

Abstract
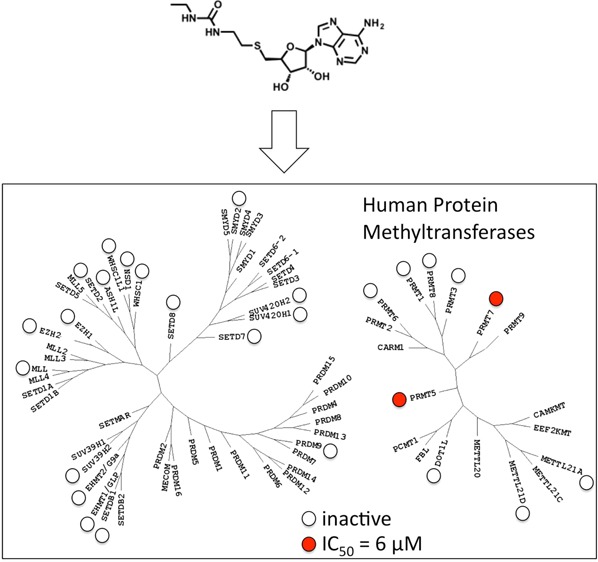
The protein arginine methyltransferases PRMT7 and PRMT5, respectively, monomethylate and symmetrically dimethylate arginine side-chains of proteins involved in diverse cellular mechanisms, including chromatin-mediated control of gene transcription, splicing, and the RAS to ERK transduction cascade. It is believed that PRMT5 and PRMT7 act in conjunction to methylate their substrates, and genetic deletions support the notion that these enzymes derepress cell proliferation and migration in cancer. Using available structures of PRMT5, we designed DS-437, a PRMT5 inhibitor with an IC50 value of 6 μM against both PRMT5 and PRMT7 that is inactive against 29 other human protein-, DNA-, and RNA-methyltransferases and inhibits symmetrical dimethylation of PRMT5 substrates in cells. This compound behaves as a cofactor competitor and represents a valid scaffold to interrogate the potential of the PRMT5–PRMT7 axis as a target for therapy.
Keywords: PRMT5, PRMT7, inhibitor, methyltransferases
Protein arginine methyltransferases are a class of enzymes that transfer a methyl group from the cofactor S-adenosylmethionine (SAM) onto the arginine omega nitrogens of substrate proteins, including histones, and can be divided into three subclasses based on their product specificity: type I, II, and III PRMTs asymmetrically dimethylate, symmetrically dimethylate, and monomethylate their substrates, respectively.1 Of the nine confirmed human PRMTs, PRMT5 is currently the only known type II PRMT, and PRMT7 the only monomethylase.2,3 PRMT9 is uncharacterized, and other PRMTs are type I enzymes.1
PRMT5 has been reported to methylate a number of substrates, both nuclear and cytoplasmic: methylation of arginine 3 of histone 4 (H4R3) by PRMT5 generates a binding site for DNMT3A and is associated with gene silencing;4 PRMT5-mediated methylation of RAF proteins regulates the RAS to ERK signal transduction cascade and promotes cell proliferation;5 methylation of Sm ribonucleoproteins (Sm) by PRMT5 mediates recruitment of SMN, and deletion of PRMT5 results in aberrant splicing/degradation of MDM4, associated with derepression of p53-mediated apoptosis.6,7 These results suggest that PRMT5 may represent an attractive cancer target. Further supporting this notion, PRMT5-targeting siRNAs lead to cell-cycle arrest, apoptosis, and loss of cell migratory activity in glioblastoma cell lines, and increase survival in mice xenograft models of glioblastoma.8
Similar to PRMT5, PRMT7 plays a role in the methylation of H3R2 as well as Sm proteins.2,9 PRMT7 only monomethylates arginine side-chains3 and interacts with PRMT5, suggesting that the two enzymes may function in conjunction to symmetrically dimethylate protein substrates.1 Genetic silencing of PRMT7 reduces symmetrical dimethylation of H4R3 (H4R3me2s), derepresses E-cadherin expression, and attenuates cell migration and invasion in breast cancer cells.10
Potent, selective, and cell-active chemical probes targeting PRMT5 and PRMT7 would be useful tools to investigate molecular pathways, phenotypic responses, and toxicity associated with the chemical inhibition of these methyltransferases. Toward this goal, we present here a low micromolar, dual PRMT5–PRMT7 inhibitor that represents a promising template for the development of potent and selective PRMT5–PRMT7 chemical probes.
The structure of human PRMT5 (hPRMT5) was recently solved in complex with a cofactor mimetic (A9145C), an H4 peptide, and MEP50, a WD repeat protein necessary for full PRMT5 catalytic activity.11 The guanidinium group of the substrate arginine H4R3 forms multiple hydrogen-bonds with the side-chains of the conserved “PRMT double-E loop” glutamates E435 and E444 in a conformation where it is poised to accept a methyl group departing from the cofactor (Figure 1). A Xenopus laevis PRMT5 (xPRMT5) structure in complex with MEP50 and the cofactor product S-adenosine homocysteine (SAH) largely recapitulates structural features observed in the hPRMT5 complex.12 In particular, the conformation of the catalytic glutamates E431 and E440 and the adenosine moiety of the cofactor are absolutely conserved. However, in the xPRMT5 complex, the H4 peptide is absent, and the methionine tail of SAH fills the section of the substrate-binding site that is occupied by the H4R3 guanidinium group in the hPRMT5 structure. Superimposing the two structures suggests that replacement of the amino-acid end of SAH with a urea would recapitulate hydrogen bonds formed between the arginine side-chain and E444 in hPRMT5 (Figure 1). Such a compound may present more interesting selectivity and physicochemical profiles than typical SAM analogues such as sinefungin or SAH. To test this hypothesis, we synthesized the corresponding compound, DS-437.
Figure 1.
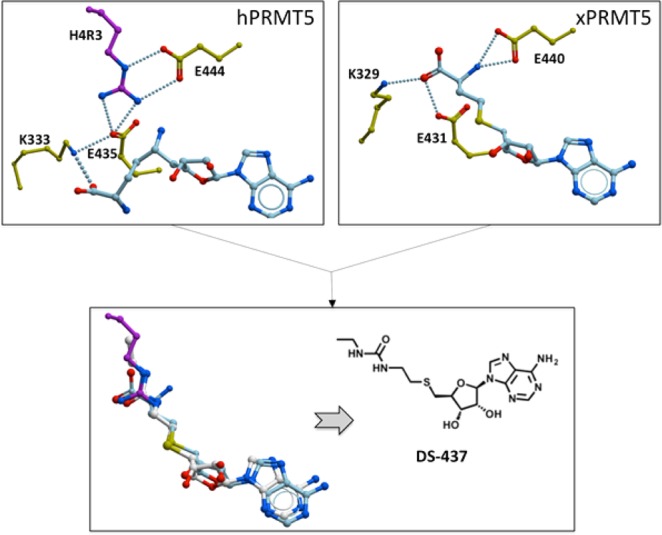
Design of a PRMT5 inhibitor. Superimposing structures of human PRMT5 in complex with a cofactor analogue and a substrate peptide (top left), and Xenopus laevis PRMT5 in complex with SAH (top right) suggests that compound DS-437 would occupy the cofactor binding site, and recapitulate interactions between the substrate arginine and a conserved glutamate side-chain (E444 in hPRMT5). Stick color-coding: yellow, PRMT5; sky blue, cofactor; magenta, substrate arginine; white, DS-437.
DS-437 was able to inhibit methylation of an H4[1–24] peptide by the PRMT5–MEP50 complex under balanced conditions (cofactor and substrate concentrations set at their respective Km values) in a dose-dependent manner with an IC50 of 5.9 ± 1.4 μM (Figure 2A). It was previously reported that the DOT1L inhibitor EPZ004777, a SAM analogue where the amino-acid end is replaced with a tert-butylphenyl-urea moiety, was a 500 nM PRMT5 inhibitor.13 This result was obtained with a PRMT5 assay devoid of MEP50, an interaction partner that is critical for full PRMT5 activity,11 and we were unable in the past to confirm it in the presence of MEP50.14 The urea tail of EPZ004777 is exploiting a cavity that is absent in the catalytically competent state of DOT1L, while the urea moiety of DS-437 is designed to mimic the guanidinium group of the substrate arginine in the PRMT5 complex. In spite of these distinct putative structural mechanisms, we were intrigued with the chemical relatedness between the two compounds and tested the response of the PRMT5–MEP50 complex to increasing concentrations of EPZ004777. We confirmed that EPZ004777 has an IC50 value higher than 20 μM with the PRMT5–MEP50 complex and only inhibits the methylation of the peptide substrate by 80%, at 750 μM (Supplementary Figure 1). An IC50 value of 30 μM can be obtained if the top and bottom of the partial curves are fixed to 100 and 0, respectively.
Figure 2.

Inhibition profile of DS-437. (A) DS-437 inhibits the PRMT5–MEP50 complex with an IC50 of 5.9 ± 1.4 μM. (B) Out of 31 human methyltransferases tested, DS-437 also significantly inhibited PRMT7, with an IC50 of 6 ± 0.5 μM (C).
To investigate the selectivity of DS-437, we profiled its activity against a panel of 27 human protein methyltransferases, three DNA methyltransferases, and the RNA methyltransferase BCDIN3D (Figure 2B and Supplementary Table 1). In the absence of an established assay for PRMT9, a close homologue of PRMT7, the activity of DS437 against this enzyme was not assessed. In spite of its chemical similarity with the pan-methyltransferase inhibitor SAH, DS-437 showed an exquisite dual-specificity against PRMT5 and PRMT7, and residual activity against DNMT3A and DNMT3B (IC50 > 50 μM, Supplementary Figure 2). The compound was able to inhibit methylation of an H2B[23–37] substrate by PRMT7 in a dose-dependent manner, with an IC50 value of 6.0 ± 0.5 μM (Figure 2C). Reflecting similarities in IC50 values, DS-437 had similar binding affinities for PRMT5–MEP50 and PRMT7 as measured by surface plasmon resonance (KD values of 25 and 37 μM, respectively. Supplementary Figure 3). Interestingly, EPZ004777 also inhibited PRMT7 activity with an IC50 value of 7.5 ± 0.5 μM (Supplementary Figure 1C).
Having established that DS-437 was a dual PRMT5–PRMT7 inhibitor, we determined its mechanism of inhibition. The IC50 of the compound for the PRMT5–MEP50 complex and PRMT7 was tested at increasing concentrations of either cofactor or substrate. As expected for a SAM competitive inhibitor, an increase in cofactor concentration resulted in higher IC50 values (Supplementary Figure 4). Conversely, increasing the concentration of the histone peptide substrate had no significant effect on the IC50 values, supporting a noncompetitive pattern toward peptide substrate (Figure 3). These results confirm that DS-437 is at least partially occupying the cofactor-binding site, but the accuracy of our design hypothesis, whereby the urea moiety would occupy the substrate arginine side-chain binding site, remains unresolved. Indeed, local conformational rearrangement of H4R3 may be sufficient to accommodate the ethyl-urea moiety of DS-437 with limited or no impact on binding of the H4[1–24] peptide. A similar mechanism of inhibition was also observed with EPZ004777 (Supplementary Figure 4).
Figure 3.

Cellular activity of DS-437. (A) Immunoblotting with a Rme2s-specific antibody shows reduced amount of symmetrical dimethylation in several cellular proteins upon DS-437 treatment (top panel). Middle panel: immunoblotting with the Y12 antibody that mostly recognizes Sm proteins with symmetrically dimethylated arginines. Bottom panel: immunoblotting of the total cellular SmD1, SmD3, and actin proteins. (B) DS-437 has little effect on the symmetrical dimethylation of histones. (C) Quantitation of symmetrical Arg methylation levels. Band intensities were normalized to SmD3, actin, or total histones.
While other PRMT inhibitors designed to occupy both substrate and cofactor binding sites have been reported,15,16 DS-437 is to our knowledge the first dual PRMT5–PRMT7 inhibitor. This refined selectivity profile is welcome, considering the suspected cellular synergy between PRMT5 and PRMT7, but it was not engineered by design. In the expected binding mode of DS-437, the urea moiety is hydrogen-bonded to E444 (Figure 1), a glutamate that is conserved in all PRMTs. This does not rationalize the lack of activity against all tested class I PRMTs (PRMT1, 3, 6, and 8). A structural feature that is unique to class I PRMTs is a conserved YFxxY motif located on the α-helix that wraps around the cofactor, makes orthogonal stacking with the cofactor adenine ring, and stabilizes a conformation of the glutamate side-chain that is distinct from that observed in PRMT5 and PRMT7 structures (Supplementary Figure 5).17 This class I specific feature is captured, for instance, in a structure of CARM1 in complex with SAH (PDB code 3B3F).18 Superimposition of PRMT5 and CARM1 structures shows that the YFxxY motif is in the immediate vicinity of the cofactor and of residues predicted to interact with DS-437, such as E444 (E267 in CARM1), which may antagonize binding of the inhibitor (Supplementary Figure 5). Similarly, the catalytic domain of DNMT3A is structurally related to PRMTs19 but lacks the α-helix of class-I PRMTs, which may underlie the weak inhibition of this enzyme.
PRMT5 and 7 have been reported to symmetrically methylate Sm ribonucleoproteins involved in splicing.9,20 Given that these enzymes are mostly cytoplasmic,21 we focused on the effect of DS-437 on the best characterized SmD and SmB proteins. We first verified biochemically that the compound could inhibit methylation of SmD3 by PRMT5 and PRMT7 (Supplementary Figure 6). Breast carcinoma MB-MDA231 cells were then exposed to increasing concentrations of the inhibitor and symmetric arginine methylation determined using two antibodies (Figure 3). Both symmetrically dimethylated arginine (Rme2s) and Y12 antibodies showed decreased signal at SmD1/D3 and SmB/B′, as well as at an unidentified p60 protein that has been reported before.9 The signal was normalized to the total levels of SmD3 and actin protein. DS-437 inhibitory activity was apparent starting at 10 μM, a concentration where the compound has no effect on cell viability (Supplementary Figure 7). In contrast, no inhibitory activity was observed against the methylation of H4R3 at 50 μM, and limited inhibition against the methylation of H3R2 was detected at 50 μM (Figure 3).
PRMT5 methylates diverse targets involved in chromatin structure maintenance, transcription, splicing, and cytoplasmic and membrane signaling.22 The knockout of PRMT5 is early embryonic lethal,23 and several tissue specific conditional knockouts indicated profound effects on the splicing machinery function.6 PRMT7, while less well characterized, has been reported to also methylate splicing associated proteins,9 regulate DNA repair associated genes,24 and play a role in breast cancer metastasis.10 The reduction of symmetrical arginine dimethylation of SmD1/3 and SmB proteins in breast cancer cells upon treatment with DS-437 is consistent with the cellular effect of PRMT5 and PRMT7 knockout/downs. PRMT5 and PRMT7 have also been found to cooperate in methylating H3R2.2 We see very limited cellular effect of DS-437 against histone marks, which may indicate longer half-life of the methylated histone (we note that DS-437 inhibits methylation of full-length histone 4 by PRMT5–MEP50 with an IC50 of 37 ± 1.2 μM in a biochemical assay (Supplementary Figure 8)). More potent analogues will be useful tools to investigate the role of the catalytic activity of these methyltransferases in PRMT5/7 mediated signaling.
In summary, we have discovered and characterized, to our best knowledge, the first dual PRMT5–PRMT7 inhibitor, a compound that reduces symmetrical dimethylation of PRMT5 substrates in cells, and propose a rationale for the observed specificity profile. This compound may serve as a scaffold for the development of potent and cell-active chemical probes to investigate the therapeutic potential of targeting the PRMT5–PRMT7 arginine methylation pathway.
Acknowledgments
We would like to thank Elisa Gibson and Taraneh Hajian for protein purification support.
Glossary
ABBREVIATIONS
- SAM
S-adenosylmethionine
- SAH
S-adenosine homocysteine
Supporting Information Available
Materials and instruments used, synthesis, and spectroscopic details of compounds and biological assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ These authors contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, Genome Canada through the Ontario Genomics Institute [OGI-055], GlaxoSmithKline, Janssen, Lilly Canada, Merck, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust [092809/Z/10/Z].
The authors declare no competing financial interest.
Supplementary Material
References
- Bedford M. T.; Clarke S. G. Protein arginine methylation in mammals: who, what, and why. Mol. Cell 2009, 33, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliori V.; Muller J.; Phalke S.; Low D.; Bezzi M.; Mok W. C.; Sahu S. K.; Gunaratne J.; Capasso P.; Bassi C.; Cecatiello V.; De Marco A.; Blackstock W.; Kuznetsov V.; Amati B.; Mapelli M.; Guccione E. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat. Struct. Mol. Biol. 2012, 19, 136–44. [DOI] [PubMed] [Google Scholar]
- Zurita-Lopez C. I.; Sandberg T.; Kelly R.; Clarke S. G. Human protein arginine methyltransferase 7 (PRMT7) is a type III enzyme forming omega-NG-monomethylated arginine residues. J. Biol. Chem. 2012, 287, 7859–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Rank G.; Tan Y. T.; Li H.; Moritz R. L.; Simpson R. J.; Cerruti L.; Curtis D. J.; Patel D. J.; Allis C. D.; Cunningham J. M.; Jane S. M. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat. Struct. Mol. Biol. 2009, 16, 304–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu-Perez P.; Esteve-Puig R.; de Torre-Minguela C.; Lopez-Fauqued M.; Bech-Serra J. J.; Tenbaum S.; Garcia-Trevijano E. R.; Canals F.; Merlino G.; Avila M. A.; Recio J. A. Protein arginine methyltransferase 5 regulates ERK1/2 signal transduction amplitude and cell fate through CRAF. Sci. Signaling 2011, 4, ra58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi M.; Teo S. X.; Muller J.; Mok W. C.; Sahu S. K.; Vardy L. A.; Bonday Z. Q.; Guccione E. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013, 27, 1903–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G.; Eggert C.; Buhler D.; Brahms H.; Kambach C.; Fischer U. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr. Biol. 2001, 11, 1990–4. [DOI] [PubMed] [Google Scholar]
- Yan F.; Alinari L.; Lustberg M. E.; Martin L. K.; Cordero-Nieves H. M.; Banasavadi-Siddegowda Y.; Virk S.; Barnholtz-Sloan J.; Bell E. H.; Wojton J.; Jacob N. K.; Chakravarti A.; Nowicki M. O.; Wu X.; Lapalombella R.; Datta J.; Yu B.; Gordon K.; Haseley A.; Patton J. T.; Smith P. L.; Ryu J.; Zhang X.; Mo X.; Marcucci G.; Nuovo G.; Kwon C. H.; Byrd J. C.; Chiocca E. A.; Li C.; Sif S.; Jacob S.; Lawler S.; Kaur B.; Baiocchi R. A. Genetic validation of the protein arginine methyltransferase PRMT5 as a candidate therapeutic target in glioblastoma. Cancer Res. 2014, 74, 1752–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsalvez G. B.; Tian L.; Ospina J. K.; Boisvert F. M.; Lamond A. I.; Matera A. G. Two distinct arginine methyltransferases are required for biogenesis of Sm-class ribonucleoproteins. J. Cell Biol. 2007, 178, 733–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao R.; Jiang H.; Ma Y.; Wang L.; Du J.; Hou P.; Gao Y.; Zhao L.; Wang G.; Zhang Y.; Liu D. X.; Huang B.; Lu J. PRMT7 induces epithelial-to-mesenchymal transition and promotes metastasis in breast cancer. Cancer Res. 2014, 74, 5656–67. [DOI] [PubMed] [Google Scholar]
- Antonysamy S.; Bonday Z.; Campbell R. M.; Doyle B.; Druzina Z.; Gheyi T.; Han B.; Jungheim L. N.; Qian Y.; Rauch C.; Russell M.; Sauder J. M.; Wasserman S. R.; Weichert K.; Willard F. S.; Zhang A.; Emtage S. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M. C.; Wilczek C.; Bonanno J. B.; Xing L.; Seznec J.; Matsui T.; Carter L. G.; Onikubo T.; Kumar P. R.; Chan M. K.; Brenowitz M.; Cheng R. H.; Reimer U.; Almo S. C.; Shechter D. Structure of the arginine methyltransferase PRMT5-MEP50 reveals a mechanism for substrate specificity. PLoS One 2013, 8, e57008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Majer C. R.; Sneeringer C. J.; Song J.; Johnston L. D.; Scott M. P.; Smith J. J.; Xiao Y.; Jin L.; Kuntz K. W.; Chesworth R.; Moyer M. P.; Bernt K. M.; Tseng J. C.; Kung A. L.; Armstrong S. A.; Copeland R. A.; Richon V. M.; Pollock R. M. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.; Chory E. J.; Wernimont A. K.; Tempel W.; Scopton A.; Federation A.; Marineau J. J.; Qi J.; Barsyte-Lovejoy D.; Yi J.; Marcellus R.; Iacob R. E.; Engen J. R.; Griffin C.; Aman A.; Wienholds E.; Li F.; Pineda J.; Estiu G.; Shatseva T.; Hajian T.; Al-Awar R.; Dick J. E.; Vedadi M.; Brown P. J.; Arrowsmith C. H.; Bradner J. E.; Schapira M. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 2012, 3, 1288. [DOI] [PubMed] [Google Scholar]
- van Haren M.; van Ufford L. Q.; Moret E. E.; Martin N. I. Synthesis and evaluation of protein arginine N-methyltransferase inhibitors designed to simultaneously occupy both substrate binding sites. Org. Biomol. Chem. 2014, 13, 549–60. [DOI] [PubMed] [Google Scholar]
- Dowden J.; Hong W.; Parry R. V.; Pike R. A.; Ward S. G. Toward the development of potent and selective bisubstrate inhibitors of protein arginine methyltransferases. Bioorg. Med. Chem. Lett. 2010, 20, 2103–5. [DOI] [PubMed] [Google Scholar]
- Schapira M.; Ferreira de Freitas R. Structural biology and chemistry of protein arginine methyltransferases. MedChemComm 2014, 5, 1779–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troffer-Charlier N.; Cura V.; Hassenboehler P.; Moras D.; Cavarelli J. Functional insights from structures of coactivator-associated arginine methyltransferase 1 domains. EMBO J. 2007, 26, 4391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert F. M.; Cote J.; Boulanger M. C.; Richard S. A proteomic analysis of arginine-methylated protein complexes. Mol. Cell Proteomics 2003, 2, 1319–30. [DOI] [PubMed] [Google Scholar]
- Herrmann F.; Pably P.; Eckerich C.; Bedford M. T.; Fackelmayer F. O. Human protein arginine methyltransferases in vivo--distinct properties of eight canonical members of the PRMT family. J. Cell Sci. 2009, 122, 667–77. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Bedford M. T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [DOI] [PubMed] [Google Scholar]
- Tee W. W.; Pardo M.; Theunissen T. W.; Yu L.; Choudhary J. S.; Hajkova P.; Surani M. A. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 2010, 24, 2772–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkhanis V.; Wang L.; Tae S.; Hu Y. J.; Imbalzano A. N.; Sif S. Protein arginine methyltransferase 7 regulates cellular response to DNA damage by methylating promoter histones H2A and H4 of the polymerase delta catalytic subunit gene, POLD1. J. Biol. Chem. 2012, 287, 29801–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsyte-Lovejoy D.; Li F.; Oudhoff M. J.; Tatlock J. H.; Dong A.; Zeng H.; Wu H.; Freeman S. A.; Schapira M.; Senisterra G. A.; Kuznetsova E.; Marcellus R.; Allali-Hassani A.; Kennedy S.; Lambert J. P.; Couzens A. L.; Aman A.; Gingras A. C.; Al-Awar R.; Fish P. V.; Gerstenberger B. S.; Roberts L.; Benn C. L.; Grimley R. L.; Braam M. J.; Rossi F. M.; Sudol M.; Brown P. J.; Bunnage M. E.; Owen D. R.; Zaph C.; Vedadi M.; Arrowsmith C. H. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 12853–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.