

Abstract

We describe the hybridization of our previously reported acyclic and cyclic CC chemokine receptor 2 (CCR2) antagonists to lead to a new series of dual antagonists of CCR2 and CCR5. Installation of a γ-lactam as the spacer group and a quinazoline as a benzamide mimetic improved oral bioavailability markedly. These efforts led to the identification of 13d, a potent and orally bioavailable dual antagonist suitable for use in both murine and monkey models of inflammation.
Keywords: Chemokine, GPCR, dual antagonist, inflammation
Over the past 15 years, both the academic and pharmaceutical communities have directed a tremendous amount of research effort toward CC chemokine receptor 2 (CCR2) and its primary ligand (CCL2, which historically was referred to as monocyte chemoattractant protein-1, or MCP-1). This has primarily been motivated by the central role that these two proteins play in the migration and activation of inflammatory monocytes,1 which results in their implication as key players in a number of preclinical disease models.2−4 A number of companies have advanced CCR2 antagonists from different chemical series into human clinical studies,5−7 but none of these compounds appear to have yet advanced to pivotal Phase 3 trials.
Much has been written about the unspectacular results observed to date with chemokine receptor antagonists in autoimmune and inflammatory diseases,8−10 and we have also commented on the nuances of the clinical results in the CCR2 field.5 In this context, we have recently become attracted to the growing body of literature that supports a role for dual antagonism of CCR2 and the closely related CCR5 for the treatment of cardiovascular and autoimmune diseases.11 Herein, we describe an important extension of our earlier work on CCR2 antagonism, which has led to the discovery of a potent and orally bioavailable CCR2/5-dual antagonist.
As described in recent publications from our laboratories, we have pursued both cyclic and acyclic antagonists of CCR2. Our early efforts led to the discovery of compounds in both series that exhibited potent activity in binding and functional assays, but only modest oral bioavailability (Figure 1). For example, cyclic compound 1 showed IC50 values of 0.3 and 1.9 nM for blockade of monocyte binding and chemotaxis, and modest oral bioavailability in mouse (F ≈ 10%).12 Acyclic compound 2 exhibited IC50 values of 3 and 24 nM in binding and chemotaxis assays, and was modestly orally bioavailable in mouse (F = 29%).13 Despite these similarities, there were key differences in the structure–activity relationships in the two series, such that compound 2 exhibited reasonable activity at mouse CCR2 (mCCR2, IC50 = 3 nM), whereas 1 did not (IC50 = 440 nM). This trend paralleled their activity in human CCR5, where binding IC50 values of 86 and 1000 nM were observed for 2 and 1, respectively.
Figure 1.

Cyclic and acyclic CCR2 antagonists from our laboratories.
We were intrigued by the possibility of hybridizing these two series, in order to benefit from structure–activity relationship (SAR) trends evident in each. We thus turned to our earlier receptor mutagenesis and homology modeling experiments14 for insights into how this might be accomplished. Three key observations guided our hypothesis (Figure 2):
-
1.
Installation of nitrogen into the cyclic chemotype to give a piperidine subseries (Figure 2, Cyclic-1) yielded an enhancement in CCR2 binding. Such compounds were shown to interact with Glu291 in transmembrane domain 7 (TMD7) of CCR2, whereas the parent cyclohexyl derivatives did not.15
-
2.
Both the amide16 and hydroxyethyl isostere (e.g., 2)13 variants of the acyclic series appeared to strongly engage this same glutamic acid in TMD7.
-
3.
The stereochemical preferences in the cyclic and acyclic series were identical. Given that the stereochemistry of the trisubstituted series (e.g., 1) pointed to a low energy conformer (Figure 2, Cyclic-2),12 the conformation of the acyclic series could be inferred (Figure 2, Acyclic).
Figure 2.
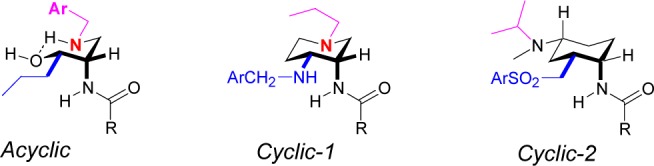
Potentially analogous binding interactions exist between acyclic and cyclic series.
The proposed model could be tested readily in the acyclic series: if the alignment was as illustrated in Figure 2, then the propyl in the acyclic series could be replaced by phenethyl; or alternatively, the N-benzyl substituent could be replaced by an alkyl group. We thus synthesized the analogues in Table 1 using the methods described in our earlier work.13,17 As described previously,13 benzyl was a poor replacement for n-propyl on the side chain (cf. 3c and 3a). Remarkably, when we extended the side chain by one carbon to phenethyl (3d), the potency was improved ∼100-fold, bringing it within range of the previously described isobutyl side chain (3b). Given that this was consistent with the model, we moved to replace the N-benzyl substituent. Relative to our expectations based on earlier CCR2 SAR (prior to 2003), we were surprised to see that the benzyl group could be replaced with a larger branched alkyl substituent (see 3e–3g, Table 1). The isobutyl group was studied further in the context of the more potent ureidobenzamide group, which confirmed that high potency could be achieved without an N-benzyl substituent (see 3h, CCR2 binding IC50 = 1.1 nM; chemotaxis IC50 < 0.45 nM). The analogous neopentyl and cyclopropylmethyl groups were also effective N-substituents, and all of the N-alkyl analogues compared favorably with the benchmark N-benzyl compound (cf. 3h–3j and 3k, Table 1). These three N-alkyl compounds also exhibited activity against CCR5, with binding IC50 values of 137, 321, and 1242 nM for compounds 3h, 3i, and 3j, respectively. Compound 3i was also dosed in an abbreviated mouse pharmacokinetic study and found to be orally bioavailable (4 h AUC ≈ 1500 nM·h at 10 mpk PO).
Table 1. Binding and Functional Activity of Acyclic Molecules Designed to Test the Hypothesis in Figure 2a.
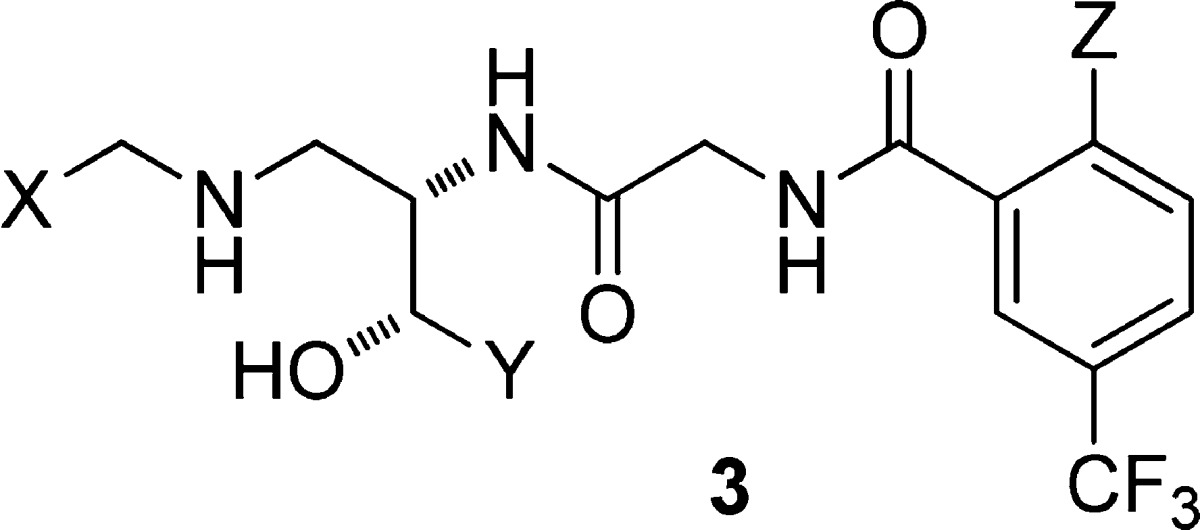
| compd | X | Y | Z | CCR2 binding IC50 (nM) | monocyte chemotaxis IC50 (nM) |
|---|---|---|---|---|---|
| 3a | 2,4-Me2Ph | CH2Et | H | 5 | 240 |
| 3b | 2,4-Me2Ph | CH2i-Pr | H | 19 | |
| 3c | 2,4-Me2Ph | CH2Ph | H | 6400 | |
| 3d | 2,4-Me2Ph | CH2CH2Ph | H | 62 | |
| 3e | i-Pr | CH2Et | H | 15 | |
| 3f | i-Bu | CH2Et | H | 337 | |
| 3g | c-Hex | CH2Et | H | 567 | |
| 3h | i-Pr | CH2Et | NHC(O)NHi-Pr | 1.1 | <0.45 |
| 3i | t-Bu | CH2Et | NHC(O)NHi-Pr | 0.8 | <0.45 |
| 3j | c-Pr | CH2Et | NHC(O)NHi-Pr | 4.9 | 3.3 |
| 3k | 2,4-Me2Ph | CH2Et | NHC(O)NHi-Pr | 4.7 | 9.9 |
Binding was performed with 0.3 nM [125I]MCP-1 and human peripheral blood mononuclear cells at room temperature (RT) (ref (17)). IC50 values reported as the average of two or more determinations. Antagonism of chemotaxis of human peripheral blood mononuclear cells was induced by 10 nM MCP-1 at 37 °C (ref (17)). Compounds 3a, 3b, 3c, and 3k were described in our earlier paper on the acyclic series (ref (13)).
The data shown in Table 1 gave us confidence that the alignment shown in Figure 2 was plausible. We next sought to capitalize on the hybridization model to enhance the properties of the cyclic series, which already had both greater potency (e.g., compare chemotaxis activity of 1 vs that of 3a) and scaffold rigidity. We specifically focused on replacing the phenylsulfonylmethyl of Cyclic-2 with the propyl group of Acyclic. From the perspective of physicochemical properties, this switch should reduce the polar surface area in the cyclic series and lead to improved bioavailability. From a pharmacological perspective, we knew that the propyl group was a key determinant of both mouse CCR2 and human CCR5 binding in the acyclic series, and hypothesized that incorporating it into the cyclic series could endow this series with greater potency for these two receptors.
In order to synthesize the cyclic analogues with nonsulfone side chains, we capitalized on our recently reported synthesis of (1R,2S,5R)-tert-butyl 2-(benzyl-oxycarbonylamino)-7-oxo-6-azabicyclo[3.2.1]octane-6-carboxylate.18 As shown in Scheme 1,19 compound 5 could be partially reduced with DIBAL-H. The subsequent hemiaminal was not purified, but rather reacted with an ylide to give the cis-olefin 6. Hydrogenation and deprotection provided the free amine, which was readily coupled with a hippuric acid to afford 7. Deprotection of the t-Bu carbamate with TFA provided the penultimate amine, which could be subjected to sequential reductive amination with acetone and formaldehyde to yield the target product 4e. Other alkyl side chains could be installed in a similar fashion. Alternatively, ether side chains were accessed from 8, the product of complete reduction from bicycle 5.18 The hydroxylmethyl group could either be methylated directly or subjected to Mitsonobu displacement to yield intermediates 9 and 10, respectively. These could be advanced to final products in a manner similar to the sequence 6 → 7 → 4e.
Scheme 1. Synthesis of Novel Cyclic Analogues.

Reagents and conditions: (a) i. DIBAL-H, CH2Cl2, −55 → 0 °C; ii. BrPh3PCH2i-Pr, KHMDS, THF, 0 °C. (b) i. H2, MeOH, 5% Pd/C, Degussa; ii. HO2CCH2NH(O)C(3-CF3Ph), BOP, i-Pr2NEt, CH2Cl2/DMF. (c) i. TFA, CH2Cl2; ii. MeOH, Me2CO, NaCNBH3, 4 h, then aq. CH2O. (d) THF, H2O, NaBH4. (e) MeI, Ag2O, DMF. (f) PhOH, Ph3P, DEAD, THF.
The structure–activity relationships are shown in Table 2 for this novel series of compounds lacking either amide or sulfone functionality in the side chain. The characterization of phenylsulfone 4h(20) in the same assay suite is provided for comparison. The targeted n-propyl compound 4a was indeed active at CCR2, albeit with lower potency than sulfone 4h. It displayed relatively low turnover in rat liver microsomes and ∼1000-fold selectivity for CCR2 antagonism relative to hERG or CYP2D6 inhibition. Furthermore, compound 4a was orally bioavailable in the mouse and showed exposures in 4 h PK studies21 (AUC/D = 2.4 μM·h) that were lower than those achieved with the acyclic compound 3i (AUC/D = 3.2 μM·h) but higher than those with the cyclic sulfone 4h (AUC/D = 1.1 μM·h).
Table 2. Binding, Functional, and Selectivity Data for Cyclic CCR2 Antagonists with Nonsulfone Side Chainsa.
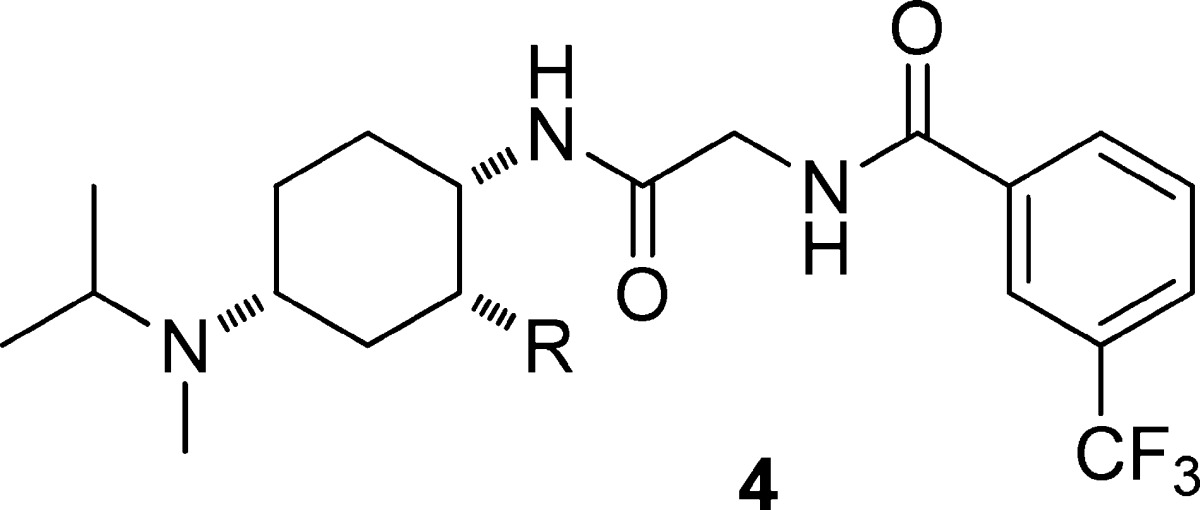
| compd | R | CCR2 binding IC50 (nM) | monocyte chemotaxis IC50 (nM) | Rat LM (rate, nmol/min/mg) | hERG flux (IC50, μM) | CYP 2D6 (IC50, μM) |
|---|---|---|---|---|---|---|
| 4a | CH2Et | 13 | 25 | 0.01 | 32 | 20 |
| 4b | CH2OMe | 33 | 60 | 0.01 | >80 | >40 |
| 4c | CH2CH2Et | 2.5 | 2.7 | 0.17 | 0.36 | 0.32 |
| 4d | CH2CH2OMe | 20 | 23 | 0.27 | >80 | >40 |
| 4e | CH2CH2i-Pr | 4.7 | 16 | 0.08 | 2.7 | 0.01 |
| 4f | CH2CH2Ph | 2.5 | 1.8 | 0.26 | 1.6 | 0.02 |
| 4g | CH2OPh | 41 | 76 | 0.29 | 5.3 | 0.73 |
| 4h | CH2SO2Ph | 0.8 | 1.3 | 0.16 | >80 | 2.7 |
See footnote to Table 1 for description of binding and chemotaxis assays. Also shown are the rate of metabolism in rat liver microsomes, inhibition of hERG activity in a high throughput ion flux assay, and inhibition of CYP2D6 activity.
It proved possible to improve the potency of 4a through simple extension of the chain (4c) or addition of hydrophobic groups (4e, 4f), but in each case, this resulted in a decrease in both metabolic stability and off-target selectivity (Table 2). Replacement of one of the side chain methylenes with an oxygen provided compounds that exhibited lower hERG and CYP inhibition than their matched analogues but that were also less potent at CCR2 and sometimes also less metabolically stable (cf. 4a/4b; 4c/4d; and 4f/4g). Given the overall profile of the compounds, we elected to focus the bulk of our follow-up efforts on the n-propyl side chain. Our research on the optimization of phenethyl 4f is described separately,22 as is our work on alkyl sulfone analogues.23
Our attempts to improve the mouse oral bioavailability and CCR2 potency of 4a capitalized on our previous structure–activity relationship studies, which suggested two approaches: replacement of the glycinamide motif with a γ-lactam20 and replacement of the benzamide with anthranilic amide derivatives.13 Chart 1 shows a pair-wise comparison of the effect of γ-lactam substitution of the glycinamide in both phenylsulfonylmethyl and n-propyl subseries. In both instances, the γ-lactam had only a modest effect on the CCR2 binding affinity but increased the exposure achieved in mouse after oral dosing.
Chart 1. Pairwise Comparison of in Vitro CCR2 Binding Activity and Dose-Normalized Mouse AUC (PO) Observed with Glycinamides and γ-Lactams in Phenylsulfonylmethyl and n-Propyl Subseries.

The high level of oral exposure observed with compounds having the combination of n-propyl side chain and γ-lactam spacer was observed in multiple instances, as highlighted in Table 3.24 Urea analogues 13a and 13b were more potent than benzamide 12 but had lower exposure in 4 h mouse pharmacokinetic studies. The introduction of the quinazoline as an alternative anthranilic acid mimetic13,25 afforded compounds 13c and 13d, both of which exhibited potent CCR2 activity with improved exposure. In the quinazoline series, the use of other side chain alkyl groups (methyl, ethyl, iso-propyl, methoxymethyl) did not offer a clear advantage over the n-propyl analogue 13d.
Table 3. Structure–Activity Relationships of Compounds Bearing Alkyl Side Chains and γ-Lactam Linkers, with Different Anthranilamide Derivativesa.
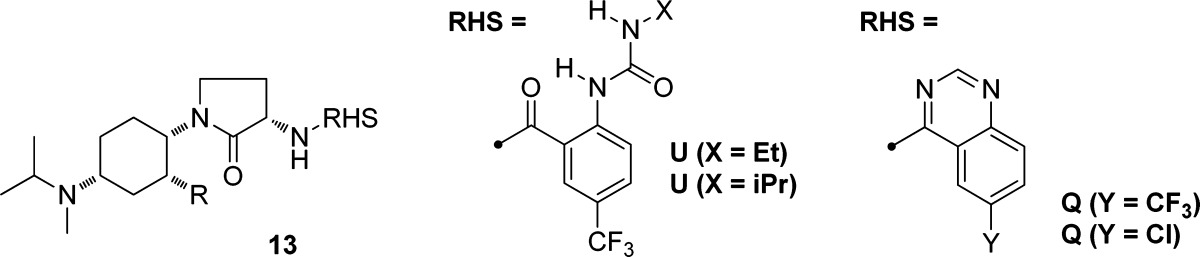
| compd | R | RHS | CCR2
binding IC50 (nM) |
monocyte chemotaxis IC50 (nM) |
CCR5 binding IC50 (nM) |
mouse PO PK (AUC/D, μM·h) |
hERG patch clamp (IC50 or %Inh) |
|---|---|---|---|---|---|---|---|
| 13a | CH2CH2CH3 | U (X = Et) | 1.0 | 0.6 | 3.4 | 6.2 | 14 μM |
| 13b | CH2CH2CH3 | U (X = i-Pr) | 1.7 | 3.3 | 4.8 | 72% @ 10 μM | |
| 13c | CH2CH2CH3 | Q (Y = Cl) | 2.2 | 2.7 | 22 | 16 | 87% @ 10 μM |
| 13d | CH2CH2CH3 | Q (Y = CF3) | 0.7 | 0.24 | 2.4 | 22 | 1.8 μM |
| 13e | CH2CH3 | Q (Y = CF3) | 0.5 | 0.5 | 3.0 | 13 | |
| 13f | CH3 | Q (Y = CF3) | 2.2 | 11 | 10% @ 1 μM | ||
| 13g | CH(CH3)2 | Q (Y = CF3) | 0.4 | 10 | 15 | 81% @ 10 μM | |
| 13h | CH2OCH3 | Q (Y = CF3) | 3.0 | 18 | 0.7 |
CCR2 binding and chemotaxis assays were performed as described in Table 1. CCR5 binding was assessed through antagonism of MIP-1β binding to HT1080 cells stably expressing CCR5. For mouse PK, the dose-adjusted 4 h AUC is listed from experiments in which mice were dosed orally with the indicated compounds (ref (21)). The final column shows activity in the hERG patch clamp assay, expressed either as estimated IC50 or % inhibition at a single test concentration.
Compound 13d was profiled further in vitro. The potent binding to CCR2 (IC50 = 0.7 ± 0.3 nM) was confirmed in two functional assays: monocyte chemotaxis (IC50 = 0.24 ± 0.16 nM) and CD11b upregulation (whole blood IC50 = 4.7 ± 0.9 nM). The potent binding of 13d to CCR5 in HT1080 cells (Table 3) was confirmed in human T-cells (IC50 = 2.3 ± 1.8 nM) and whole blood CCR5-mediated CD11b upregulation (IC50 = 4.3 ± 4.4 nM). The dual activity at CCR2 and CCR5 was maintained against the mouse receptors, where it likewise exhibited affinities <2 nM.
Compound 13d was studied further and found to exhibit high selectivity against other chemokine receptors and GPCRs, showing >1000-fold separation for everything but members of the muscarinic family. The Ki values for M1, M2, and M4 receptors were 341, 4010, and 865 nM, respectively. As indicated in Table 3, compound 13d was ca. 2000-fold selective for binding CCR2 over hERG. It exhibited a similar level of selectivity (ca. 5000-fold) relative to the Na+ channel. In Sprague–Dawley rats, it displayed a clearance of 40 mL/min/kg (2.5 mg/kg, IV) and an oral bioavailability of 68% (AUC = 9294 nM·h after a 25 mg/kg PO dose). In Cynomolgus monkey, compound 13d was cleared at 25 mL/min/kg (1.0 mg/kg, IV) and had an oral bioavailability of 46% (AUC = 862 nM·h after a 1.4 mg/kg PO dose).
The synthesis of compound 13d is provided as representative of the series (Scheme 2).24 Partial reduction of compound 5(18) with DIBAL-H produced the hemiaminal, which was reacted with the propyl ylide to produce olefin 14. Introduction of the γ-lactam followed a 4-step procedure analogous to that reported by us previously.20 Conversion of the bis-carbamate 16 to the diamine 17 was accomplished in good yield through sequential TFA-mediated Boc removal, one-pot double reductive amination, and HBr-mediated Cbz removal. The primary amine was reacted with the quinazoline chloride24 in refluxing ethanol to obtain 13d in 52% yield after crystallization.
Scheme 2. Synthesis of Key Compound 13d.

Reagents and conditions: (a) i. DIBAL-H, CH2Cl2, 0 °C; ii. BrPh3PCH2Et, KHMDS, THF, 0 °C. (b) i. H2, MeOH, 5% Pd/C, Degussa; ii. (S)-Cbz-methionine, BOP, i-Pr2NEt, CH2Cl2/DMF. (c) i. Neat MeI; ii. Cs2CO3, DMF. (d) i. TFA, CH2Cl2; ii. MeOH, Me2CO, NaCNBH3, 4 h, then aq. CH2O; iii. neat 30% HBr/AcOH. (e) 4-Cl-6-CF3-quinazoline, Et3N, EtOH, 75 °C.
In order to secure the stereochemistry of 13d and explore its solution conformation, we conducted a detailed NMR study. We were able to confirm the relative stereochemical relationships using coupling constant analysis and nuclear Overhauser effect spectroscopy (NOESY) studies (see Supporting Information). Furthermore, these studies showed the axial/equatorial disposition of cyclohexyl substituents to be analogous to that previously hypothesized12 (Figure 2, Cyclic-2): the γ-lacatm was oriented axially, while the propyl and amine groups were equatorial.
In summary, we have described the hybridization of our previously reported acyclic and cyclic CCR2 antagonists. These studies confirmed our binding model and led to the introduction of potent CCR5 activity into our CCR2-selective cyclic series. The oral bioavailability of the new series was improved through the combined replacement of the glycine spacer and benzamide capping group with a γ-lactam and quinazoline, respectively. These studies led to the identification of 13d, a potent and orally bioavailable dual antagonist of CCR2 and CCR5. Subsequent manuscripts from our laboratories will detail the characterization of the activity of this molecule in multiple animal models of acute and chronic inflammation.
Acknowledgments
We gratefully acknowledge other team members who synthesized analogues in related chemical series, as well as Dr. Arvind Mathur and the Department of Chemical Synthesis and the Biocon/BMS Research Center for their synthesis of 13d to support large scale in vivo studies that will be disclosed separately. The support of our colleagues in Lead Evaluation, Lead Profiling, and Bioanalytical Sciences is also much appreciated.
Glossary
ABBREVIATIONS
- AUC
area under the curve
- CCR2
CC chemokine receptor 2
- CCL2
CC chemokine ligand 2
- CCR5
CC chemokine receptor 5
- PK
pharmacokinetics
Supporting Information Available
Synthetic procedures and complete characterization data for compound 13d; NMR study on 13d; and protocols for assays listed in Tables 1–3. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript contains contributions from all authors.
This research was funded by Bristol-Myers Squibb Company.
The authors declare no competing financial interest.
Supplementary Material
References
- Gordon S.; Taylor P. R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5 (12), 953–964. [DOI] [PubMed] [Google Scholar]
- Kang Y. S.; Cha J. Jo.; Hyun Y. Y.; Cha D. R. Novel C-C chemokine receptor 2 antagonists in metabolic disease: a review of recent developments. Expert Opin. Invest. Drugs 2011, 20 (6), 745–756. [DOI] [PubMed] [Google Scholar]
- Charo I. F.; Ransohoff R. M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 2006, 354 (6), 610–621. [DOI] [PubMed] [Google Scholar]
- Feria M.; Díaz-González F. The CCR2 receptor as a therapeutic target. Expert Opin. Ther. Pat. 2006, 16 (1), 49–57. [Google Scholar]
- Carter P. H. Progress in the discovery of CC chemokine receptor 2 antagonists, 2009 - 2012. Expert Opin. Ther. Pat. 2013, 23 (5), 549–568. [DOI] [PubMed] [Google Scholar]
- Struthers M.; Pasternak A. CCR2 antagonists. Curr. Top. Med. Chem. 2010, 10 (13), 1278–1298. [DOI] [PubMed] [Google Scholar]
- Xia M.; Sui Z. Recent developments in CCR2 antagonists. Expert Opin. Ther. Pat. 2009, 19 (3), 295–303. [DOI] [PubMed] [Google Scholar]
- Schall T. J.; Proudfoot A. E. I. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat. Rev. Immunol. 2011, 11 (5), 355–363. [DOI] [PubMed] [Google Scholar]
- Gladue R. P.; Brown M. F.; Zwillich S. H. CCR1 antagonists: what we have learned from clinical trials. Curr. Top. Med. Chem. 2010, 10 (13), 1268–1277. [DOI] [PubMed] [Google Scholar]
- Horuk R. Chemokine receptor antagonists: overcoming developmental hurdles. Nat. Rev. Drug Discovery 2009, 8 (1), 23–33. [DOI] [PubMed] [Google Scholar]
- Zhao Q. Dual targeting of CCR2 and CCR5: therapeutic potential for immunologic and cardiovascular diseases. J. Leukoc. Biol. 2010, 88 (1), 41–55. [DOI] [PubMed] [Google Scholar]
- Cherney R. J.; Mo R.; Meyer D. T.; Voss M. E.; Lo Y. C.; Yang G.; Miller P. B.; Scherle P. A.; Tebben A. J.; Carter P. H.; Decicco C. P. Novel sulfone-containing di- and trisubstituted cyclohexanes as potent CC chemokine receptor 2 (CCR2) antagonists. Bioorg. Med. Chem. Lett. 2009, 19 (13), 3418–3422. [DOI] [PubMed] [Google Scholar]
- Carter P. H.; Brown G. D.; King S. R.; Voss M. E.; Tebben A. J.; Cherney R. J.; Mandlekar S.; Lo Y. C.; Yang G.; Miller P. B.; Scherle P. A.; Zhao Q.; Decicco C. P. Discovery of an orally-bioavailable CC chemokine receptor 2 antagonist derived from an acyclic diaminoalcohol backbone. Bioorg. Med. Chem. Lett. 2012, 22 (9), 3311–3316. [DOI] [PubMed] [Google Scholar]
- Carter P. H.; Tebben A. J. Chapter 12. The use of receptor homology modeling to facilitate the design of selective chemokine receptor antagonists. Methods Enzymol. 2009, 461, 249–279. [DOI] [PubMed] [Google Scholar]
- Cherney R. J.; Nelson D. J.; Lo Y. C.; Yang G.; Scherle P. A.; Jezak H.; Solomon K. A.; Carter P. H.; Decicco C. P. Synthesis and evaluation of cis-3,4-disubstituted piperidines as potent CC chemokine receptor 2 (CCR2) antagonists. Bioorg. Med. Chem. Lett. 2008, 18 (18), 5063–5065. [DOI] [PubMed] [Google Scholar]
- Carter P. H.; Brown G. D.; Friedrich S. R.; Cherney R. J.; Tebben A. J.; Lo Y. C.; Yang G.; Jezak H.; Solomon K. A.; Scherle P. A.; Decicco C. P. Capped diaminopropionamide-glycine dipeptides are inhibitors of CC chemokine receptor 2 (CCR2). Bioorg. Med. Chem. Lett. 2007, 17 (19), 5455–5461. [DOI] [PubMed] [Google Scholar]
- For the details on the synthesis and biological testing of compounds 3e–3j, see: Carter P. H. PCT Int. Appl WO 2005021499.
- Campbell C. L.; Hassler C.; Ko S. S.; Voss M. E.; Guaciaro M. A.; Carter P. H.; Cherney R. J. Enantioselective synthesis of benzyl (1S,2R,4R)-4-(tert-butoxycarbonylamino)-2-(hydroxymethyl)cyclohexylcarbamate using an iodolactam-ization as the key step. J. Org. Chem. 2009, 74 (16), 6368–6370. [DOI] [PubMed] [Google Scholar]
- For more synthetic details around the preparation of compounds 4a–4g, see: Carter P. H.; Cherney R. J.; Batt D. G.; Brown G. D.; Duncia J. V.; Gardner D. S.; Yang M. G. PCT Int. Appl. WO 2005020899.
- Cherney R. J.; Mo R.; Meyer D. T.; Voss M. E.; Yang M. G.; Santella J. B. 3rd; Duncia J. V.; Lo Y. C.; Yang G.; Miller P. B.; Scherle P. A.; Zhao Q.; Mandlekar S.; Cvijic M. E.; Barrish J. C.; Decicco C. P.; Carter P. H. gamma-Lactams as glycinamide replacements in cyclohexane-based CC chemokine receptor 2 (CCR2) antagonists. Bioorg. Med. Chem. Lett. 2010, 20 (8), 2425–2430. [DOI] [PubMed] [Google Scholar]
- Compound was administered orally to mice at a nominal concentration of 10 mg/kg. The area under the compound concentration curve from 0 to 4 h was normalized to the actual dose, as determined by measurement of the dosing solution. AUC/D = AUC(10/actual dose), where AUC has units of μM·h and the actual dose has units of mg/kg.
- Batt D. G.; Clark C. M.; et al. Carboxylic acid incorporation improves the selectivity of potent CC Chemokine Receptor 2 (CCR2) antagonists against inhibition of potassium and sodium ion channels. Bioorg. Med. Chem. Lett. To be submitted. [Google Scholar]
- Cherney R. J.; Mo R.; Yang M. G.; Xiao Z.; Zhao Q.; Mandlekar S.; Cvijic M. E.; Charo I. F.; Barrish J. C.; Decicco C. P.; Carter P. H. Alkylsulfone-containing trisubstituted cyclohexanes as potent and bioavailable chemokine receptor 2 (CCR2) antagonists. Bioorg. Med. Chem. Lett. 2014, 24 (7), 1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For more synthetic details around the preparation of compounds 12a and 13a–13h, see: Carter P. H.; Cherney R. J.; Batt D. G.; Duncia J. V.; Gardner D. S.; Ko S. S.; Srivastava A. S.; Yang M. G. PCT Int. Appl. WO 2005021500.
- Subasinghe N. L.; Lanter J.; Markotan T.; Opas E.; McKenney S.; Crysler C.; Hou C.; O’Neill J.; Johnson D.; Sui Z. A novel series of N-(azetidin-3-yl)-2-(heteroarylamino)-acetamide CCR2 antagonists. Bioorg. Med. Chem. Lett. 2013, 23 (4), 1063–1069. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.