 [7-methoxycoumarin-4-acetic acid (Mca)-Lys-Lys-Val-Ala-Pro-Tyr-Pro-Met-Glu-Dpa(Dnp)-NH2] (Fig. S5) and pupylated 3-methyl-2-oxobutanoate hydroxymethyltransferase (PanB) are described in SI Experimental Procedures.
For peptide degradation assays, 500 ng of purified 20S CP was used per reaction, and triplicate reactions were set up for each condition. The 20S CP was incubated at 37 °C in activity assay reaction buffer (50 mM Tris, pH 8.0, 5 mM MgCl2) containing 100 μM Suc-LLVY-AMC or at room temperature in the presence of 20 μM LF-2. Where indicated, increasing amounts of purified PafE or Mpa were added. Reaction mixtures using Mpa also included 3 mM ATP. Peptide degradation was assessed by measuring the change in fluorescence over time (Suc-LLVY-AMC: λex = 380, λem = 460; LF-2: λex = 340, λem = 405), and reaction rates were calculated by determining relative fluorescence units (RFU) generated per minute.
For protein-degradation assays, 150-μL reactions were set up in activity assay reaction buffer, including 1.5 μg of 20S CPs, 15 μg of PafE, and 1.5 μg of substrate. The 20S CPs and PafE were mixed first and incubated for 30 min at 37 °C and then cooled to room temperature (∼25 °C), at which point substrate was added, and the reaction was incubated at either room temperature, 37 °C, or 45 °C, as indicated. At the indicated time points after substrate addition, a portion of the reaction was removed and quenched by mixing with reducing and denaturing sample buffer. Samples were separated by SDS/PAGE, and substrate degradation was assessed by either Coomassie Brilliant Blue staining (Bio-Rad) for HspR or by monitoring in-gel fluorescence for FITC–β-casein (Sigma-Aldrich) and Pup∼PanB by using a Typhoon Trio imager (GE Healthcare). Fluorescence was quantified by densitometry of a nonsaturated exposure using ImageQuant software.
Construction of the M. tuberculosis pafE Mutant.
A plasmid encoding Rv3781–3783 under control of the operon’s presumed native promoter upstream of Rv3779 was generated by sewing overlap extension PCR (73) and cloned into the vector pMV306-kan, which integrates into the mycobacterial L5 attB site. This plasmid was introduced into M. tuberculosis H37Rv by electroporation (74), and integration was confirmed by PCR using primers Rv3779_promoter_XbaI_F and Rv3783_HindIII_R (Table S5).
To delete and disrupt pafE, ∼700 base pairs of sequence both upstream and downstream of its coding sequence was PCR amplified with primers pafE_pYUB_5′_StuI_F and pafE pYUB 5′ XbaI for the upstream region and pafE pYUB 3′ HindIII F and pafE pYUB 3′ XhoI R for the downstream region (Table S5), and each was cloned into the vector pYUB854 (75) flanking a hygromycin resistance cassette. Cloning was performed to delete the majority of the pafE gene, but left intact the first 30 and the final 65 base pairs of the coding sequence. After the sequence was confirmed, the plasmid was linearized by PacI digestion and introduced into the parental M. tuberculosis strain by electroporation. Putative mutants were selected by plating on hygromycin and kanamycin, and deletion of pafE was confirmed by immunoblotting and by PCR using primers pafE_promoter_HindIII_F and pafE_HindIII_R (Table S5). Complementation of the pafE strain was accomplished by introducing a single copy of the pafE gene under control of its presumed native promoter at the chromosomal Tweety attB site using the integrating vector pAJF381.
Mouse Infections.
Mouse infections were performed as previously described with a few modifications (38, 39, 76). The 6- to 8-wk-old C57BL6/J female mice (Jackson Laboratories) were infected by using an Inhalation Exposure System (Glas-Col) to administer ∼200–400 colony-forming units (CFU) per mouse; however, due to unknown circumstances, we unintentionally infected the mice with ∼4,000 CFU per mouse in our experiments. Mice were humanely killed at days 1 (n = 6), 21 (n = 8), and 56 (n = 8) after infection, and bacteria were harvested from homogenized lungs and spleens and inoculated onto Middlebrook 7H11 agar to enumerate CFU. All procedures were performed with the approval of the New York University Institutional Animal Care and Use Committee. Statistical analysis was performed by using a nonparametric Student’s two-tailed t test.
RNA Extraction and Quantitative Real-Time PCR.
Three biological replicate cultures of the indicated M. tuberculosis strains were grown to an OD580 ∼ 1.0, and RNA was purified and analyzed as described (44).
Heat Stress Sensitivity Assays.
M. tuberculosis cultures were grown to an OD580 ∼ 1.0 and diluted to OD = 0.08 in fresh Middlebrook 7H9 medium. A total of 1 mL was transferred to a 2-mL O-ringed tube and incubated for 24 h at 45 °C, and bacterial survival was assessed by plating serial dilutions on Middlebrook 7H11 agar to enumerate CFU. Statistical analysis was performed by using a nonparametric Student’s two-tailed t test.
Note.
While this manuscript was under review, Delley et al. reported their independent discovery of PafE as an activator of proteasomal degradation (77). However, Delley et al. concluded that PafE forms hexamers whereas we concluded it forms dodecamers.
Supplementary Material
Acknowledgments
We thank Alfred Goldberg, Olga Kandror, and Tatos Akopian for suggesting the use of the LF-2 peptide; Michael S. Glickman and Allison J. Faye for sharing pAJF381; Stevan Hubbard and Ching-Shin Huang for assistance with the SEC-MALS analysis; Andrew Darwin, Ian Mohr, and Victor Torres for helpful discussions; and Charlie Rice and The Rockefeller University for space and support during the 15 months after Hurricane Sandy. J.B.J. was supported by NIH Grants T32 AI007180 and F30 AI110067. H.L. was supported by NIH Grant AI070285. C.C. was supported by NIH Grant GM110430. K.H.D. was supported by NIH Grant AI088075 and the Irma T. Hirschl Charitable Trust, and holds an Investigators in the Pathogenesis of Infectious Diseases Award from the Burroughs Wellcome Fund.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1423319112/-/DCSupplemental.
References
- 1.Kish-Trier E, Hill CP. Structural biology of the proteasome. Annu Rev Biophys. 2013;42:29–49. doi: 10.1146/annurev-biophys-083012-130417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomko RJ, Jr, Hochstrasser M. Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem. 2013;82:415–445. doi: 10.1146/annurev-biochem-060410-150257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Groll M, et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386(6624):463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 4.Löwe J, et al. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science. 1995;268(5210):533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 5.Stadtmueller BM, Hill CP. Proteasome activators. Mol Cell. 2011;41(1):8–19. doi: 10.1016/j.molcel.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arrigo AP, Tanaka K, Goldberg AL, Welch WJ. Identity of the 19S ‘prosome’ particle with the large multifunctional protease complex of mammalian cells (the proteasome) Nature. 1988;331(6152):192–194. doi: 10.1038/331192a0. [DOI] [PubMed] [Google Scholar]
- 7.Chu-Ping M, Vu JH, Proske RJ, Slaughter CA, DeMartino GN. Identification, purification, and characterization of a high molecular weight, ATP-dependent activator (PA700) of the 20 S proteasome. J Biol Chem. 1994;269(5):3539–3547. [PubMed] [Google Scholar]
- 8.Walz J, et al. 26S proteasome structure revealed by three-dimensional electron microscopy. J Struct Biol. 1998;121(1):19–29. doi: 10.1006/jsbi.1998.3958. [DOI] [PubMed] [Google Scholar]
- 9.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt M, et al. The HEAT repeat protein Blm10 regulates the yeast proteasome by capping the core particle. Nat Struct Mol Biol. 2005;12(4):294–303. doi: 10.1038/nsmb914. [DOI] [PubMed] [Google Scholar]
- 11.Ustrell V, Hoffman L, Pratt G, Rechsteiner M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 2002;21(13):3516–3525. doi: 10.1093/emboj/cdf333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumoi K, et al. An archaeal homolog of proteasome assembly factor functions as a proteasome activator. PLoS ONE. 2013;8(3):e60294. doi: 10.1371/journal.pone.0060294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao Y, et al. Structural and functional characterizations of the proteasome-activating protein PA26 from Trypanosoma brucei. J Biol Chem. 1999;274(48):33921–33930. doi: 10.1074/jbc.274.48.33921. [DOI] [PubMed] [Google Scholar]
- 14.Dubiel W, Pratt G, Ferrell K, Rechsteiner M. Purification of an 11 S regulator of the multicatalytic protease. J Biol Chem. 1992;267(31):22369–22377. [PubMed] [Google Scholar]
- 15.Ma CP, Slaughter CA, DeMartino GN. Identification, purification, and characterization of a protein activator (PA28) of the 20 S proteasome (macropain) J Biol Chem. 1992;267(15):10515–10523. [PubMed] [Google Scholar]
- 16.Realini C, et al. Characterization of recombinant REGalpha, REGbeta, and REGgamma proteasome activators. J Biol Chem. 1997;272(41):25483–25492. doi: 10.1074/jbc.272.41.25483. [DOI] [PubMed] [Google Scholar]
- 17.Whitby FG, et al. Structural basis for the activation of 20S proteasomes by 11S regulators. Nature. 2000;408(6808):115–120. doi: 10.1038/35040607. [DOI] [PubMed] [Google Scholar]
- 18.Förster A, Masters EI, Whitby FG, Robinson H, Hill CP. The 1.9 A structure of a proteasome-11S activator complex and implications for proteasome-PAN/PA700 interactions. Mol Cell. 2005;18(5):589–599. doi: 10.1016/j.molcel.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 19.Rabl J, et al. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol Cell. 2008;30(3):360–368. doi: 10.1016/j.molcel.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith DM, et al. Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s alpha ring opens the gate for substrate entry. Mol Cell. 2007;27(5):731–744. doi: 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu Y, et al. Interactions of PAN’s C-termini with archaeal 20S proteasome and implications for the eukaryotic proteasome-ATPase interactions. EMBO J. 2010;29(3):692–702. doi: 10.1038/emboj.2009.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groettrup M, et al. The interferon-gamma-inducible 11 S regulator (PA28) and the LMP2/LMP7 subunits govern the peptide production by the 20 S proteasome in vitro. J Biol Chem. 1995;270(40):23808–23815. doi: 10.1074/jbc.270.40.23808. [DOI] [PubMed] [Google Scholar]
- 23.Groettrup M, et al. A role for the proteasome regulator PA28alpha in antigen presentation. Nature. 1996;381(6578):166–168. doi: 10.1038/381166a0. [DOI] [PubMed] [Google Scholar]
- 24.Murata S, et al. Immunoproteasome assembly and antigen presentation in mice lacking both PA28alpha and PA28beta. EMBO J. 2001;20(21):5898–5907. doi: 10.1093/emboj/20.21.5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong S, et al. The REGγ proteasome regulates hepatic lipid metabolism through inhibition of autophagy. Cell Metab. 2013;18(3):380–391. doi: 10.1016/j.cmet.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, et al. REGγ deficiency promotes premature aging via the casein kinase 1 pathway. Proc Natl Acad Sci USA. 2013;110(27):11005–11010. doi: 10.1073/pnas.1308497110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barton LF, et al. Immune defects in 28-kDa proteasome activator gamma-deficient mice. J Immunol. 2004;172(6):3948–3954. doi: 10.4049/jimmunol.172.6.3948. [DOI] [PubMed] [Google Scholar]
- 28.Li X, et al. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGgamma-proteasome pathway. Mol Cell. 2007;26(6):831–842. doi: 10.1016/j.molcel.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 29.Li X, et al. The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell. 2006;124(2):381–392. doi: 10.1016/j.cell.2005.11.037. [DOI] [PubMed] [Google Scholar]
- 30.Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, Darwin KH. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science. 2008;322(5904):1104–1107. doi: 10.1126/science.1163885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Striebel F, et al. Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat Struct Mol Biol. 2009;16(6):647–651. doi: 10.1038/nsmb.1597. [DOI] [PubMed] [Google Scholar]
- 32.Darwin KH, Lin G, Chen Z, Li H, Nathan CF. Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol Microbiol. 2005;55(2):561–571. doi: 10.1111/j.1365-2958.2004.04403.x. [DOI] [PubMed] [Google Scholar]
- 33.Striebel F, Hunkeler M, Summer H, Weber-Ban E. The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup’s N-terminus. EMBO J. 2010;29(7):1262–1271. doi: 10.1038/emboj.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang T, Darwin KH, Li H. Binding-induced folding of prokaryotic ubiquitin-like protein on the Mycobacterium proteasomal ATPase targets substrates for degradation. Nat Struct Mol Biol. 2010;17(11):1352–1357. doi: 10.1038/nsmb.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang T, et al. Structural insights on the Mycobacterium tuberculosis proteasomal ATPase Mpa. Structure. 2009;17(10):1377–1385. doi: 10.1016/j.str.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu G, et al. Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol Microbiol. 2006;59(5):1417–1428. doi: 10.1111/j.1365-2958.2005.05036.x. [DOI] [PubMed] [Google Scholar]
- 37.Lin G, et al. Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity. Mol Microbiol. 2006;59(5):1405–1416. doi: 10.1111/j.1365-2958.2005.05035.x. [DOI] [PubMed] [Google Scholar]
- 38.Cerda-Maira FA, et al. Molecular analysis of the prokaryotic ubiquitin-like protein (Pup) conjugation pathway in Mycobacterium tuberculosis. Mol Microbiol. 2010;77(5):1123–1135. doi: 10.1111/j.1365-2958.2010.07276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302(5652):1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 40.Gandotra S, Schnappinger D, Monteleone M, Hillen W, Ehrt S. In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat Med. 2007;13(12):1515–1520. doi: 10.1038/nm1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gandotra S, Lebron MB, Ehrt S. The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLoS Pathog. 2010;6(8):e1001040. doi: 10.1371/journal.ppat.1001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell. 2003;11(3):671–683. doi: 10.1016/s1097-2765(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 43.Thompson A, et al. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75(8):1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 44.Festa RA, et al. Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis [corrected] PLoS ONE. 2010;5(1):e8589. doi: 10.1371/journal.pone.0008589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pearce MJ, et al. Identification of substrates of the Mycobacterium tuberculosis proteasome. EMBO J. 2006;25(22):5423–5432. doi: 10.1038/sj.emboj.7601405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franceschini A, et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(Database issue):D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234(4773):179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 48.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 49.Lee ME, Baker TA, Sauer RT. Control of substrate gating and translocation into ClpP by channel residues and ClpX binding. J Mol Biol. 2010;399(5):707–718. doi: 10.1016/j.jmb.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith DM, et al. ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol Cell. 2005;20(5):687–698. doi: 10.1016/j.molcel.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 51.Sauer RT, Baker TA. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 52.Dange T, et al. Blm10 protein promotes proteasomal substrate turnover by an active gating mechanism. J Biol Chem. 2011;286(50):42830–42839. doi: 10.1074/jbc.M111.300178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kusmierczyk AR, Kunjappu MJ, Kim RY, Hochstrasser M. A conserved 20S proteasome assembly factor requires a C-terminal HbYX motif for proteasomal precursor binding. Nat Struct Mol Biol. 2011;18(5):622–629. doi: 10.1038/nsmb.2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Griffin JE, et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7(9):e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48(1):77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 56.Pham TT, Jacobs-Sera D, Pedulla ML, Hendrix RW, Hatfull GF. Comparative genomic analysis of mycobacteriophage Tweety: Evolutionary insights and construction of compatible site-specific integration vectors for mycobacteria. Microbiology. 2007;153(Pt 8):2711–2723. doi: 10.1099/mic.0.2007/008904-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bandyopadhyay B, Das Gupta T, Roy D, Das Gupta SK. DnaK dependence of the mycobacterial stress-responsive regulator HspR is mediated through its hydrophobic C-terminal tail. J Bacteriol. 2012;194(17):4688–4697. doi: 10.1128/JB.00415-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Das Gupta T, Bandyopadhyay B, Das Gupta SK. Modulation of DNA-binding activity of Mycobacterium tuberculosis HspR by chaperones. Microbiology. 2008;154(Pt 2):484–490. doi: 10.1099/mic.0.2007/012294-0. [DOI] [PubMed] [Google Scholar]
- 59.Gur E, Ottofueling R, Dougan DA. Machines of destruction - AAA+ proteases and the adaptors that control them. Subcell Biochem. 2013;66:3–33. doi: 10.1007/978-94-007-5940-4_1. [DOI] [PubMed] [Google Scholar]
- 60.Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol. 2013;14(10):630–642. doi: 10.1038/nrm3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grandvalet C, de Crécy-Lagard V, Mazodier P. The ClpB ATPase of Streptomyces albus G belongs to the HspR heat shock regulon. Mol Microbiol. 1999;31(2):521–532. doi: 10.1046/j.1365-2958.1999.01193.x. [DOI] [PubMed] [Google Scholar]
- 62.Stewart GR, et al. Overexpression of heat-shock proteins reduces survival of Mycobacterium tuberculosis in the chronic phase of infection. Nat Med. 2001;7(6):732–737. doi: 10.1038/89113. [DOI] [PubMed] [Google Scholar]
- 63.Chung CH, Goldberg AL. The product of the lon (capR) gene in Escherichia coli is the ATP-dependent protease, protease La. Proc Natl Acad Sci USA. 1981;78(8):4931–4935. doi: 10.1073/pnas.78.8.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gottesman S, Zipser D. Deg phenotype of Escherichia coli lon mutants. J Bacteriol. 1978;133(2):844–851. doi: 10.1128/jb.133.2.844-851.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kowit JD, Goldberg AL. Intermediate steps in the degradation of a specific abnormal protein in Escherichia coli. J Biol Chem. 1977;252(23):8350–8357. [PubMed] [Google Scholar]
- 66.Shineberg B, Zipser D. The ion gene and degradation of beta-galactosidase nonsense fragments. J Bacteriol. 1973;116(3):1469–1471. doi: 10.1128/jb.116.3.1469-1471.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Phillips TA, VanBogelen RA, Neidhardt FC. lon gene product of Escherichia coli is a heat-shock protein. J Bacteriol. 1984;159(1):283–287. doi: 10.1128/jb.159.1.283-287.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sadre-Bazzaz K, Whitby FG, Robinson H, Formosa T, Hill CP. Structure of a Blm10 complex reveals common mechanisms for proteasome binding and gate opening. Mol Cell. 2010;37(5):728–735. doi: 10.1016/j.molcel.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li D, et al. Structural basis for the assembly and gate closure mechanisms of the Mycobacterium tuberculosis 20S proteasome. EMBO J. 2010;29(12):2037–2047. doi: 10.1038/emboj.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gallagher S, Winston SE, Fuller SA, Hurrell JGR. In: Short Protocols in Molecular Biology. Ausubel FR, et al., editors. Wiley; New York: 2002. pp. 10.12–10.53. [Google Scholar]
- 71.Tian J, et al. Mycobacterium tuberculosis appears to lack alpha-ketoglutarate dehydrogenase and encodes pyruvate dehydrogenase in widely separated genes. Mol Microbiol. 2005;57(3):859–868. doi: 10.1111/j.1365-2958.2005.04741.x. [DOI] [PubMed] [Google Scholar]
- 72.Blum H, Beier H, Gross HJ. Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis. 1987;8(2):93–99. [Google Scholar]
- 73.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77(1):51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 74.Larsen MH, Biermann K, Tandberg S, Hsu T, Jacobs WR., Jr 2007. Genetic manipulation of Mycobacterium tuberculosis. Curr Prot Microbiol Chap 10(Unit 10A): 12. [DOI] [PubMed]
- 75.Bardarov S, et al. Specialized transduction: An efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology. 2002;148(Pt 10):3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- 76.Festa RA, et al. A novel copper-responsive regulon in Mycobacterium tuberculosis. Mol Microbiol. 2011;79(1):133–148. doi: 10.1111/j.1365-2958.2010.07431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Delley CL, et al. Bacterial proteasome activator Bpa (rv3780) is a novel ring-shaped interactor of the mycobacterial proteasome. PLoS ONE. 2014;9(12):e114348. doi: 10.1371/journal.pone.0114348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McWilliam H, et al. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 2013;41(Web Server issue):W597–W600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.