

Abstract

The preclinical characterization of novel phenyl(piperazin-1-yl)methanones that are histamine H3 receptor antagonists is described. The compounds described are high affinity histamine H3 antagonists. Optimization of the physical properties of these histamine H3 antagonists led to the discovery of several promising lead compounds, and extensive preclinical profiling aided in the identification of compounds with optimal duration of action for wake promoting activity. This led to the discovery of two development candidates for Phase I and Phase II clinical trials.
Keywords: Histamine H3 antagonists, phenyl(piperazin-1-yl)methanones, histamine
Following the discovery that the histamine H3 receptor is abundantly expressed in the brain, numerous research groups have investigated the effects of modulation of the histamine H3 receptor on brain function. These efforts have demonstrated that histamine H3 receptors regulate the sleep-wake cycle and that histamine H3 antagonists promote wake and have potential utility for the treatment of a variety of indications, including excessive daytime sleepiness, narcolepsy, and attention deficit hyperactivity disorder (ADHD), among others.1 More recently, several pharmaceutical companies embarked on medicinal chemistry efforts to advance histamine H3 antagonists into the clinic for a variety of indications, and numerous clinical trials have been conducted over the past 5–10 years. The details of these efforts have been reviewed multiple times.1−6
Select examples of compounds that have been reported to advance into the clinic include the GSK compound GSK-189,254 (1), Pfizer’s PF-03654746 (2),7 A-331440 (3) from Abbott, and JNJ-39220675 (4).8 Several of these compounds have advanced into Phase II trials for therapeutic intervention in narcolepsy, ADHD, and allergic rhinitis. To date, the most advanced candidate appears to be pitolisant (5).9
Janssen’s efforts toward the preclinical characterization and clinical development of histamine H3 antagonists have been ongoing for quite some time. During this research a variety of potent histamine H3 antagonists were discovered, and several compounds advanced into preclinical toxicology studies. However, while advancing these compounds it was noted that some of the compounds induced varying degrees of phospholipidosis, a phenomenon that has also been noted by other researchers in the field.7,10

As it is well-known that phospholipidosis can be associated with cationic amphiphilic drugs, efforts to generate optimized histamine H3 antagonists with respect to physical properties and duration of action were pursued in order to advance compounds into the clinic devoid of phospholipidosis potential while maintaining a duration of action appropriate for a compound that promotes wakefulness. This report describes one aspect of the progress toward those goals with a series of phenyl(piperazin-1-yl)methanones.
These piperazine amides shown were synthesized by one of the two routes shown in Scheme 1. As such, 4-formylbenzoic acid (6) was combined with tert-butyl piperazine-1-carboxylate to form 7, which was then condensed with an amine to form 8. Removal of the protecting group followed by a reductive amination gave the desired compounds 11. Alternately, 4-formylbenzoic acid reacts with a substituted piperazine to directly install R1, and the resulting aldehyde 9 is converted to the desired compounds 11 via reductive amination. The required piperazines can be made from tert-butyl piperazine-1-carboxylate by known methods.
Scheme 1. Synthesis of Compounds 11.

Reagents and conditions: (a) (dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride, 1-hydroxybenzotriazole hydrate, N-methylmorpholine, tert-butyl piperazine-1-carboxylate, CH2Cl2; (b) R2R3NH, HOAc, NaBH(OAc)3, CH2Cl2; (c) trifluoroacetic acid, CH2Cl2; (d) various aldehydes, HOAc, NaBH(OAc)3, CH2Cl2; (e) (dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride, 1-hydroxybenzotriazole hydrate, various piperazines, CH2Cl2.
The initial set of structures that were prepared were high affinity compounds with human binding affinities of <100 nM (11a–11d, Table 1).11 The histamine H3 receptor affinity could be further improved by installing larger/more lipophilic substituents on the benzyl amine (i.e., 11e–g); however, efforts focused on the smallest/most polar groups that provided sufficient histamine H3 affinity to move forward. This work led to the preparation of the morpholine derivatives 11h–11j, all of which are high affinity human histamine H3 antagonists. In addition, since one focus of these efforts was to modify the physical properties of our leads, a few 4-fluoropiperidines were made (11k–11m) and were also found to be high affinity antagonists. The morpholines 11h–j and the 4-fluoropiperidines 11k–m were expected to have pKas lower than the piperidine 11g, and the 4-fluoropiperidines have the highest A log Ps as shown in Table 1. As two examples, comparing 11g to 11h (piperidine vs morpholine) installation of the morpholine lowers the A log P and the pKa of the benzyl nitrogen, whereas comparing 11h to 11k (piperidine vs 4-fluoropiperidine) the A log P is higher for the 4-fluoropiperidine and the calculated pKa of the benzyl nitrogen is slightly lower.12
Table 1. Binding Data for the Human H3 Receptor for Compounds 11.
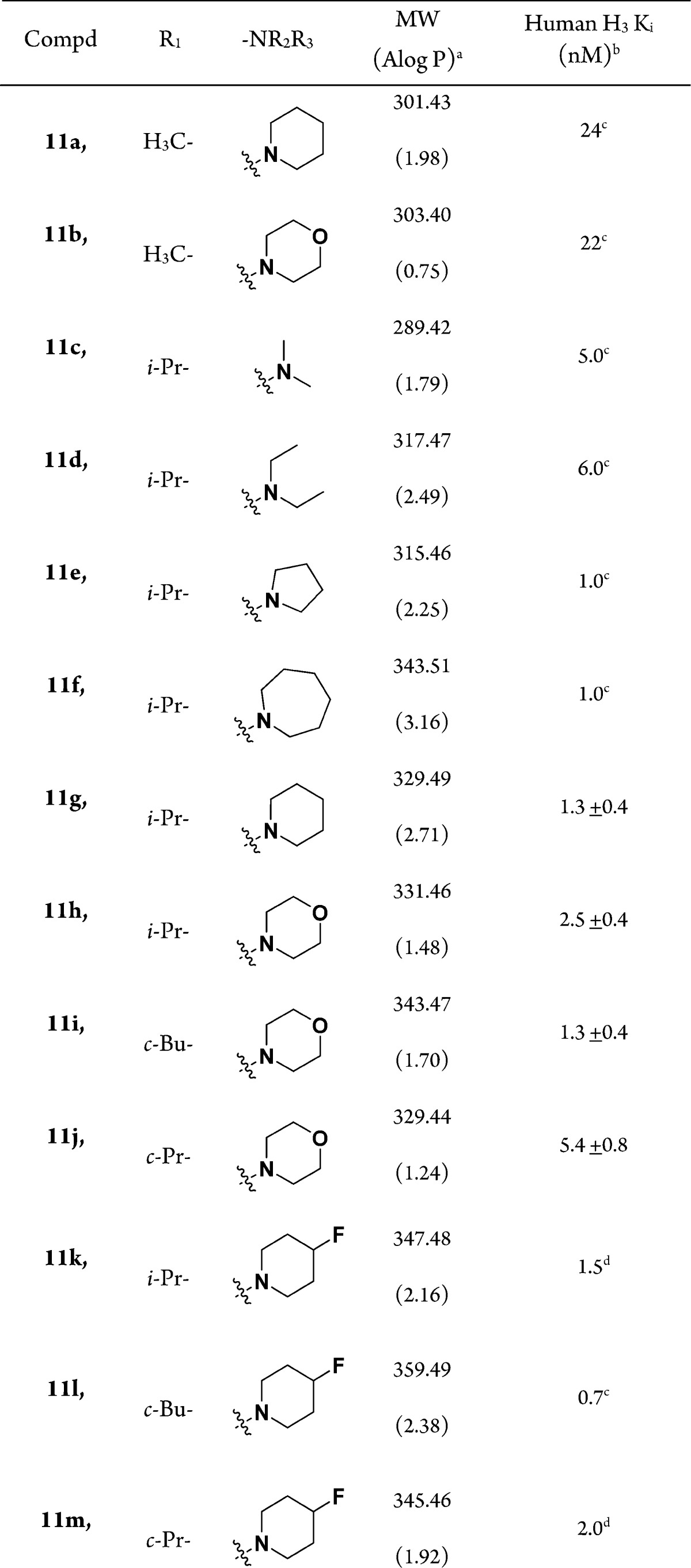
Calculated A log P.
Kis are the mean of at least three experiments in triplicate, unless otherwise stated.
n = 1 (in triplicate),
n = 2 (in triplicate); Ki ± SD is reported.
Next, focusing on the basicity of the piperazine nitrogen, several cyclopropyl and cyclobutyl piperazines were prepared and profiled. Again, high affinity compounds were obtained (11i–11j, 11l–11m), and again, these changes (i.e., isopropyl to cyclopropyl or cyclobutyl) were expected to modulate the pKa of the piperazine nitrogen to which these groups were attached.
Rat affinity data for a select group of the compounds in Table 1 as well as pA2 data in rat and human, which demonstrate that the key compounds in this series are all high affinity histamine H3 antagonists, are detailed in Table S1 in the Supporting Information.
Table 2 shows liver microsomal stability, hERG data, and Caco-2 data for some of the more interesting compounds that were prepared. The compounds have moderate to high liver microsomal stability, none have significant interactions with the hERG channel, and all compounds tested were highly permeable with low to moderate efflux potential. Consequently, these compounds were tested in vivo for oral exposure, duration of action, and target engagement as measured by ex vivo autoradiography.
Table 2. RLM and HLM Stability, and in Vivo Data for Compounds 11g–m.
| compd | RLM stabilitya | HLM stabilityb | hERGc | Caco-2d |
|---|---|---|---|---|
| 11g | n.d. | n.d. | >30 μM | 12.4/14.8 |
| 11h | n.d. | 65.0% | >10 μM | 22.4/20.9 |
| 11i | 49.6% | 85.4% | >10 μM | 71.2/22.4 |
| 11j | 44.1% | 96.1% | >10 μM | 108/37.8 |
| 11k | 50.0% | 84.8% | >10 μM | n.d. |
| 11l | n.d. | n.d. | >10 μM | n.d. |
| 11m | 26.8% | 83.6% | >10 μM | n.d. |
Stability in rat liver microsomes. Data reported as percent remaining after a 15 min incubation.
Stability in human liver microsomes. Data reported as percent remaining after a 15 min incubation.
hERG IC50 as measured in an [3H]-astemizole competition binding assay in HEK-293 cells expressing the hERG channel.
Data shown are Papp A–B/B–A (×10–6) cm/s.
In order to further demonstrate the specificity of these compounds, 11g–j were also tested in a commercial panel of 50 ion channel, receptor, and transporter assays (Eurofins-CEREP, http://www.eurofins.com/). None of the compounds had any significant affinity (at 1 μM) for any of these targets.
Table 3 shows rat PK parameters for compounds 11g–m. The i.v. t1/2s trend as expected. Comparing 11g with 11h, a change from piperidine to morpholine substantially reduced the half-life, and as expected, this appears to be driven primarily by a reduction in Vss, which can be attributed to a reduction in log P and pKa. Similarly, comparing 11h to 11j, the half-life is reduced as the expected pKa is reduced even further.
Table 3. Rat Pharmacokinetic Parameters for Compounds 11g–m.
| compd | Cl (mL/min/kg)a | Vss (L/kg)b | t1/2 (h)c | %Fd |
|---|---|---|---|---|
| 11g | 26.1 ± 5.3 | 39.4 ± 8.0 | 20.0 ± 4.7 | 52 ± 26 |
| 11h | 12.8 ± 5.5 | 3.6 ± 1.6 | 3.2 ± 0.1 | 62 ± 1 |
| 11i | 34.9 ± 12.9 | 6.4 ± 2.9 | 2.1 ± 0.1 | 85 ± 26 |
| 11j | 21.6 ± 1.8 | 1.7 ± 0.1 | 0.9 ± 0.1 | 112 ± 35 |
| 11k | 44.2 ± 12.5 | 29.2 ± 10.2 | 7.6 ± 1.1 | 17 ± 7 |
| 11l | 113.3 ± 13.6 | 20.3 ± 5.2 | 2.1 ± 0.6 | 31 ± 41 |
| 11m | 41.4 ± 17.1 | 3.9 ± 1.7 | 1.1 ± 0.2 | 14 ± 10 |
Clearance.
Volume of distribution at steady state.
i.v. half-life.
All data are mean ± SD.
The PK profile of both cyclobutyl compounds (11i and 11l) appears to be influenced by a combination of log P, pKa, and increased clearance, possibly due to oxidation of the cyclobutyl group. Similar trends are observed with the 4-fluoropiperidine compounds 11k–m.
The pKas, log Ps and log D7.4s of select compounds were calculated in order to confirm earlier estimations. A summary of this data is shown in Table 4.
Table 4. Physical Chemical Data for Compounds 11h–j.
| compd | pKa (calcd) | pKa (meas) | log P | log D7.4 |
|---|---|---|---|---|
| 11h | 8.5, 6.2 | 7.6, 6.2 | 0.55 | 0.13 |
| 11i | 7.8, 6.2 | 7.4, 6.5 | 2.09 | 1.77 |
| 11j | 8.5, 6.2 | 6.5, 5.3 | 0.89 | 0.86 |
The measured data confirmed the assumption that a cyclopropyl substituent would substantially reduce the pKa with little effect on log P, although as shown the calculated pKa did not predict this observation.13 There is at least one literature report that suggests that cyclopropyl amines are approximately ten times less basic than aliphatic amines;14 however no direct experimental evidence has been reported (although one reported calculation corroborates the literature suggestion).15 Illustrating the point, the measured pKa values for 11j were significantly lower than that predicted through calculation (using the Pallas software package from CompuDrug, Inc.) and also were significantly lower than that measured for 11h and 11i. The data here corroborate literature suggestions.
Next, key compounds were profiled for their ability to distribute into the rat brain and bind to the histamine H3 receptor. To do this, drug was administered and drug concentrations in the brain and plasma were measured along with histamine H3 receptor occupancy. These data are shown in Table 5. As shown, all the compounds were able to penetrate the brain and occupy the histamine H3 receptor.16 Distribution into the brain varied, but the brain concentrations measured for all compounds was high, and with the exception of compound 11m, all of the compounds in Table 5 showed good receptor occupancy.
Table 5. Drug Levels in Rat Brain and Plasma and Levels of ex Vivo H3 Receptor Occupancy for Compounds 11.
| compd | conc. (brain)a,c | R.O.b,c |
|---|---|---|
| 11g | 0.48 ± 0.14 | 90 ± 2 |
| 11h | 5.37 ± 1.09 | 86 ± 1 |
| 11i | 8.10 ± 0.53 | 96 ± 2 |
| 11j | 2.54 ± 0.36 | 58 ± 8 |
| 11k | 1.82 ± 0.51 | 92 ± 2 |
| 11m | 0.54 ± 0.14 | 25 ± 25 |
Concentration in μM at 1 h in brain following a 3 mg/kg p.o. dose.
Receptor Occupancy at 1 h Following a 3 mg/kp p.o. Dose.
Mean ± SD.
Considering the overall rat PK and distribution parameters observed, compounds 11g–k and 11m were all eventually chosen for further profiling. Toward that end, time course ex vivo H3 receptor occupancy data in rat was generated for these compounds and are shown in Figure 1. This data provides an indication of duration of action. Dose response related to receptor occupancy is shown in Figure S1 and PK parameters for canine and mouse are shown in Table S2 (detailed in the Supporting Information). All of the compounds show robust target engagement, and as expected based on the half-lives reported in Table 3, the compounds had varying duration of action. In addition, the PK trends in canine and mouse are similar to those observed in rat. As an example, focusing on a comparison of 11h and 11j, volume, clearance, and t1/2 were reduced in both cases for 11j, which results in a reduced half-life observed for 11j. Figure 1 compares compounds 11h and 11j demonstrating the shorter duration of action for the cyclopropyl compound 11j.
Figure 1.

Ex vivo H3 receptor occupancy with compounds 11g–k and 11m at 3 mg/kg in rat striatum: time dependency after oral administration.
Compounds 11a–11h were the first set of compounds to be prepared and profiled; of that set it appeared that 11h had the most desirable balance of potency, PK, target engagement, and duration of action. Therefore, this was the first compound in this series to be advanced into rat and dog safety toleration studies.
In order to support preclinical toleration studies and other first in human enabling studies, compound 11h was prepared on larger scale by a slightly different synthetic route than the one used to enable medicinal chemistry profiling. The synthesis is described in detail in the Supporting Information.
Compound 11h is a BCS Class I nonhygroscopic crystalline solid as the bis-maleate salt. It has a very high free fraction in human plasma (% bound 12.4%), and no CYP interactions were noted (IC50s all >40 μM at CYP 1A2, 2C9, 2C19, 2D6, and 3A4). Numerous additional in vitro and in vivo safety studies were completed in order to support potential first in human studies, and a summary of those studies is reported here.
In rats, after oral administration, 11h penetrates the blood–brain barrier and occupied histamine H3 receptor sites. As expected for this class of compound, administration of 11h in rats during the light sleep phase produced a significant increase in time spent awake (∼1 h) during the first 6 h post-treatment, and this was associated with a decrease in NREM sleep duration while REM sleep was not affected (Figure S2 in the Supporting Information). This occurred in the absence of any increase in locomotor activity and differentiates the compound from amphetamine like stimulants. Administration of 11h also increased acetylcholine (ACh) levels in the frontal cortex of freely moving rats and in a 7-trial passive avoidance model, 11h decreased the number of trials needed to increase the latency to enter the aversive chamber, consistent with pro-cognitive effects (data not shown).
Compound 11h had no effect in models predictive of cardiovascular toxicity, including hERG channel binding and models of QTc prolongation. Overall, the compound was well tolerated in the mouse, rat, and dog. Administration of up to 250 mg/kg of 11h orally to rats for up to 28 days induced mild and sporadic salivation and decreased activity. Minor decreases in body weight, food consumption, and clinical pathology alterations were considered related to 11h treatment but nonadverse. The rat NOAEL was 25 mg/kg/day. In dogs, administration of 11h for 5 days within the dose range of 10 to 75 mg/kg/day resulted in no adverse findings. These data supported progression to first in human studies. In human, the mean elimination half-life for the 2 and 5 mg dose groups of 11h was 49.1 and 56.1 h, respectively. Given these data, and that fact that significant insomnia was reported upon dose escalation, the team looked for back-up compounds with significantly reduced duration of action.
One such compound, 11j, appeared promising since this compound had significantly reduced duration of action in all preclinical species (Tables 3 and S2 (Supporting Information), and Figure 1). Compound 11j is also a BCS Class I nonhygroscopic crystalline solid as the bis-HCl salt. In human liver microsomes, 11j is not a potent inhibitor of the various CYPs. In rats, 11j penetrates the blood–brain barrier after systemic administration and effectively occupies histamine H3 receptor sites (>80% receptor occupancy at 10 mg/kg SC, corresponding to a maximum plasma concentration [Cmax] of 1933 ng/mL) (Figure S1, Supporting Information). Therefore, the anticipated plasma concentration for maximal receptor occupancy in humans was anticipated to be approximately 200 ng/mL as the potency of 11j is about 10-fold higher for the human receptor than for the rat receptor.
In the rat, the administration of 11j resulted in a significant increase in time spent awake, an increase in acetylcholine (ACh) levels in frontal cortex, and pro-cognitive effects in a 7-trial passive avoidance model.
Compound 11j was devoid of any cardiovascular liability in preclinical species including QTc prolongation. The compound (Bavisant) was also well tolerated in rats and dogs and the safety margins supported first in human dosing for this compound. No evidence of phospolipidosis was observed in these studies. Following single oral doses, 11j was rapidly absorbed with peak concentrations observed 0.5 to 4 h postdose. The Cmax increased dose-proportionally over 100 to 400 mg. The area under the concentration–time curve extrapolated to infinity (AUCinf) was dose-proportional between 10 and 30 mg and in the 100 to 400 mg dose range. The mean t1/2 ranged from 13.9 to 22.1 h. The reduced human t1/2 observed with 11j validates our assertion that reduced duration of action in human could be achieved by modification of the physical properties of this series of H3 antagonists.
In conclusion, we identified a series of novel, potent histamine H3 antagonists that demonstrated that improved human pharmacokinetic profiles can be obtained by tuning the physical properties of these ligands in order to reduce tissue retention time and t1/2 in vivo. Optimization of these ligands led to the discovery of Bavisant, a histamine H3 ligand useful for clinical investigation of the utility of histamine H3 antagonists.
Glossary
ABBREVIATIONS
- ADHD
attention deficit hyperactivity disorder
- i-Pr
isopropyl
- c-Pr
cyclopropyl
- c-Bu
cyclobutyl
- CYPs
cytochrome P450s
Supporting Information Available
Experimental details and analytical data for compounds 11a–m, additional Figures and Tables. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Esbenshade T. A.; Browman K. E.; Bitner R. S.; Strakhova M.; Cowart M. D.; Brioni J. D. The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br. J. Pharmacol. 2008, 154, 1166–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemkow M. J.; Davenport A. J.; Harich S.; Ellenbroek B. A.; Cesura A.; Hallett D. The histamine H3 receptor as a therapeutic drug target for CNS disorders. Drug Discovery Today 2009, 14, 509–515. [DOI] [PubMed] [Google Scholar]
- Sander K.; Kottke T.; Stark H. Histamine H3 receptor antagonists go to clinics. Biol. Pharm. Bull. 2008, 31 (12), 2163–2181. [DOI] [PubMed] [Google Scholar]
- Stocking E. M.; Letavic M. A. Histamine H3 antagonists as wake-promoting and pro-cognitive agents. Curr. Top. Med. Chem. 2008, 8 (11), 988–1002. [DOI] [PubMed] [Google Scholar]
- Singh M.; Jadhav H. R. Histamine H3 Receptor Function and Ligands: Recent Developments. Mini-Rev. Med. Chem. 2013, 13, 47–57. [PubMed] [Google Scholar]
- Szabadi E. Selective targets for arousal-modifying drugs: implications for the treatment of sleep disorders. Drug Discovery Today 2014, 19 (5), 701–708. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Pettersen b. A.; Schmidt A. W.; Spracklin D. K.; Mente S.; Butler T. W.; Howard H.; Lettiere D. J.; Rubitski D. M.; Wond D. F.; Nedza F. M.; Nelson F. R.; Rollema H.; Raggon J. W.; Aubrecht J.; Freeman J. K.; Marcek J. M.; Cianfrogna J.; Cook K. W.; James L. C.; Chatman L. A.; Iredale P. A.; Banker M. J.; Homiski M. L.; Munzere J. B.; Chandrasekaran R. Y. Discovery of two clinical histamine H3 receptor antagonists: trans-N-ethyl-3-fluoro-3-[3-fluoro-4-(pyrrolidinylmethyl)-phenyl]cyclobutanecarbox-amide (PF-03654746) and trans-3-fluoro-3-[3-fluoro-4-(pyrrolidin-1-ylmethyl)phenyl]-N-(2-methylpropyl)cyclo-butanecarboxamide (PF-03654764). J. Med. Chem. 2011, 54, 7602–7620. [DOI] [PubMed] [Google Scholar]
- Letavic M. A.; Aluisio L.; Atack J.; Bonaventure P.; Carruthers N. I.; Dugovic C.; Everson A.; Feinstein M. A.; Fraser I. C.; Hoey K.; Jiang X.; Keith J. M.; Koudriakova T.; Leung P.; Lord B.; Lovenberg T. W.; Ly K. S.; Morton K. L.; Motley S. T.; Nepomuceno D.; Rizzolio M.; Rynberg R.; Sepassi K.; Shelton J. E. Characterization of aryloxypyridine amides as histamine H3 receptor antagonists: Identification of candidates for clinical development. Bioorg. Med. Chem. Lett. 2010, 20, 4210–4214. [DOI] [PubMed] [Google Scholar]
- Dauvilliers Y.; Bassetti C.; Lammers G. J.; Arnulf I.; Mayer G.; Rodenbeck A.; Lehert P.; Ding C.-L.; Lecomte J.-M.; Schwartz J.-C. Pitolisant versus placebo or modafinil in patients with narcolepsy: a double-blind, randomized trial. Lancet Neurol. 2013, 12, 1068–1075. [DOI] [PubMed] [Google Scholar]
- Rodriguez Sarmiento R. M.; Nettekoven M. H.; Taylor S.; Plancher J.-M.; Richter H.; Roche O. Selective naphthalene H3 receptor inverse agonists with reduced potential to induce phospholipidosis and their quinoline analogs. Bioorg. Med. Chem. Lett. 2009, 19, 4495–4500. [DOI] [PubMed] [Google Scholar]
- Detailed procedures for the determination of affinity can be found in:Letavic M. A.; Keith J. M.; Ly K. S.; Bonaventure P.; Feinstein M. A.; Lord B.; Miller K. L.; Motley S. T.; Nepomuceno D.; Sutton S. W.; Carruthers N. I. 2-Aryloxymethylmorpholine histamine H3 antagonists. Bioorg. Med. Chem. Lett. 2008, 18, 5796–5799. [DOI] [PubMed] [Google Scholar]
- The calculated pKas for piperidine, morpholine, and 4-fluoropiperidine are 10.59, 9.31, and 7.16, respectively.
- Measured pKa, log P, and log D values were determined by pION, Inc. (Woburn, Mass).
- Zaragoza F.; Stephensen H.; Knuddsen S. M.; Pridal L.; Wulff B. S.; Rimvall K. 1-Alkyl-4-acylpiperazines as a new class of imidazole-free histamine H3 receptor antagonists. J. Med. Chem. 2004, 47 (11), 2833–2838. [DOI] [PubMed] [Google Scholar]
- Love P.; Cohen R. B.; Taft R. W. Polar substituent effects in gas-phase Lewis acid-base equilibriums. I. Intrinsic basicity of amines. J. Am. Chem. Soc. 1968, 90 (10), 2455–2462. [Google Scholar]
- The detailed procedures used to determine receptor occupancy can be found in:Barbier A. J.; Aluisio L.; Lord B.; Qu Y.; Wilson S. J.; Boggs J. D.; Bonaventure P.; Miller K.; Fraser I.; Dvorak L.; Pudiak C.; Dugovic C.; Shelton J.; Mazur C.; Letavic M. A.; Carruthers N. I.; Lovenberg T. W. Pharmacological characterization of JNJ-28583867, a histamine H3 receptor antagonist and serotonin reuptake inhibitor. Eur. J. Pharmacol. 2007, 675, 43–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.