

Abstract

A novel series of 2-aminopyridopyrimidinone based JNK (c-jun N-terminal kinase) inhibitors were discovered and developed. Structure–activity relationships (SARs) were systematically developed utilizing biochemical and cell based assays and in vitro and in vivo drug metabolism and pharmacokinetic (DMPK) studies. Through the optimization of lead compound 1, several potent and selective JNK inhibitors with high oral bioavailability were developed. Inhibitor 13 was a potent JNK3 inhibitor (IC50 = 15 nM), had high selectivity against p38 (IC50 > 10 μM), had high potency in functional cell based assays, and had high stability in human liver microsome (t1/2 = 76 min), a clean CYP-450 inhibition profile, and excellent oral bioavailability (%F = 87). Moreover, cocrystal structures of compounds 13 and 22 in JNK3 were solved at 2.0 Å. These structures elucidated the binding mode (Type-I binding) and can pave the way for further inhibitor design of this pyridopyrimidinone scaffold for JNK inhibition.
Keywords: JNK3, kinase inhibitor, Parkinson’s disease (PD), pyridopyrimidinone, mitochondrial dysfunction
As a member of the mitogen-activated protein kinase (MAPK) family, the c-Jun N-terminal kinases (JNKs) are activated (dual phosphorylation on threonine and tyrosine) via an upstream kinase signaling cascade initiated by environmental stress and culminate in effects on both nuclear and mitochondrial function.1−3 It is well-known that there are three human JNK isoforms, JNK1, JNK2, and JNK3.4 JNK1 and JNK2 are ubiquitously expressed in a variety of human tissues.2,5 Recent studies showed that JNK1 and JNK2 play an important role in the development of diabetes, obesity, arthritis, cancer, and heart disease. JNK1 seems to be involved in the development of obesity induced insulin resistance, which implies inhibition of JNK1 might be an effective way of treating type-2 diabetes.6,7 JNK2 has been implicated to play an important role in many autoimmune disorders such as rheumatoid arthritis, asthma, and cancer, as well as in a broad range of diseases with an inflammatory component.5,8 JNK3 is primarily expressed in the central nervous system (CNS) and plays an important role in Alzheimer’s disease,9 Parkinson’s disease, and stroke.3,10,11 Therefore, JNK inhibitors may have implications in many therapeutic areas, and development of JNK inhibitors as therapeutic agents has gained considerable interest over the past few years.12−17
Given the significant amount of evidence supporting the role of JNK3 in neurodegenerative disorders, our interest is in discovering potent, selective JNK3 inhibitors with good in vivo pharmacokinetics (PK) profiles as potential therapeutics for CNS disease.3,10,11,18−20 The pyridopyrimidinone scaffold based compound 1 (Figure 1) was identified in our preliminary medicinal chemistry efforts as an ATP competitive pan-JNK inhibitor with an IC50 of 58 nM against JNK3 and 18 nM over both JNK1 and JNK2. This scaffold (pyridopyrimidinone) has long been shown to be good for kinase inhibition.21 While genetic evidence suggests that JNK1 inhibition is not required for efficacy in many CNS applications, there is no solid evidence to demonstrate that it has counter effects and/or other side effects either.
Figure 1.
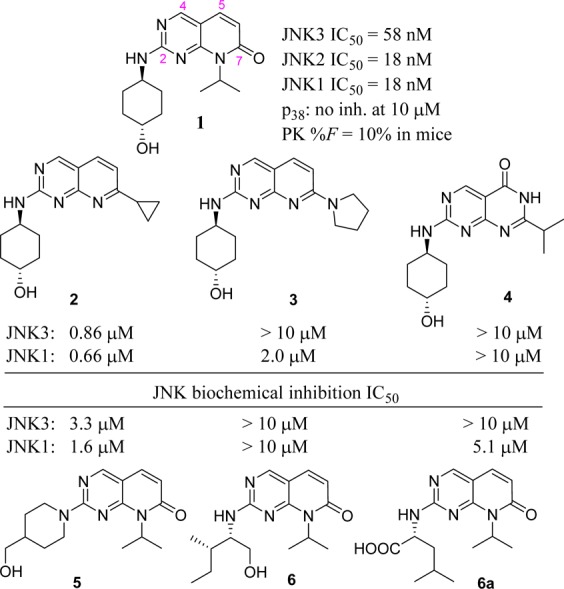
Structure of lead compound 1 and its SAR studies.
Inhibitor 1 had excellent selectivity against its closely related MAP kinase p38 and four selected p450 isoforms (its inhibition against four p450 isoforms 1A2/2C9/2D6/3A4 at 10 μM was 12, 18, −6, and 13%, respectively). In addition, compound 1 had good stability in human and mouse liver microsomes (t1/2 was >120 and 23 min, respectively). However, the oral bioavailability of this JNK inhibitor (1) was low (F = 10% in mice). Herein, we report the structure–activity relationship (SAR) studies for this novel pyridopyrimidinone scaffold of JNK inhibitors and the cocrystal structures of optimized lead inhibitors 13 and 22 in JNK3. Systematic optimizations of inhibitor 1 successfully led to potent JNK inhibitors with much improved oral bioavailability (F = 43–100%) while still maintaining the high JNK inhibitory potency and cell activity in c-Jun phosphorylation assays and in 6-OHDA-induced mitochondrial membrane depolarization assays.
The synthesis, purification, and characterization of all JNK inhibitors (analogues 1–23) are described in the Supporting Information. The initial SAR strategy was to modify the core structure of inhibitor 1 (the fused ring system). Our first attempt was to replace the carbonyl group at C-7 of the pyrido[2,3-d]pyrimidinone core. As shown in Figure 1, any modifications to the C-7 carbonyl group greatly reduced the JNK potency. Significant loss of potency occurred with the replacement by an alkyl group (2, cyclopropyl), by a tertiary amino group (3, pyrrolidine-), and by its regio-isomer (4). These results indicated that the C-7 carbonyl group was part of a key H-bonding interaction and/or the N′-alkyl group might be involved in hydrophobic interactions critical for an efficient ligand binding.
We next explored modifications to the cyclohexyl-amino group attached to C-2 of the pyridopyrimidinone core. As shown in Figure 1, replacing the secondary amine (cyclohexylamine) with a tertiary amine (piperidinoamine) resulted in much higher IC50 values (5). In addition, the amino alcohol and amino acid based structures (6 and 6a) also showed significant loss of JNK inhibitory activity. The JNK inhibition data for these three compounds were reasonable since this 2-amino group was involved in hinge binding interactions (as an H-bond donor, demonstrated in the cocrystal structure shown in Figure 2). Unlike the H-bond formed by −OH of the cyclohexanol in 1, which is helpful for ligand binding in JNKs (see discussions for Table 1), the extra H-bonding interactions from the −OH (6) or the −COOH (6a) group might have disturbed the optimal hinge binding, and the open alkyl chains in 6 and 6a could not exhibit hydrophobic interactions as strong as the cyclohexyl ring of 1. Results in Figure 1 indicated that both the pyrido[2,3-d]pyrimidinone core and the 2-cyclohexyl amino (secondary) moiety were critical for an efficient ligand binding in JNKs for this scaffold.
Figure 2.
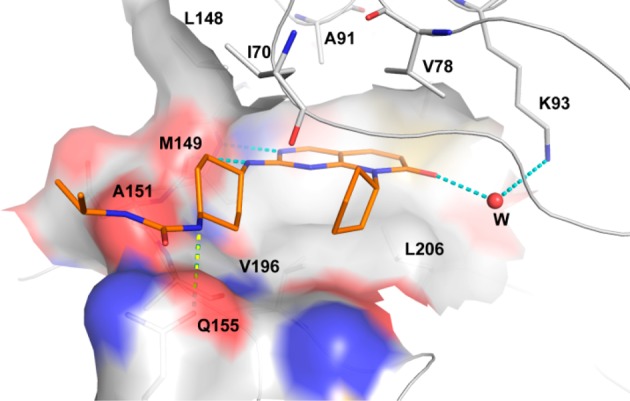
Overlay X-ray crystal structure of 22 in JNK3 (2.0 Å).
Table 1. SAR Studies for the trans-Cyclohexanol Moiety.

IC50 values are the mean of ≥2 experiments with errors within 40% of the mean.
No inhibition at 10 μM.
Not determined.
We then focused SAR studies on the cyclohexanol group, where a series of 6-membered saturated ring derivatives were used to replace the cyclohexanol moiety in compound 1. As shown in Table 1, approximately a 9-fold loss of potency was observed when the cyclohexanol moiety was replaced by a tetrahydro-2H-pyran group (7). Furthermore, application of an N-methylpiperidine ring (8) resulted in complete loss of JNK inhibitory activity. These results demonstrated that an H-bond donor in the ligand around this area (compare the structures among 1, 7, and 8) might be necessary to preserve a tight ligand binding. However, removal of the N-methyl group (9), which produced a new H-bond donor moiety, did not yield potent JNK inhibition either, indicating that the H-bond donor moiety in 9 might not be in the optimal position to exhibit strong H-bonding interactions. Thus, an NH2 group was used to replace the −OH moiety on the trans-cyclohexanol ring, and indeed, the resulting compound 10 picked up some JNK inhibitory activity although it was not as good as that of 1.
Comparison between 10 and 1 indicated that a mono H-bond donor moiety might be preferred. Thus, compounds having substituents on −NH2 were evaluated. Indeed, an isopropyl-amide substitution on NH2 yielded JNK inhibitor 11 having much greater potency (JNK3: 31 nM, Table 1) compared to 10. In addition, analogues 12–14, which were the urea counterparts of amide 11, also exhibited strong JNK inhibition (IC50: 15–81 nM). Among these three JNK inhibitors, compound 13 showed the best potency (IC50: 15 nM for JNK3).
Interesting JNK inhibition data were obtained for the cis-cyclohexyl isomers. Amide 15 exhibited a lower JNK inhibitory potency compared to its trans counterpart inhibitor 11 (Table 1), which was probably due to the unfavorable positioning of the amide NH on 15 (opposite direction compared to 11). This NH direction may not be able to yield optimal H-bonding interactions. However, urea inhibitor 16 had a JNK inhibition profile similar to that of its trans counterpart analogue 12. It is speculated that optimal H-bonding interactions critical to ligand binding were from the other urea NH group in 16 thus resulting in strong JNK inhibitions. However, because of the unfavorable microsomal stability of 16 (see data in Table 3), inhibitors composed of the cis-cyclohexane moiety will not be explored in further optimizations.
Table 3. Data for p450 inhibition, microsomal stability, and c-Jun phosphorylation for lead JNK inhibitors.
| CYP-450% inh.a | mic.
stability t1/2 (min) |
|||
|---|---|---|---|---|
| cmpd | 1A2/2C9/2D6/3A4 | human | mouse | c-Jun phosphorylation IC50 (nM)b |
| 11 | 11/28/18/33 | 44 | 19 | 1232 |
| 12 | 1/19/2/16 | 46 | 17 | 1015 |
| 13 | 10/–26/–58/14 | 76 | 22 | 1733 |
| 16 | 38/47/15/50 | 16 | 8 | ndc |
| 18 | –12/10/–20/–2 | 65 | 14 | 990 |
| 22 | –5/–7/–30/53 | 7 | 5 | ndc |
Percent inhibition at 10 μM.
Data were the average of ≥2 experiments performed in SHSY5Y cells.
Not determined.
Structural modifications were also applied to the 4-ylpiperidine based compound 9 in Table 1. As expected, a simple amide formation to the piperidine secondary amine yielded a compound (17) that had only moderate JNK inhibition due to the lack of an H-bond donor moiety. However, urea based structures 18 and 19, both having a free NH from the urea moiety, exhibited a much higher JNK potency (Table 1). These results indicated that an H-bond donor moiety in this area was critical for an efficient ligand binding to JNK.
Data in Figure 1 demonstrated that the N′-substitution on the pyridoamide moiety might be involved in hydrophobic interactions critical for potent ligand–JNK interactions. Therefore, SAR studies on this region might yield important information for JNK inhibition by the pyridopyrimidinone scaffold. Various substitutions were applied, and representative compounds are summarized in Table 2. Compound 20, which was unsubstituted on the amide, was a weak JNK inhibitor having IC50 values in the micromolar range. A simple methyl substitution (21) immediately picked up some JNK inhibition potency. However, the IC50 values for 21 were still much lower than 13, the isopropyl-substituted analogue (Table 2). A larger alkyl substitution, such as a cyclopentyl group (22), led to even greater JNK inhibitions mainly due to stronger hydrophobic interactions. Interestingly, the tetrahydropyran-substituted analogue 23 was a much less potent inhibitor of all three JNK isoforms compared to inhibitors 13 and 22. The weaker inhibition observed for 23 might be due to plausible H-bonding interactions from the tetrahydropyran ring oxygen atom, which might have disturbed the optimal inhibitor binding conformation.
Table 2. SAR Studies for the N-Substitutions.

IC50 values are the mean of ≥2 experiments with errors within 40% of the mean.
To help explain the binding mode and to aid in future design of JNK inhibitors from this class, the cocrystal structures of lead inhibitors 13 and 22 with human JNK3 were pursued. The 2.0 Å crystal structure showed that JNK inhibitors from this pyridopyrimidinone class had a typical Type-1 kinase binding mode, as demonstrated by the structure of JNK3-22 shown in Figure 2. Key interactions from this structure included multiple H-bonds and tight hydrophobic interactions (similar interactions were also found in the JNK3-13 cocrystal, see SI and TOC graphic). The 2-aminopyrimidine head formed two H-bonds to hinge residue M149 (Figure 2); the urea NH (attached to the cyclohexane moiety) formed an H-bond to the side chain amide carbonyl group of residue Q155; and more importantly, the amide carbonyl group from the pyridopyrimidinone core interacted with the side chain NH2 group of K93 by forming a water bridged H-bond (Figure 2). A total of 10 hydrophobic and one Lys residues were involved in hydrophobic interactions with the ligand. The pyridopyrimidinone ring along with the cyclopentyl group of 22 (or the isopropyl in 13) were nested near a pocket against residues V78, A91, K93, I124, M146, L148, M149, V196, and L206. In addition, the cyclohexyl ring was sandwiched between side chains of residues I70 and A151 (Figure 2, also see Supporting Information).
The binding motif exhibited in this cocrystal structure explained very well the observed SAR. For example, the water bridged H-bonding interactions rationalized the low JNK inhibition for compounds 2–4 (Figure 1); the hinge H-bonding (by the 2-NH group) clarified the much weaker JNK inhibitory activity of compound 5, and the involvement of the cyclohexyl ring in hydrophobic interactions explained why 6 and 6a were not good JNK inhibitors (Figure 1); the H-bond formed between urea or amide NH and Q155 clarified why compounds 7–9, 15, and 17 did not give optimal JNK inhibitions (Table 1, the amide NH on 15 points to the wrong direction); and the contribution of hydrophobic interactions from the N-substitution to a tight ligand binding justified why inhibitors 13 and 22 exhibited much better JNK inhibitory activity than compounds 20 and 21 (Table 2).
Several lead JNK inhibitors were subjected to additional assays against p38 and four P450 isoforms to evaluate the selectivity of these pyridopyrimidinone based JNK inhibitors. Counter screening demonstrated that all of these compounds had low inhibition against p38 (IC50 > 5 μM). In addition, data in Table 3 also showed that most of these pyridopyrimidinone based JNK inhibitors had generally low inhibition for P450 isoforms with slightly higher inhibition for 16 and 22 against 3A4 (inhibition at 10 μM was 50% and 53%, respectively). The stability of these compounds in both human and mouse liver microsomes were also evaluated. Table 3 shows a trans-1,4-disubstitution for the cyclohexyl group was clearly favored over its cis-configuration for metabolic stability (12 vs 16), and an isopropyl group for pyrido-N-substitution was superior to the larger cyclopentyl substitution in terms of microsomal stability (13 vs 22). In addition, data for compound 18 revealed that piperidine based JNK inhibitors had better human microsomal stability but a slightly lower stability in mice as compared to its counterpart cyclohexane based JNK inhibitor (12).
Lead JNK inhibitors were tested in vitro for their ability to inhibit the phosphorylation of JNK substrate phospho-c-jun in neuronal SHSY5Y cells.10,14,22,23 The in-cell Western blot assay for c-Jun phosphorylation is a direct measure of JNK activity and was employed to investigate the ability of lead compounds to inhibit JNK phosphorylation of c-Jun. As shown in Table 3, these lead compounds gave an IC50 of around 1 μM in this assay, indicating that they were reasonable JNK inhibitors.23,24 It is not clear why there was a large right shift between JNK biochemical potency and cell based potency in phosphorylation cell assays for these pyrimidinone derivatives, but it could be due to the presumed higher intracellular ATP concentration than those used in biochemical assays. The cell potency of lead JNK inhibitors was also evaluated in 6-OHDA-induced mitochondrial membrane depolarization, which is regulated and promoted by JNK activation.25 As shown in Figure 3 for compounds 11 and 13, much better cell potency was obtained in this mitochondrial potential assays (IC50 < 50 nM) as compared to the phosphorylation assay. One potential explanation for this finding is that the in-cell Western read out is directly related to the intracellular ATP concentration where the functional readout for mitochondrial dysfunction is not as dependent on ATP concentrations. Indeed, perhaps a small amount of JNK inhibition has a large contribution to protecting the mitochondrial membrane potential.19,26 Similar cell potency was also obtained for other lead compounds in Table 3, such as compounds 12 and 18, indicating this observation was consistent among the class.
Figure 3.

Inhibition of 6-OHDA-induced mitochondrial membrane depolarization in SHSY5Y cells. Cells were treated with 35 μM 6-OHDA for 5 h, and mitochondrial membrane depolarization was measured by normalized MitoTracker Orange CMTMRos fluorescence in the presence or absence of JNK inhibitors. Statistical significance (p < 0.05) between control, untreated group, and 6-OHDA treated group is shown by *, and between 6-OHDA-treated groups and different concentrations of the JNK inhibitors, and 6-OHDA-treated groups is shown by **.
Finally, in vivo pharmacokinetics (PK) were assessed in mice for lead JNK inhibitors in order to uncover whether we achieved the goal of obtaining potent JNK inhibitors with improved PK properties especially with high oral bioavailability. As shown in Table 4, all four JNK inhibitors 11–13 and 18 exhibited fair to excellent in vivo PK properties in mice. At an i.v. dosage of 0.5 mg/kg, these compounds gave good to excellent Cmax, AUC, t1/2, and Vd values. The clearance (Cl) for these compounds was also fair except for inhibitor 12, which had a high Cl value of 72 mL/min.kg. The much better PK profile for 13 as compared to that of 12 demonstrated that a small structural change from ethyl to isopropyl substitution on the terminal urea NH group could lead to significant differences of in vivo PK properties. It is important to point out that all four JNK inhibitors had much higher oral bioavailability in mice (66–100% for 11–13, and 43% for 18) than the lead compound 1 (only 10%).
Table 4. In Vivo PK Data in Mice for Selected Lead JNK Inhibitorsa.
| cmpd | Cmax (μM, i.v.) | AUC (μM·h, i.v.) | t1/2 (h, i.v.) | Cl (mL/min.kg, i.v.) | Vd (L/kg, i.v.) | %F |
|---|---|---|---|---|---|---|
| 11 | 1.0 | 0.7 | 0.5 | 33 | 1.3 | 66 |
| 12 | 0.6 | 0.3 | 0.3 | 72 | 1.9 | 100 |
| 13 | 0.9 | 0.8 | 1.1 | 28 | 1.8 | 87 |
| 18 | 1.1 | 0.7 | 0.4 | 33 | 1.0 | 43 |
Data were generated from three determinations and dosed at 0.5 mg/kg for i.v. and at 3 mg/kg for p.o.
In summary, we have developed a novel class of pyridopyrimidinone based JNK inhibitors through systematic SAR optimization from lead compound 1. The optimized inhibitors had potent pan-JNK inhibition (JNK1/2/3), were selective against p38 kinase, were potent inhibitors of mitochondrial dysfunction, had good in vitro and in vivo DMPK properties, and more importantly, had much higher oral bioavailability compared to 1. The overall best JNK inhibitor obtained from this scaffold was compound 13, which had an IC50 = 15 nM for JNK3 and 21 nM for JNK1, a clean P450 inhibition profile, and good microsomal stability (t1/2 = 76 and 22 min in human and mouse microsomes, respectively). Most importantly, it had excellent in vivo PK properties with an oral bioavailability of 87% in mice. The cocrystal structures of 13 and 22 bound to human JNK3 confirmed that these inhibitors were type-1 kinase binders. Future optimizations for these pyridopyrimidinone based pan-JNK inhibitors will mainly be focused on further reducing the in vivo clearance (Cl) and enhancing the brain penetration for CNS applications. These studies will be reported in due course.
Glossary
ABBREVIATIONS
- JNK
c-jun N-terminal kinases
- MAPK
mitogen activated protein kinase
- ATF2
activating transcription factor 2
- ATP
adenosine triphosphate
- 6-OHDA
6-hydroxydopamine
- MMP
mitochondrial membrane potential
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- ALS
amyotrophic lateral sclerosis
- AUC
pharmacokinetic area under curve
- Cl
pharmacokinetic clearance
- Cmax
pharmacokinetic maximum concentration
- F%
PK oral bioavailability
- Vd
volume of distribution
- SAR
structure–activity relationship
- DMPK
drug metabolism and pharmacokinetics
Supporting Information Available
Details for the synthesis of 2–23; LC purity data; H NMR data; LC–MS data; procedures for bioassays and DMPK studies; cocrystal structures of 13 and 22 (2.0 Å) in human JNK3. The coordinates of JNK3:22 and JNK3:13 complexes were deposited under PDB ID 4Y46 and 4Y5H. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# Korea Research Institute of Chemical Technology (KRICT), 141 Gajeongro, Yuseong 305–600, Korea.
Author Contributions
⊥ These authors contributed equally to this work.
This work was supported by DOD grant W81XWH-12-1-0431 and NIH grant GM103825 (to P.L.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- LoGrasso P.; Kamenecka T. Inhibitors of c-jun-N-terminal kinase (JNK). Mini-Rev. Med. Chem. 2008, 8, 755–66. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Barrett T.; Whitmarsh A. J.; Cavanagh J.; Sluss H. K.; Derijard B.; Davis R. J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [PMC free article] [PubMed] [Google Scholar]
- Chambers J. W.; Howard S.; LoGrasso P. V. Blocking c-Jun N-terminal kinase (JNK) translocation to the mitochondria prevents 6-hydroxydopamine-induced toxicity in vitro and in vivo. J. Biol. Chem. 2013, 288, 1079–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derijard B.; Hibi M.; Wu I. H.; Barrett T.; Su B.; Deng T. L.; Karin M.; Davis R. J. Jnk1 - a protein-kinase stimulated by UV-light and Ha-Ras that binds and phosphorylates the C-Jun activation domain. Cell 1994, 76, 1025–1037. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Shi X. Q.; Zhang Q. J.; Hampong M.; Paddon H.; Wahyuningsih D.; Pelech S. Nocodazole-induced p53-dependent c-Jun N-terminal kinase activation reduces apoptosis in human colon carcinoma HCT116 cells. J. Biol. Chem. 2002, 277, 43648–43658. [DOI] [PubMed] [Google Scholar]
- Chang L. F.; Jones Y.; Ellisman M. H.; Goldstein L. S. B.; Karin M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev. Cell 2003, 4, 521–533. [DOI] [PubMed] [Google Scholar]
- Hirosumi J.; Tuncman G.; Chang L. F.; Gorgun C. Z.; Uysal K. T.; Maeda K.; Karin M.; Hotamisligil G. S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [DOI] [PubMed] [Google Scholar]
- Blease K.; Lewis A.; Raymon H. K. Emerging treatments for asthma. Expert Opin. Emerg. Drugs 2003, 8, 71–81. [DOI] [PubMed] [Google Scholar]
- Yoon S. O.; Park D. J.; Ryu J. C.; Ozer H. G.; Tep C.; Shin Y. J.; Lim T. H.; Pastorino L.; Kunwar A. J.; Walton J. C.; Nagahara A. H.; Lu K. P.; Nelson R. J.; Tuszynski M. H.; Huang K. JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron 2012, 75, 824–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers J. W.; Pachori A.; Howard S.; Ganno M.; Hansen D. Jr.; Kamenecka T.; Song X.; Duckett D.; Chen W.; Ling Y. Y.; Cherry L.; Cameron M. D.; Lin L.; Ruiz C. H.; Lograsso P. Small molecule c-jun-N-terminal kinase (JNK) inhibitors protect dopaminergic neurons in a model of Parkinson’s disease. ACS Chem. Neurosci. 2011, 2, 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker C. E.; Khan S.; Cameron M. D.; Robertson H. A.; Robertson G. S.; LoGrasso P. JNK inhibition protects dopamine neurons and provides behavioral improvement in a rat 6-hydroxydopamine model of Parkinson’s disease. ACS Chem. Neurosci. 2011, 2, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J. R.; Gao H.; Bemis G.; Salituro F.; Ledeboer M.; Harrington E.; Wilke S.; Taslimi P.; Pazhanisamy S.; Xie X. L.; Jacobs M.; Green J. Structure-based design and parallel synthesis of N-benzyl isatin oximes as JNK3 MAP kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2891–2895. [DOI] [PubMed] [Google Scholar]
- He Y. J.; Kamenecka T. M.; Shin Y. S.; Song X. Y.; Jiang R.; Noel R.; Duckett D.; Chen W. M.; Ling Y. Y.; Cameron M. D.; Lin L.; Khan S.; Koenig M.; LoGrasso P. V. Synthesis and SAR of novel quinazolines as potent and brain-penetrant c-jun N-terminal kinase (JNK) Inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 1719–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenecka T.; Habel J.; Duckett D.; Chen W. M.; Ling Y. Y.; Frackowiak B.; Jiang R.; Shin Y. S.; Song X. Y.; LoGrasso P. Structure-activity relationships and X-ray structures describing the selectivity of aminopyrazole inhibitors for c-Jun N-terminal kinase 3 (JNK3) over p38. J. Biol. Chem. 2009, 284, 12853–12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenecka T.; Jiang R.; Song X. Y.; Duckett D.; Chen W. M.; Ling Y. Y.; Habel J.; Laughlin J. D.; Chambers J.; Figuera-Losada M.; Cameron M. D.; Lin L.; Ruiz C. H.; LoGrasso P. V. Synthesis, biological evaluation, X-ray structure, and pharmacokinetics of aminopyrimidine c-jun-N-terminal kinase (JNK) inhibitors. J. Med. Chem. 2010, 53, 419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenitsky V. P.; Delgado M.; Nadolny L.; Sahasrabudhe K.; Ayala L.; Clareen S. S.; Hilgraf R.; Albers R.; Kois A.; Hughes K.; Wright J.; Nowakowski J.; Sudbeck E.; Ghosh S.; Bahmanyar S.; Chamberlain P.; Muir J.; Cathers B. E.; Giegel D.; Xu L.; Celeridad M.; Moghaddam M.; Khatsenko O.; Omholt P.; Katz J.; Pai S.; Fan R.; Tang Y.; Shirley M. A.; Benish B.; Blease K.; Raymon H.; Bhagwat S.; Henderson I.; Cole A. G.; Bennett B.; Satoh Y. Aminopurine based JNK inhibitors for the prevention of ischemia reperfusion injury. Bioorg. Med. Chem. Lett. 2012, 22, 1427–1432. [DOI] [PubMed] [Google Scholar]
- Swahn B. M.; Xue Y.; Arzel E.; Kallin E.; Magnus A.; Plobeck N.; Viklund J. Design and synthesis of 2′-anilino-4,4′-bipyridines as selective inhibitors of c-Jun N-terminal kinase-3. Bioorg. Med. Chem. Lett. 2006, 16, 1397–401. [DOI] [PubMed] [Google Scholar]
- Borsello T.; Forloni G. JNK signalling: A possible target to prevent neurodegeneration. Curr. Pharm. Des. 2007, 13, 1875–1886. [DOI] [PubMed] [Google Scholar]
- Zheng K.; Iqbal S.; Hernandez P.; Park H.; LoGrasso P.; Feng Y. Design and synthesis of highly potent and isoform selective JNK3 inhibitors: SAR studies on aminopyrazole derivatives. J. Med. Chem. 2014, 57, 10013–10030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y.; Chambers J. W.; Iqbal S.; Koenig M.; Park H.; Cherry L.; Hernandez P.; Figuera-Losada M.; LoGrasso P. V. A small molecule bidentate-binding dual inhibitor probe of the LRRK2 and JNK kinases. ACS Chem. Biol. 2013, 8, 1747–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookfield F.; Dillon M. P.; Goldstein D. M.; Gong L.; Ha X.; Hogg J. H.; Park J.; Reuter D. C.; Sjogren E. B.. Pyrimidinyl Pyridone Inhibitors of Kinase. US 20090270389, 2009.
- Chambers J. W.; Cherry L.; Laughlin J. D.; Figuera-Losada M.; Lograsso P. V. Selective inhibition of mitochondrial JNK signaling achieved using peptide mimicry of the Sab kinase interacting motif-1 (KIM1). ACS Chem. Biol. 2011, 6, 808–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Wang S. Y.; Clampit J. E.; Gum R. J.; Haasch D. L.; Rondinone C. M.; Trevillyan J. M.; Abad-Zapatero C.; Fry E. H.; Sham H. L.; Liu G. Discovery of a new class of 4-anilinopyrimidines as potent c-Jun N-terminal kinase inhibitors: Synthesis and SAR studies. Bioorg. Med. Chem. Lett. 2007, 17, 668–672. [DOI] [PubMed] [Google Scholar]
- Alam M.; Beevers R. E.; Ceska T.; Davenport R. J.; Dickson K. M.; Fortunato M.; Gowers L.; Haughan A. F.; James L. A.; Jones M. W.; Kinsella N.; Lowe C.; Meissner J. W.; Nicolas A. L.; Perry B. G.; Phillips D. J.; Pitt W. R.; Platt A.; Ratcliffe A. J.; Sharpe A.; Tait L. J. Synthesis and SAR of aminopyrimidines as novel c-Jun N-terminal kinase (JNK) inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3463–7. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Herdegen T. Cerebral ischemia provokes a profound exchange of activated JNK isoforms in brain mitochondria. Mol. Cell. Neurosci. 2009, 41, 186–195. [DOI] [PubMed] [Google Scholar]
- Park H.; Iqbal S.; Hernandez P.; Mora R.; Zheng K.; Feng Y.; LoGrasso P. Structural basis for JNK2/3 isoform selective aminopyrazoles. Sci. Rep. 2015, 5, 8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.