

Abstract

Eighteen different bisphosphonates, including four clinically used bisphosphonate acids and their phosphoesters, were studied to evaluate how the bisphosphonate structure affects binding to bone. Bisphosphonates with weak bone affinity, such as clodronate, could not bind to hydroxyapatite after the addition of one ester group. Medronate retained its ability to bind after the addition of one ester group, and hydroxy-bisphosphonates could bind even after the addition of two ester groups. Thus, several bisphosphonate esters are clearly bone binding compounds. The following conclusions about bisphosphonate binding emerge: (1) a hydroxyl group in the geminal carbon takes part in the binding process and increases the bisphosphonate’s ability to bind to bone; (2) the bisphosphonate’s ability to bind decreases when the amount of ester groups increases; and (3) the location of the ester groups affects the bisphosphonate’s binding ability.
Keywords: Bisphosphonates, hydroxyapatite, nuclear magnetic resonance, etidronate, alendronate
Bisphosphonates (BPs) are synthetic molecules containing a P–C–P backbone (Table 1). They are stable analogues of naturally occurring pyrophosphate that contains a P–O–P backbone. When in the bloodstream, BPs are rapidly taken up by the bone tissue where they can prevent the resorption of bone.1 This rare ability to bind specifically to bone forms the basis of their clinical use. BPs have several medical applications: treatment of osteoporosis and other skeletal disorders and bone imaging when linked to a gamma-emitting radioisotope. In addition, BPs are used to reduce bone metastasis associated complications, such as bone pain, and to maintain and improve the bone health and mobility of cancer patients.2 BPs are proposed to have some direct anticancer activities as well.3 The problem with the use of BPs for targets outside of the skeletal system, however, is their high affinity for bone tissue. BPs are also highly ionized at physiological pH, which leads to their low oral bioavailability and poor cellular uptake in soft tissues.4
Table 1. Structures and in Vitro Stabilities of BP Esters from Different Publicationsa.
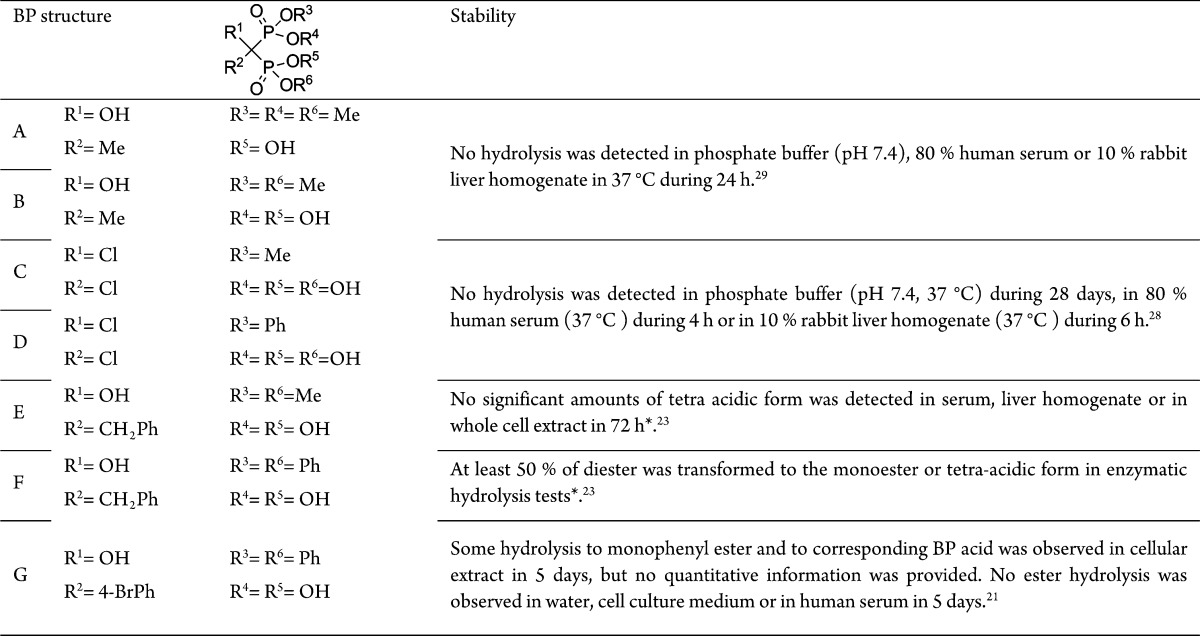
* = data not shown in the publication.
BPs’ affinity for bone depends highly on their structure.5 The binding is mainly mediated by the two phosphonate groups, but other properties are thought to contribute to the binding affinity as well, for example, a hydroxyl group as substituent R1 or a primary amine group in the substituent R2.6 Several different methods have been utilized to assess BPs’ affinity for hydroxyapatite (HAP) and bone powder.7 These methods include solid-state NMR spectroscopy,8 constant composition kinetic studies of crystal growth5,9 and fast performance liquid chromatography with HAP columns.10 Also, both 31P and 1H NMR spectroscopy11,12 have been used to determine BP binding to HAP. NMR has also been exploited to study how BPs bind to bone. For example, on the basis of their NMR investigations, Mukherjee et al. proposed that BPs could bind irrotationally to bone and displace orthophosphate from the bone mineral matrix.13
It has been suggested that BPs and their analogues with lower affinity for bone mineral could be useful in the treatment of skeletal14,15 and nonskeletal22 tumors. One obvious solution to reduce the bone affinity and increase the bioavailability of BPs is to introduce ester groups to BPs’ phosphonate groups to partly or fully mask the negative charge of BPs and to increase their lipophilicity. The structure of the ester groups has a significant influence to the metabolic stability of BP esters. Therefore, this class of compounds includes both prodrugs16−18 and metabolically stable compounds that are active as such. The prodrug strategy relies on the hydrolysis of the ester bonds and the release of free BP acids, which are known to have direct cellular effects. This strategy has been utilized in order to improve the oral bioavailability of BPs19 or to improve the cellular uptake of BPs in soft tissues.20
Both partially and fully esterified BPs have shown anticancer activity in in vitro and in vivo studies. For example, BP P,P′-phenylesters can inhibit the viability of hepatocarcinoma cells21 and the tumor growth, angiogenesis, and metastasis formation in nude mice with breast cancer cell xenografts.22 BP alkylesters also have displayed anticancer properties. A BP P,P′-dimethylester showed both antitumoral and antiangiogenic activity in nude mice with human epidermoid carcinoma xenografts.23 Interestingly, a BP tetraisopropylester, apomine, was found to inhibit the growth of several different tumor cell lines when used in micromolar concentrations.24 It reached an open-label, nonrandomized phase II trial in 2001 for the treatment of metastatic melanoma.25 Additionally, several other BP tetraethylesters have shown promising anticancer activity in vitro in studies with multiple tumor cell lines.26,27
The stability of different BP esters has been investigated in vitro to evaluate their resistance toward enzymatic hydrolysis (Table 1). Both BP alkyl- (A, B, and C) and phenylesters (D) are reported to be stable in human serum and in 10% rabbit liver homogenate.28,29 The stability of the aforementioned BP dimethylester (E) having both antitumor and antiangiogenic activity was investigated in vitro to evaluate its resistance toward enzymatic hydrolysis, but no significant amount of the corresponding BP acid was detected, though no data was shown to support this finding.23 This indicates the methylester to be stable against enzymatic hydrolysis and that it could induce anticancer mechanisms as such. It was also stated that when the enzymatic hydrolysis tests were conducted with the BP diphenylester (F), at least 50% of the diphenylester was hydrolyzed into its corresponding monophenylester or tetra acid form. Additionally, a BP P,P′-diphenylester (G) has been defined as a prodrug.21 It was suggested to be converted into the free BP acid by phosphodiesterases inside the cells. The prodrug proposal was supported by the fact that the addition of a known phosphodiesterase inhibitor reversed the BP ester’s effect on the cell viability. This suggests that the BP phenylesters might be more suitable when developing prodrug-like BPs, while the simple alkylesters seem to be stable against hydrolysis and should be considered as individual drugs.
However, BP tetraesters that contain a hydroxyl group as substituent R1 are determined to be unstable in solutions.30 They can undergo a rearrangement from the P–C(OH)–P structure into a P–C–O–P structure even at pH 7.4 in phosphate buffer.29 This prevents the use of these kinds of compounds in vitro or in vivo as such. Some metabolites of nonhydroxy BP tetraesters have been observed and identified from bile and urine samples collected from rats after an oral administration of the BP tetraesters.31,32 One of the metabolites in the first of these two studies was identified as the triester of the studied tetraethyl BP. This suggests that at least a partial hydrolysis of BP alkyl esters is possible in vivo, although it is still insufficient for them to be considered as prodrugs.
The BP esters can be prodrugs or active compounds since there are several possible cellular mechanisms they can act through. However, these compounds are rather stable in bloodstream, and therefore, their affinity for bone should be systemically studied. The bone affinity of BP esters has not been studied before, to our knowledge, although it can have a significant effect on the BPs’ distribution in the body. Therefore, we have studied 18 different BPs, including BP acids and their phosphoesters, to determine their ability to bind to bone. The aim of the study was to investigate if the different BP esters have any affinity for HAP and to clarify the structural requirements important for BP binding to bone.
Four clinically used BPs etidronate (1), medronate (8), clodronate (12), and alendronate (15) and their phosphoesters (Figure 1) were systematically studied to evaluate their bone binding properties. Additionally, we used diacetylated alendronate (18) to compare these changes with another type of potential BP prodrug. HAP was used as a synthetic representation of bone. The BPs’ binding to HAP was determined after 1 h of mixing with HAP. This seemed to represent a realistic time point for BPs’ binding to bone since it has been described in the literature that BPs’ half-life in plasma after intravenous administration is approximately 1–2 h.4 Statistical analysis was done by using the t test. Differences were considered statistically significant at P < 0.05.
Figure 1.

Structures of the BP esters and the corresponding BP acids used in this study.
The results of this study are collected in Figure 2. As expected, etidronate, medronate, and alendronate were bound to HAP to the greatest extent, and for all of them the amount of BP bound to HAP was at least 70%. Medronate’s bone binding abilities have not been extensively studied before since medronate is mainly used as a complexing agent with 99mTc in bone imaging.33 Clodronate is known to have poor affinity for HAP, and it had the lowest binding to HAP (41.9%) among the BP acids used in our study.
Figure 2.

HAP binding of BP acids and their esters. The amount of HAP-bound BP is stated in percentages. It is calculated from the concentration difference between BP solution before and after incubation with HAP.
In the case of etidronate, the size of the ester group had little effect on the HAP binding of the studied monoesters since etidronate monomethyl- and monophenyl-esters (2 and 3) were bound to HAP in similar amounts (55.6 and 48.0%). Also the asymmetric P,P-dimethylester and P,P-di-isopropylester (5 and 6) were binding in similar amounts (12.6 and 11.4%). The symmetric P,P′-dimethylester of etidronate (4) had low binding to HAP (3.8%), and the trimethylester was not bound to HAP at all. The asymmetric dimethylester had higher binding than the symmetric one, though the difference between the two diesters was not significant. There are several studies13 indicating that BPs are displacing orthophosphate, while they bind to HAP. Thus, as our results also show, one could expect that the mono- and asymmetric esters would have better HAP binding abilities than the symmetric one since one of the phosphonate groups is left free for binding in the asymmetric BP ester.
In the case of medronate, the monomethylester (9) was able to bind to HAP (18.6%), but the amount was lower when compared to etidronate and alendronate monomethylesters. This was expected since the hydroxyl group in the central carbon is known to increase the BP’s affinity for HAP.34 The di- and trimethylesters of medronate were not able to bind to HAP. For clodronate, the addition of one ester group was enough to prevent the binding to HAP completely (13 and 14).
For alendronate the acetylation of the hydroxyl group on the central carbon and the amine group (compound 18, 47.7%) had a smaller effect on the HAP binding than the addition of one methyl ester group (compound 16, 31.0%). The binding of the asymmetric dimethylester (compound 17, 21.7%) was lower when compared to the monomethylester, but the difference was not significant.
The amount of HAP bound BP should not be confused with the BPs’ affinity for HAP. It is likely that this kind of method represents a combination of affinity, the kinetic rate of the binding, and HAP’s different capacity for different BPs, rather than just the actual affinity.
To show the relative effect the ester groups have for the HAP binding of each studied BP, the binding of each BP ester was compared with the binding of the corresponding BP acid (Figure 3). The calculation was done by dividing the bound amount of each BP ester by the bound amount of the corresponding BP acid.
Figure 3.

HAP binding of different BP esters proportioned to the HAP binding of the corresponding BP acid.
One ester group appears to affect the HAP binding of etidronate less when compared to alendronate and medronate. For clodronate one ester group was enough to prevent the HAP binding completely. However, in the case of asymmetric diester, the two ester groups affect the HAP binding of alendronate less than etidronate. It is possible that the amino group’s effect on the HAP binding is highlighted when the phosphonate groups become more hindered as it is known that the amino group can participate in the HAP binding.5 This shows that the BPs’ ability to bind to HAP after the addition of ester moieties to the phosphonate groups depends highly on the original structure (the R1 and R2 groups) of the BP. Thus, the addition of one ester group prevented the binding of nonhydroxy BPs more when compared to hydroxy-BPs since the hydroxyl group is known to enhance the BPs’ affinity for HAP. Also, the binding to HAP was still possible for the hydroxy-BPs with two ester groups regardless of the size of the ester group. The acetylation of hydroxyl and amino groups of alendronate also caused a decrease in the HAP binding. Even though the decrease was not as big as the one caused by monomethylation, our results suggest that BP esters have to be considered as bone seeking compounds.
The results presented here can be utilized when designing new BPs with modified bone affinity, for example, BPs for cancer therapy or BP prodrugs.
Acknowledgments
Authors would like to thank Maritta Salminkoski for her skillful technical assistance.
Supporting Information Available
Experimental procedures and references regarding the synthesis of each compound used in the present study. This material is available free of charge via the Internet at http://pubs.acs.org.
Research has been supported by the strategic funding of the University of Eastern Finland and the Academy of Finland’s project 132070.
The authors declare no competing financial interest.
Supplementary Material
References
- Cremers S.; Papapoulos S. Pharmacology of bisphosphonates. Bone 2011, 49, 42–49. [DOI] [PubMed] [Google Scholar]
- Gralow J.; Tripathy D. Managing metastatic bone pain: the role of bisphosphonates. J. Pain Symptom Manage. 2007, 33, 462–472. [DOI] [PubMed] [Google Scholar]
- Clézardin P. Bisphosphonates’ antitumor activity: an unravelled side of a multifaceted drug class. Bone 2011, 48, 71–79. [DOI] [PubMed] [Google Scholar]
- Lin J. Bisphosphonates: a review of their pharmacokinetic properties. Bone 1996, 18, 75–85. [DOI] [PubMed] [Google Scholar]
- Nancollas G.; Tang R.; Phipps R.; Henneman Z.; Gulde S.; Wu W.; Mangood A.; Russell R.; Ebetino F. Novel insights into actions of bisphosphonates on bone: differences in interactions with hydroxyapatite. Bone 2006, 38, 617–627. [DOI] [PubMed] [Google Scholar]
- Ironside M. S.; Duer M. J.; Reid D. G.; Byard S. Bisphosphonate protonation states, conformations, and dynamics on bone mineral probed by solid-state NMR without isotope enrichment. Eur. J. Pharm. Biopharm. 2010, 76, 120–126. [DOI] [PubMed] [Google Scholar]
- Shinoda H.; Adamek G.; Felix R.; Fleisch H.; Schenk R.; Hagan P. Structure-activity relationships of various bisphosphonates. Calcif. Tissue Int. 1983, 35, 87–99. [DOI] [PubMed] [Google Scholar]
- Grossmann G.; Grossmann A.; Ohms G.; Breuer E.; Chen R.; Golomb G.; Cohen H.; Hägele G.; Classen R. Solid-state NMR of bisphosphonates adsorbed on hydroxyapatite. Magn. Reson. Chem. 2000, 38, 11–16. [Google Scholar]
- Henneman Z. J.; Nancollas G. H.; Ebetino F. H.; Russell R. G. G.; Phipps R. J. Bisphosphonate binding affinity as assessed by inhibition of carbonated apatite dissolution in vitro. J. Biomed. Mater. Res., Part A 2008, 85, 993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson M.; Xia Z.; Barnett B.; Triffitt J.; Phipps R.; Dunford J.; Locklin R.; Ebetino F.; Russell R. Differences between bisphosphonates in binding affinities for hydroxyapatite. J. Biomed. Mater. Res. B 2010, 92, 149–155. [DOI] [PubMed] [Google Scholar]
- Juillard A.; Falgayrac G.; Cortet B.; Vieillard M.; Azaroual N.; Hornez J.; Penel G. Molecular interactions between zoledronic acid and bone: an in vitro Raman microspectroscopic study. Bone 2010, 47, 895–904. [DOI] [PubMed] [Google Scholar]
- Jahnke W.; Henry C. An in vitro assay to measure targeted drug delivery to bone mineral. ChemMedChem 2010, 5, 770–776. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; Song Y.; Oldfield E. NMR investigations of the static and dynamic structures of bisphosphonates on human bone: a molecular model. J. Am. Chem. Soc. 2008, 130, 1264–1273. [DOI] [PubMed] [Google Scholar]
- Fournier P. G.; Daubine F.; Lundy M. W.; Rogers M. J.; Ebetino F. H.; Clezardin P. Lowering bone mineral affinity of bisphosphonates as a therapeutic strategy to optimize skeletal tumor growth inhibition in vivo. Cancer Res. 2008, 68, 8945–8953. [DOI] [PubMed] [Google Scholar]
- Coxon F. P. An update on the pharmacology of bisphosphonates and analogues with lower bone affinity. IBMS BoneKEy 2008, 5, 357–369. [Google Scholar]
- Vepsalainen J. J. Bisphosphonate prodrugs. Curr. Med. Chem. 2002, 9, 1201–1208. [DOI] [PubMed] [Google Scholar]
- Saari W.; Anderson P.. Acyloxymethyl esters of bisphosphonic acids as bone resorption inhibitors. European patent office, EP0416689 A2, 1991.
- Ebetino F. H.; Mazur A. W.; Dobson R. L. M.. Bisphosphonate compounds. U.S. Pat. Appl, US20110098251 A1, 2011.
- Niemi R.; Vepsäläinen J.; Taipale H.; Järvinen T. Bisphosphonate prodrugs: synthesis and in vitro evaluation of novel acyloxyalkyl esters of clodronic acid. J. Med. Chem. 1999, 42, 5053–5058. [DOI] [PubMed] [Google Scholar]
- Webster M. R.; Zhao M.; Rudek M. A.; Hann C. L.; Freel Meyers C. L. Bisphosphonamidate clodronate prodrug exhibits otent anticancer activity in non-small-cell lung cancer cells. J. Med. Chem. 2011, 54, 6647–6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteil M.; Migianu-Griffoni E.; Sainte-Catherine O.; Di Benedetto M.; Lecouvey M. Bisphosphonate prodrugs: Synthesis and biological evaluation in HuH7 hepatocarcinoma cells. Eur. J. Med. Chem. 2014, 77, 56–64. [DOI] [PubMed] [Google Scholar]
- Abdelkarim M.; Guenin E.; Sainte-Catherine O.; Vintonenko N.; Peyri N.; Perret G. Y.; Crepin M.; Khatib A.; Lecouvey M.; Di Benedetto M. New symmetrically esterified m-bromobenzyl non-aminobisphosphonates inhibited breast cancer growth and metastases. PloS One 2009, 4, e4685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux D.; Hamma-Kourbali Y.; Di Benedetto M.; Foucault-Bertaud A.; Oudar O.; Sainte-Catherine O.; Lecouvey M.; Kraemer M. A new dimethyl ester bisphosphonate inhibits angiogenesis and growth of human epidermoid carcinoma xenograft in nude mice. Anticancer Drugs 2006, 17, 479–485. [DOI] [PubMed] [Google Scholar]
- Flach J.; Antoni I.; Villemin P.; Bentzen C. L.; Niesor E. J. The mevalonate/isoprenoid pathway inhibitor apomine (SR-45023A) is antiproliferative and induces apoptosis similar to farnesol. Biochem. Biophys. Res. Commun. 2000, 270, 240–246. [DOI] [PubMed] [Google Scholar]
- Lewis K. D.; Thompson J. A.; Weber J. S.; Robinson W. A.; O’Day S.; Lutzky J.; Legha S. S.; Floret S.; Ruvuna F.; Gonzalez R. A phase II open-label trial of apomine (SR-45023A) in patients with refractory melanoma. Invest. New Drugs 2006, 24, 89–94. [DOI] [PubMed] [Google Scholar]
- Abdou W. M.; Barghash R. F.; Sediek A. A. Design of new arylamino-2-ethane-1, 1-diyl-and benzoxazole-2-methylene-bisphosphonates vs cytotoxicity and chronic inflammation diseases. From hydrophobicity prediction to synthesis and biological evaluation. Eur. J. Med. Chem. 2012, 57, 362–372. [DOI] [PubMed] [Google Scholar]
- Kamel A. A.; Geronikaki A.; Abdou W. M. Inhibitory effect of novel S,N-bisphosphonates on some carcinoma cell lines, osteoarthritis, and chronic inflammation. Eur. J. Med. Chem. 2012, 51, 239–249. [DOI] [PubMed] [Google Scholar]
- Niemi R.; Taipale H.; Ahlmark M.; Vepsäläinen J.; Järvinen T. Simultaneous determination of clodronate and its partial ester derivatives by ion-pair reversed-phase high-performance liquid chromatography coupled with evaporative light-scattering detection. J. Chromatogr. B 1997, 701, 97–102. [DOI] [PubMed] [Google Scholar]
- Niemi R.; Turhanen P.; Vepsäläinen J.; Taipale H.; Järvinen T. Bisphosphonate prodrugs: synthesis and in vitro evaluation of alkyl and acyloxymethyl esters of etidronic acid as bioreversible prodrugs of etidronate. Eur. J. Pharm. Sci. 2000, 11, 173–180. [DOI] [PubMed] [Google Scholar]
- Fitch S. J.; Moedritzer K. NMR study of the PC (OH)-P to PCOP rearrangement: Tetraethyl 1-hydroxyalkylidenediphosphonates. J. Am. Chem. Soc. 1962, 84, 1876–1879. [Google Scholar]
- Slatter J. G.; Feenstra K. L.; Hauer M. J.; Kloosterman D. A.; Parton A. H.; Sanders P. E.; Scott G.; Speed W. Metabolism of the bisphosphonate ester U-91502 in rats. Drug Metab. Dispos. 1996, 24, 65–73. [PubMed] [Google Scholar]
- Slatter J. G.; Feenstra K. L.; Hauer M. J.; Sanders P. E.; Vrbanac J. J.; Scott G.; Speed W. Metabolism of a bisphosphonate ester (PNU-91638) in rat. Xenobiotica 1997, 27, 1039–1051. [DOI] [PubMed] [Google Scholar]
- Pauwels E.; Blom J.; Camps J.; Hermans J.; Rijke A. A comparison between the diagnostic efficacy of 99mTc-MDP, 99mTc-DPD and 99mTc-HDP for the detection of bone metastases. Eur. J. Nucl. Med. 1983, 8, 118–122. [DOI] [PubMed] [Google Scholar]
- Russell R. G. G. Determinants of structure–function relationships among bisphosphonates. Bone 2007, 40, S21–S25. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.