

Summary
The flagellar motor switch complex protein FliG plays an essential role in flagella biosynthesis and motility. In most motile bacteria, only one fliG homologue is present in the genome. However, several spirochete species have two putative fliG genes (referred to as fliG1 and fliG2) and their roles in flagella assembly and motility remain unknown. In this report, the Lyme disease spirochete Borrelia burgdorferi was used as a genetic model to investigate the roles of these two fliG homologues. It was found that fliG2 encodes a typical motor switch complex protein that is required for the flagellation and motility of B. burgdorferi. In contrast, the function of fliG1 is quite unique. Disruption of fliG1 did not affect flagellation and the mutant was still motile but failed to translate in highly viscous media. GFP-fusion and motion tracking analyses revealed that FliG1 asymmetrically locates at one end of cells and the loss of fliG1 somehow impacted one bundle of flagella rotation. In addition, animal studies demonstrated that the fliG1− mutant was quickly cleared after inoculation into the murine host, which highlights the importance of the ability to swim in highly viscous media in the infectivity of B. burgdorferi and probably other pathogenic spirochetes.
Introduction
Spirochetes are a group of medically and ecologically important but poorly understood bacteria. Spirochetes can be classified into free-living, symbiotic and pathogenic groups based on their habitats and pathogenicity (Charon and Goldstein, 2002; Rosa et al., 2005). Pathogenic spirochetes can cause a variety of human and animal diseases, some of which are quite prevalent and can have grave consequences. For example, Lyme disease, caused by the spirochete Borrelia burgdorferi, is the most prevalent vector-borne disease in the USA (Steere et al., 2004; Rosa et al., 2005). Many Leptospira species cause leptospirosis. This potentially fatal water-borne zoonosis has many possible clinical manifestations and occurs worldwide (Levett, 2001; Mcbride et al., 2005). Treponema pallidum causes the dreaded sexually transmitted disease, syphilis, which is a major disease in developing countries worldwide (Weinstock et al., 1998; Chen et al., 2007). Brachyspira spp. can cause both human and animal gastrointestinal diseases (Mikosza and Hampson, 2001). The symbiotic spirochetes that dwell in the hind-guts of termites provide their insect host with essential nutrients via acetogenesis and nitrogen fixation, which is one of the most striking examples of the extraordinary biodiversity achieved by spirochetes (Leadbetter et al., 1999; Warnecke et al., 2007).
Spirochetes are very diverse but they constitute a monophlytic group of bacteria that share some common characteristics: unique helical or planar wave-like cell morphology and an unusual means of motility (Holt, 1978; Paster and Dewhirst, 2000). Spirochetes have a protoplasmic cell cylinder surrounded by an outer membrane sheath. In the periplasm, between the peptidoglycan layer and the outer membrane, are periplasmic flagella (PFs). In many spirochete species, PFs are long enough to overlap in the centre of the cell with those extending from the opposite ends. The number of PFs at each end varies from species to species (Charon and Goldstein, 2002). PFs are clearly the spirochetes’ organelles of motility since mutations that inhibited the synthesis of PFs resulted in non-motile phenotype (Motaleb et al., 2000; Li et al., 2008; Sal et al., 2008). Such unique cell morphology and structure give spirochetes an unusual means of motility: spirochetes can swim in a highly viscous, gel-like medium, such as that found in connective tissue, that inhibits the motility of most other motile bacteria (Greenberg and Canale-Parola, 1977a; Berg and Turner, 1979; Charon and Goldstein, 2002). In fact, the speed of spirochetes actually accelerates as viscosity increases. For example, the speed of Treponema denticola increases from less than 1 μm s−1 in liquid media to 19 μm s−1 in the presence of 1% methylcellulose (Ruby and Charon, 1998).
It is widely believed that this unique means of motility is essential for the pathogenesis of spirochetes, e.g. it may empower the spirochetes to penetrate into tissues, and help these organisms to escape from the innate immune response and rapidly disseminate in mammalian hosts (Kimsey and Spielman, 1990; Lux et al., 2000; Charon and Goldstein, 2002). Consistently, clinical evidence has shown that these pathogenic spirochetes are highly invasive in hosts. For example, B. burgdorferi can be recovered from the vitreous humour of the eye, brain tissues, endomyocardial tissue and other tissues of Lyme disease patients (Stanek et al., 1990; Kuiper et al., 1994; Nocton et al., 1996). Leptospira interrogans can be isolated from cerebrospinal fluid and the kidneys of patients (Brown et al., 2003; Palaniappan et al., 2007). However, due to the difficulty in obtaining the mutants that only fail to swim in highly viscous media, the biology about this unique means of motility and its role in the infections are poorly understood.
Among these pathogenic spirochetes, B. burgdorferi is one of the best understood spirochetes and one for which genetic tools have rapidly evolved in the past few years (Rosa et al., 2005). For example, genetic targeting mutagenesis mediated by allelic exchange and a whole genome transposon mutagenesis were successfully developed (Samuels et al., 1994; 1995; Stewart et al., 2004; Botkin et al., 2006). In addition, multiple antibiotic selection markers and a number of shuttle plasmids have been developed to inactivate genes, complement inactivated genes or express green fluorescent protein (GFP) and luciferase in B. burgdorferi (Eggers et al., 2002; Elias et al., 2003; Blevins et al., 2007). Most importantly, animal models of Lyme disease have been well established and extensively used to study the pathogenesis of B. burgdorferi (Masuzawa et al., 1992; Barthold, 1995). Theses advantages make this spirochete an ideal model to study the unique means of motility and its role in the process of disease.
The genome of B. burgdorferi encodes 59 motility and chemotaxis genes, which constitutes approximately 5% of its total genes (Fraser et al., 1997; Li et al., 2000; Charon and Goldstein, 2002). In our recent studies, several genes involved in motility and chemotaxis have been studied, which include two flagella genes (flaB and flgE) and three chemotaxis genes (cheA1, cheA2 and cheX). The results show that the flaB and flgE genes are essential for the flagella synthesis and motility, and the cheA2 and cheX genes are critical for the chemotaxis of B. burgdorferi (Motaleb et al., 2000; 2005; Li et al., 2002; Sal et al., 2008). Following previous studies, more motility and chemotaxis genes have recently been inactivated. Among these genes, fliG, a gene encoding a flagellar motor switch complex protein, plays an essential role in flagella assembly and motility in enteric bacteria and other flagellated bacteria (Lloyd et al., 1996). For instance, the fliG null mutants of Escherichia coli and Salmonella enterica are aflagellated and non-motile. In most motile bacteria, only one fliG is present in their genomes. However, in several spirochete species including B. burgdorferi, there are two fliG homologues (referred to as fliG1 and fliG2). The functions of these two fliG genes remain unknown. In this report, these two genes were inactivated and their roles in flagella assembly and motility were evaluated. In addition, the role of motility in the virulence of B. burgdorferi was investigated by analysis of the fliG1 mutant in the mouse model of Lyme disease.
Results
Sequence alignment reveals that FliG2, but not FliG1, is a typical motor switch complex protein
As mentioned above, FliG proteins play a key role in bacterial flagella biosynthesis and motility (Irikura et al., 1993; Lloyd et al., 1996). In most flagellated bacteria, there is only one fliG gene. However, there are two fliG genes in the sequenced spirochete genomes. In B. burgdorferi, fliG1 (BB0221) and fliG2 (BB0290) encode a 405-amino-acid protein with a predicted molecular weight of 47.5 kDa and a 344-amino-acid protein with a predicted molecular weight of 34.3 kDa, respectively (Fraser et al., 1997). BLAST analysis showed that FliG2 shares a 32% identity and a 55% similarity to E. coli FliG, and FliG1 shares a 17% identity and a 45% similarity to that of E. coli. Prior to this study, it was unknown whether these two genes had redundant or unique functions in flagella assembly and motility.
Previous functional and structural analyses have shown that the C-terminal sequences of FliG proteins are extremely conserved and contain 20 key residues that are required for their functions or structures (Lloyd et al., 1999). To reveal the potential roles of FliG1 and FliG2, the C-terminal sequences of these two proteins were aligned with their counterparts from other bacteria. As shown in Fig. 1, the C-terminus of FliG2 is well conserved, and it contains all key residues. In contrast, although the C-terminus of FliG1 shares certain identity to its counterparts, it lacks seven key residues. This comparison suggests that FliG2 is a typical motor switch protein that may have the similar function as its counterparts, whereas FliG1 is an atypical motor switch protein and its function may be unique.
Fig. 1.

Multiple sequence alignment of the C-terminus of FliG proteins. The numbers show the positions of amino acids of E. coli and Salmonella typhimurium FliG proteins, and diamonds represent the key residues required for the function and structure of FliG proteins. GenBank accession numbers for the aligned proteins are: S. typhimurium FliG (NP_456531), E. coli FliG (NP_310705), B. burgdorferi FliG2 (NP_212424), T. pallidum FliG2 (NP_218840), Bacillus subtilis FliG (NP_389504), B. burgdorferi FliG1 (ZP_03086805) and T. pallidum FliG1 (NP_218466). The alignments were performed using the program MacVector 10.6.
Generations of fliG1− and fliG2− mutants
To disclose the roles of fliG1 and fliG2, these two genes were inactivated by targeted mutagenesis as described in Fig. 2. For fliG1, nine kanamycin-resistant colonies appeared 17 days after plating, and for fliG2, 11 kanamycin-resistant colonies appeared 24 days after plating. The time required for the appearance of these two mutant colonies is considerably longer than the time required for the appearance of colonies of the wild type (10–14 days).A similar phenotype was observed in the flaB and flgE mutants of B. burgdorferi (Motaleb et al., 2000; Sal et al., 2008). For each mutant, one of clones, which was, respectively, referred to as fliG1− or fliG2−, was selected for detailed characterization. A PCR analysis described before (Motaleb et al., 2000; Sal et al., 2008) showed the kan-resistant cassette was inserted within and transcribed as the same direction of fliG1 or fliG2 as expected (data not shown). Western blot analysis with an antiserum against FliG1 or FliG2 further confirmed that the cognate gene products were, respectively, inhibited in the mutants. As shown in Fig. 3B and C, a single band of 48 kDa or 35 kDa protein product was detected in the wild type, but they were absent in fliG1− or fliG2−. Taken together, these results demonstrated that fliG1 and fliG2 were expressed and the cognate gene product was disrupted in the respective mutant due to the targeted mutagenesis.
Fig. 2. Constructing plasmids for the inactivation of fliG1 and fliG2 genes and the complementation of fliG1− mutant.
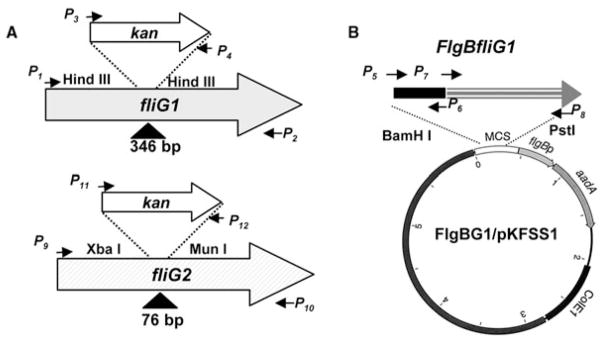
A. The constructs to inactivate fliG1 and fliG2 genes.
B. The plasmid FlgBG1/pKFSS1 was used to complement the fliG1− mutant in trans.
Arrows indicate the relative positions of PCR primers for constructing these plasmids, and the sequences of these primers were listed in Table 4; the symbols ‘Δ’ show the DNA fragments deleted from the genes, and the numbers are the sizes of deleted DNA fragments.
Fig. 3. Western blot analysis of the fliG mutants and their complemented strains.

A. Flagella filament protein FlaA and FlaB are absent in the fliG2−mutant, but still present in the fliG1− mutant.
B and C. The cognate gene products were absent in the respective mutants and were restored in the complemented strains.
FliG2, but not FliG1, is essential for flagella synthesis
In enteric bacteria, the fliG genes are essential for flagella assembly and motility. The null fliG mutants are aflagellated and non-motile. To detect the influence of fliG1 and fliG2 on the synthesis of PFs, the mutants were analysed by electron microscopy. In the micrographs of outer membrane disrupted cells, two bundles of PFs were observed at two ends of the wild-type and fliG1− cells. However, such structures were absent in the fliG2− cells (Fig. 4, top panel). In the micrographs of thin sections, there were 9–11 PFs present in the periplasm of the wild type and fliG1−. In contrast, the fliG2− mutant completely lacked PFs (Fig. 4, low panel). Western blot further confirmed that two flagellar filament proteins FlaA and FlaB were absent in fliG2− but present in fliG1− (Fig. 3A). These results demonstrated that FliG2 is a typical motor switch protein that is essential for the flagella synthesis. In contrast, FliG1 was shown as an atypical motor switch protein since it was not critical for the flagella synthesis of B. burgdorferi.
Fig. 4.

Electron microscopic analyses of the fliG1− and the fliG2− mutants. The upper panels are the electron micrographs of outer membrane-disrupted B. burgdorferi cells, and only one end of the cells was illustrated; the lower panel is the thin-section electron micrographs of B. burgdorferi cells. Arrows point to the PFs.
The fliG2− mutant is totally non-motile whereas the fliG1− mutant only fails to translate in a highly viscous medium
The impact of fliG1 and fliG2 on the motility of B. burgdorferi was evaluated by different methods, which include microscopic observation, swarm plate assay and bacterial tracking analysis (Bakker et al., 2006; Motaleb et al., 2007; Li et al., 2008; Sal et al., 2008). Dark-field microscopic analysis revealed that the fliG2− mutant cells were rod-shaped and often grew in chains, and were completely non-motile (data not shown). The observed phenotype is the same as previously reported B. burgdorferi aflagellated mutants (Motaleb et al., 2000; Sal et al., 2008). In contrast to the phenotype of fliG2−, the fliG1−mutant did not have significant changes in terms of cell morphology and swimming behaviour: the mutant still had a flat-wave morphology (data not shown) and the mutant was still motile in liquid BSK-II medium (Goldstein et al., 1994; Li et al., 2002).
Swarm plate assays further confirmed that fliG2− is non-motile. Interestingly, the swarm plate assays revealed that the motility of fliG1− was partially decreased, and the complemented strain fliG1−/+ restored the full motility as the wild type (Fig. 5), indicating that inactivation of fliG1 somehow reduced the ability of the mutant to swim on soft agarose plates. To quantitatively measure the motility, the mutants were analysed by a previously developed computer-based bacterial tracking system (Li et al., 2002; Bakker et al., 2006). Previous studies have revealed that spirochetes, including B. burgdorferi, do not translate very well in low viscous media such as BSK-II, and their swimming ability (velocity) is accelerated in highly viscous media such as 1% methylcellulose. As expected, both the wild-type and the fliG1− mutant cells were unable to efficiently displace in BSK-II medium and their velocities were less than 1 μm s−1 (Table 1). In 1% methylcellulose medium, the velocity of wild-type cells (Video S1) increased to approximately 10 μm s−1 whereas the fliG1−mutant completely failed to displace (Video S2, and Table 1). Taken together, these results showed that the inactivation of fliG1 somehow abrogated the ability of spirochete cells to displace in a highly viscous medium.
Fig. 5.

Swarm plate assays. The swarm plate assays were carried out on 0.35% agarose with 1:10 diluted BSK-II medium as previously described (Li et al., 2002; Sal et al., 2008). Diameters of swarms (centimetres) after 72 h of incubation: wild-type 2.1; fliG1−, 1.2; fliG1−/+, 2.0; fliG2−, 0.9.
Table 1.
Measuring the velocity of B. burgdorferi strains.
| Strains | Velocity (μm s−1)
|
|
|---|---|---|
| BSK-II | 1% methylcellulose | |
| B31A | Motilea | 10.2 ± 2.2 |
| fliG1− | Motile | No translational motility |
| fliG1−/+ | Motile | 9.3 ± 3.9 |
| fliG2− | Non-motile | Non-motile |
Velocity is less than 1 μm s−1. The numbers are the average velocity calculated from at least 20 cells.
One end of the fliG1− mutant cells failed to gyrate
Previous studies have shown that two bundles of PFs, one at each end of the spirochete cells, can rotate and generate torque that leads to swimming. The co-ordinated rotation of two bundles of PFs is essential for the motility of spirochetes (Goldstein et al., 1994; Charon and Goldstein, 2002; Li et al., 2002). To further disclose the mechanism involved in the above observed phenotype, the fliG1− mutant cells in BSK-II medium were videotaped under high-magnification dark-field microscopy (100×) as previously described (Li et al., 2002). This method allows us to closely observe the motion at both ends of the cells. As shown in Video S3, for the wild-type cells, both ends could actively rotate and took turns being a leading end. However, for the fliG1− mutant cells, only one end was actively rotating and the other end failed to be a leading end (Video S4), suggesting that the flagella motors at one end of the cells may be paralysed due to the inactivation of fliG1.
FliG1 locates at one end whereas FliG2 resides at two ends of B. burgdorferi cells
The above studies showed that the inactivation of fliG1 only altered the rotation of PFs at one end of cells, implying that the FliG1 protein may locate at one end of the cells. To address this speculation, GFP-fusion technique was applied to determine the cellular location of FliG1. A vector that expresses GFP–FliG1 fusion protein was constructed (Fig. 6A). In our previous studies, the strength of different promoters identified in B. burgdorferi was evaluated by GFP reporter assay, and we found that the flgB promoter could control the expression of GFP at moderate levels (Yang and Li, 2009). As shown in Fig. 6B, the GFP–FliG1 fusion protein was successfully expressed in the fliG1− mutant. Some degraded products were detected by monoclonal GFP antibody, which is commonly observed in this study (Sourjik and Berg, 2000). However, such degraded products could not be detected using FliG1 antiserum as a probe (data not shown), indicating that the degradation occurred at the portion of GFP. In addition, the strain expressing GFP–FliG1 restored the ability to swim in 1% methylcellulose, indicating GFP–FliG1 was able to substitute the function of FliG1 (data not shown). Under fluorescent microscopy, we found that GFP–FliG1 only located at one end of the cells (Fig. 6D). This pattern was not observed in the control that only expresses GFP (Fig. 6C). This result indicated that FliG1 asymmetrically locates at one end of B. burgdorferi cells.
Fig. 6. Localization of FliG1 in B. burgdorferi.

A. Constructing a vector that expresses the N-terminal GFP-fusion proteins. The flgB promoter element was fused to gfp with a five-glycine linker, and SphI cloning site was engineered at the 3′ end of gfp. A target gene, such as fliG1, will be fused to gfp at the SphI site. A T7 transcription terminator was inserted.
B. Detection of GFP–FliG1 fusion protein using Western blot. A monoclonal antibody against GFP (Invitrogen) was used to detect GFP and GFP–FliG1. GFP is about 27 kDa and GFP–FliG1 is about 75 kDa.
C. The fluorescence micrograph (40×) of the fliG1 mutant that was transformed with the plasmid pFBgfp/T7t, which only expresses GFP.
D. The fluorescence micrograph (40×) of the fliG1 mutant that was transformed with the plasmid pFBgfp–fliG1/T7t, which expresses GFP–FliG1 fusion protein. The brighter ends represent the location of GFP–FliG1.
The same method was used to determine the cellular location of FliG2. However, neither FliG2 nor GFP–FliG2 could complement fliG2− (data not shown). Immunofluorescence microscopy has been extensively applied to investigate protein cellular locations, including bacterial flagella and chemotaxis proteins (Sourjik and Berg, 2000). As such, this method was conducted to localize FliG2. For this assay, we used a primary antibody specifically reacted with FliG2 and a secondary antibody coupled with Texas red. As shown in Fig. 7, brightest spots were observed at the two poles of the wild-type cells but not in the fliG2− mutant, demonstrating that FliG2 symmetrically resides at both ends of B. burgdorferi cells.
Fig. 7.
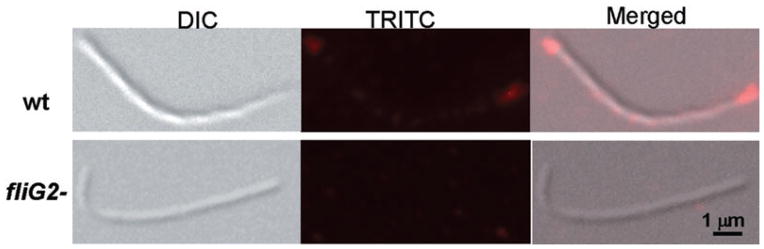
Localization of FliG2 in B. burgdorferi by immunofluorescence microscopy. The wild-type and the fliG2− mutant cells were fixed with methanol, stained with anti-FliG2 and counterstained with anti-rat Texas red-coupled antibody, as described in Experimental procedures. Photo (DIC) and fluorescent (TRITC) micrographs (100×) were, respectively, taken under a Zeiss Axioimager Z1 Axiophot microscope, and merged. For each strain, more than 50 cells were examined.
The fliG1− mutant is unable to survive in the mammalian host
The motility of spirochetes is accelerated in highly viscous media such as connective tissues that either slow down or stop most externally flagellated bacteria (Greenberg and Canale-Parola, 1977a,b; Charon and Goldstein, 2002). It is believed that such ability is essential for the infections of spirochetes (Kimsey and Spielman, 1990; Lux et al., 2000). Above studies showed that the fliG1− mutant failed to swim in a highly viscous medium. Thus, this mutant can be used to test whether the ability to swim in highly viscous media is essential for the infection. However, the above mutant was constructed in an avirulent background, and cannot be tested in animal studies. To solve this problem, the mutant was re-constructed in B31-A3, a low passage virulent strain (Elias et al., 2002; Grimm et al., 2004). Three positive clones were obtained, which were confirmed by PCR and Western blot analyses as described above (data not shown), and two of them had the full plasmid content of B31-A3. One clone, vfliG1−, was further complemented. Although 19 transformants were obtained, only one clone, vfliG1−/+, had the full plasmid content as its parental strain, B31-A3 (Fig. S1). Microscopic observation, swarm plate assay and bacterial tracking analysis confirmed that these two newly constructed strains exhibited the similar phenotype as their counterparts derived from the B31A strain (data not shown).
The wild-type B31-A3, the mutant vfliG1− and the complemented strain vfliG1−/+ were inoculated into immunocompetent BALB/c mice. Four weeks later, heart, tibiotarsal joint and skin specimens were aseptically collected for spirochete culture. The presence of spirochete cells were used to evaluate the infectivity of these three strains. As shown in Table 2, the wild-type and the complemented strain infected all inoculated mice. In contrast, the mutant could not be recovered from any tissue of inoculated mice, indicating that the ability to swim in highly viscous media is essential for mammalian infection.
Table 2.
The vfliG1− mutant was unable to infect mice.a
| Clone | No. of cultures positive/Total no. of specimens examined
|
|||
|---|---|---|---|---|
| Heart | Joint | Skin | All sites | |
| B31-A3 | 5/5 | 5/5 | 5/5 | 15/15 |
| vfliG1 −/+ | 5/5 | 5/5 | 5/5 | 15/15 |
| vfliG1− | 0/5 | 0/5 | 0/5 | 0/15 |
Groups of five BALB/c mice were inoculated with 104 spirochetes of the B31-A3, vfliG1−/+ or vfliG1− strains. Mice were sacrificed 4 weeks post inoculation; heart, tibiotarsal joint and skin specimens were harvested for spirochete culture in BSK-II medium.
After being deposited in skin following tick bites, B. burgdorferi must traverse the intercellular matrix, penetrate the vascular endothelial cell lining and become haematogenous (Kimsey and Spielman, 1990; Charon and Goldstein, 2002). It is believed that the ability to swim in highly viscous media, such as connective tissues, facilitates the accomplishment of the above process and consequently helps the spirochete to escape from host innate immunity and establish systematic infection. Thus, the failure of the vfliG1− mutant to establish a systemic infection in immunocompetent mice could be due to the loss of motility, which may be required for dissemination and/or for evasion of both innate and adaptive immune responses. To investigate these issues, the three strains were inoculated into SCID mice. As shown in Table 3, the vfliG1− mutant was cleared from all of the inoculation sites within 24 h. In contrast, the wild-type B31-A3 and vfliG1−/+ were recovered from each of the inoculation sites at all time points, indicating that the motility is required for the basic survival of B. burgdorferi in the murine host.
Table 3.
The vfliG1− mutant was quickly cleared from murine skin.a
| Clone | No. of sites positive/Total no. of sites examined at post-inoculation hours
|
||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| B31-A3 | 6/6 | 6/6 | 6/6 |
| vfliG1−/+ | 6/6 | 6/6 | 6/6 |
| vfliG1− | 0/6 | 0/6 | 0/6 |
Groups of nine SCID mice each received two intradermal/ subcutaneous injections of the B31-A3, vfliG1−/+ or vfliG1− strains. Approximately 104 organisms were administered in each inoculation; two inoculation sites were at least 2 cm apart. Three animals from each group were euthanized at 24, 48 and 72 h post inoculation; skin specimens were harvested from inoculation sites and cultured for spirochetes in BSK-II medium.
Discussion
The flagellar motor switch protein FliG plays a very important role in the flagella biosynthesis and motility (Irikura et al., 1993; Lloyd et al., 1996). The FliG proteins are well conserved among different bacteria and their functions and structures have been well studied in the enteric bacteria such as E. coli (Lloyd et al., 1999; Brown et al., 2002). Except for spirochetes, only one copy of the fliG gene has been identified in most sequenced bacterial genomes. Interestingly, there are two homologues of fliG genes in the sequenced spirochete genomes such as T. denticola, T. pallidum and L. interrogans (Fraser et al., 1997; 1998; Ren et al., 2003; Seshadri et al., 2004). The functions of these two fliG genes remain unknown. In this report, different approaches have been applied to elucidate the functions of these two FliG proteins in B. burgdorferi. First, the sequence alignment showed that FliG2 shares higher identity (32%) with its counterparts from the enteric bacteria than FliG1 (17%); FliG2 contains all the key residues that are required for its function and structure, but FliG1 lacks seven key residues (Fig. 1). Second, the genetic studies demonstrated that the fliG2−mutant shares the similar phenotype as its counterparts of the enteric bacteria: the mutant is aflagellated and non-motile (Irikura et al., 1993). In contrast, the phenotype of the fliG1− mutant is quite different: the mutant still synthesized flagella and only failed to translate in highly viscous medium such as 1% methylcellulose (Figs 3–5 and Table 1). Taken together, these studies indicated that FliG2 is a typical motor switch protein that is essential for the flagella synthesis and motility, and FliG1 is an atypical motor switch protein since it is not required for flagella synthesis and the loss of fliG1 only depleted the ability of the spirochete to swim in highly viscous media.
Flagella patterns (e.g. the number, length and cellular location of flagella) vary among different bacterial species (Aldridge and Hughes, 2002; Chevance and Hughes, 2008). For instance, Vibrio cholera has a single flagellum at one end of the cell, and Pseudomonas aeruginosa has a single flagellum at both ends. In contrast, enteric bacteria such as E. coli have multiple flagella that randomly locate on the cell surface. These patterns are essential for motility, e.g. alternations of number, location and length of flagella can either change swimming behaviours or totally disrupt motility (Klose and Mekalanos, 1998; Dasgupta et al., 2000; Correa et al., 2005; Murray and Kazmierczak, 2006). In this regard, the failure to swim in highly viscous medium can be due to the alternation of flagellum pattern in the fliG1− mutant, e.g. the number, length and location of PFs. However, the electron microscopic analysis revealed that the fliG1− mutant shares the same pattern as the wild type with respect to the number, length and location of PFs (Fig. 4), implying the failure to swim in highly viscous medium is most likely caused by other factors such as flagellar motors.
Borrelia burgdorferi cells have two swimming behaviours: run and flex (Li et al., 2002; Motaleb et al., 2005; Bakker et al., 2006). During run (translational motility), the two bundles of PFs have to be rotating asymmetrically: the leading end rotates counterclockwise (CCW), while the other end simultaneously rotates clockwise (CW). Cells would flex or tumble if both bundles of PFs rotate in the same direction. The leading end can switch from one end to the other when cells reverse (Video S3). Motion analysis demonstrated that the swimming behaviour of the fliG1− mutant is quite unique: one end of cells constantly gyrated and often switched direction, whereas the other end failed to gyrate (Video S4). The gyration is generated by the rotation of PFs, which is propelled by the flagellar motors. Thus, the failure of gyration is most likely caused by the dysfunction of the flagellar motors at the end with FliG1. Consistently, FliG1 was found to asymmetrically locate at one end of the cells. Previous studies indicate that both bundles of PFs rotate during the translational motility. Therefore, in the fliG1− mutant, the torque generated by one bundle of PFs may be insufficient to propel the cells to displace in highly viscous media such as 1% methylcellulose. However, the molecular mechanism involved still remains unknown. We are currently applying different approaches to elucidate the role of FliG1 in the flagellar motors. For example, cryo-EM has recently been applied to study B. burgdorferi cell and motor structures (Charon et al., 2009; Liu et al., 2009). This approach can be utilized to determine the location of FliG1 in the flagella motors. Co-immunoprecipitation and affinity blot analysis can be conducted to investigate the molecular partners of FliG1, e.g. the protein(s) that interacts with FliG1 (Toker and Macnab, 1997). The information obtained from these assays can help us to further disclose the molecular mechanism involved in the function of FliG1.
In enteric bacteria, flagella randomly locate on cell surface. Consistently, flagella motor switch complex proteins such as FliM and FliG appear as discrete spots along the sides of the cells (Sourjik and Berg, 2000). However, in spirochetes, two bundles of PFs are attached subterminally to the ends of the cells. Since the motors are associated with flagella, it is believed that flagella motor complex proteins locate at the cell poles (Charon and Goldstein, 2002; Li et al., 2002). In this report, immunofluorescence analysis demonstrated that FliG2 locates at the two ends of the cells, which is consistent with the polar location of PFs in spirochete cells. In contrast to FliG2, the asymmetrical location of FliG1 is quite unique. Asymmetrical localization of specific proteins and structures have been discovered in several bacterial species (Shapiro and Losick, 2000; Lybarger and Maddock, 2001; Thanbichler and Shapiro, 2008). For instance, the stalk structure localizes at one end of the cell in Caulobacter crescentus at a site previously occupied by the flagellum. The ActA protein of Listeria monocytogenes and the IcsA protein of Shigella flexneri localize at one of the cell poles in each of these species, but the mechanisms involved in their asymmetric localization are different. The localization of IcsA depends on direct targeting to a specific cell pole, whereas ActA is excluded from the newly synthesized cell pole. The asymmetrical localization of FliG1 can be achieved by one of these two mechanisms, e.g. FliG1 can be directly targeted to a new cell pole or be excluded from the new cell pole and consequently resides at the old cell pole.
Pathogenic spirochetes share some common pathogenic aspects, despite the fact that the clinical manifestations of the diseases caused by them are quite diverse (Norris et al., 2001; Antal et al., 2002; Steere et al., 2004; Rosa et al., 2005; LaFond and Lukehart, 2006; Palaniappan et al., 2007; Vijayachari et al., 2008). After encountering a mammalian host, microorganisms typically enter into the host either by ingress (e.g. inhalation and ingestion) or by penetration (e.g. insect bites, cuts and wounds on skin). For penetration, microorganisms have to cross epithelial barriers, enter into tissues, disseminate in hosts, and finally cause systemic infections. The spirochetes generally enter mammalian hosts by penetration after crossing epithelial barriers, which are mediated by either tick bite, sexual contact or through cuts and wounds on the skin. After being deposited on the skin or mucous membranes, these spirochetes must traverse the intercellular matrix, in some cases penetrate vascular endothelial cell lining, and finally spread in a host and cause systemic infections (Szczepanski et al., 1990; Charon and Goldstein, 2002; Steere et al., 2004; Palaniappan et al., 2007). It has been believed that motility is essential for the invasion and dissemination of spirochetes (Kimsey and Spielman, 1990; Charon and Goldstein, 2002; Rosa et al., 2005). However, the relevant evidence is quite limited.
Sadziene et al. (1991) isolated a non-flagella mutant of B. burgdorferi (HB19−) and found that this mutant failed to invade human tissues. However, HB19− is a spontaneous mutant and real mutation remains unknown. Botkin et al. constructed 33 B. burgdorferi mutants using transposon mutagenesis. Single mouse inoculations followed by culture of four tissue sites showed that three of these mutants were non-infectious, including a fliG1 insertion mutant (Botkin et al., 2006), which is consistent with this report. However, the phenotype of this mutant is not well characterized and the mutant is not complemented. In this communication, the phenotype of the fliG1− mutant is well characterized and the mutation was complemented. Thus, this mutant is a perfect candidate to determine whether motility is required for the infection of B. burgdorferi.
We first tested the fliG1− mutant in immunocompetent mice and found that the mutant failed to establish a systemic infection. In the process of B. burgdorferi infection, the spirochete is first deposited in mammalian skin tissue via tick bite and establishes a local infection. In this process, the spirochete has to escape from the innate immune responses, e.g. phagocytosis mediated by macrophages or neutrophils. It hypothesizes that motility facilitates the spirochete in traversing connective tissues, penetrating vascular endothelial cell linings and becoming haematogenous, and consequently helping spirochetes to escape the innate immune response (Greenberg and Canale-Parola, 1977a,b; Charon and Goldstein, 2002). Thus, the failure to establish a systemic infection in immunocompetent mice could be due to: the mutant fails to disseminate but retains the ability to establish local infection at inoculation sites; and/or the mutant can effectively evade innate immunity but is cleared by adaptive immune responses; and/or the mutant is simply eliminated by the innate immune system immediately after inoculation. To address these possibilities, the mutant was further tested in SCID mice. The result showed that the mutant cells were cleared from all inoculation sites within 24 h, thus ruling out the possibility of the involvement of adaptive immunity in eliminating the mutant. The most likely mechanism could be that the inability to swim provides the innate immune system, including phagocytes, an opportunity. Consequently, non-motile spirochetes are quickly eliminated. Taken together, this report has demonstrated that motility is an important virulent factor that plays an essential role in spirochetal pathogenesis.
Experimental procedures
Bacterial strains and growth conditions
The high-passage B. burgdorferi sensu stricto strain B31A and the low-passage strain B31-A3 have been previously described (Elias et al., 2002; Li et al., 2002). Cells were grown at 34°C in BSK-II liquid or on plates in 3% carbon dioxide. Mutants derived from B31A or B31-A3 were grown in the presence of kanamycin (300 μg ml−1), streptomycin (80 μg ml−1) or gentamicin (40 μg ml−1). E. coli JM109 and M15 cells were grown in Luria–Bertani broth with the appropriate antibiotic.
Construction of plasmids for the targeted mutagenesis
A previously described targeted mutagenesis that is mediated by allelic exchange recombination was applied to inactivate motility and chemotaxis genes in B. burgdorferi (Li et al., 2002; Sal et al., 2008; Yang and Li, 2009). Here, the fliG1 was used as an example to describe how these mutants were constructed. As illustrated in Fig. 2, to construct a vector for targeted mutagenesis, the fliG1 gene and a kanamycin resistance gene (kan) were amplified by PCR (primers P1–P4, Table 4), and the resultant PCR products were cloned into a pGEM-T vector (Promega, Madison, WI), and then kan was inserted into the fliG1 gene at two HindIII sites, resulting in 346 bp deletion. The resultant plasmid was treated with NotI to linearize the plasmid, and the obtained product (approximately 5 μg) was electroporated into competent B. burgdorferi B31A cells. After 14–21 days of incubation, antibiotic-resistant colonies were picked and grown in BSK-II liquid medium for further analysis. The mutants were confirmed by PCR and the loss of cognate gene product was further detected by Western blot as previously described (Li et al., 2002; Sal et al., 2008; Yang and Li, 2009). The same strategy was used to construct the plasmids for the targeted mutagenesis of fliG2.
Table 4.
Oligonucelotide primers used in this study.
| Primer | Description | Sequences |
|---|---|---|
| P1 | fliG1 (F), inactivation | 5′-AGATTCAAGGCTCTATGC-3′ |
| P2 | fliG1 (R), inactivation | 5′-TTTAGCAGTCTCTGTATCG-3′ |
| P3 | kan (F), fliG1 inactivation | 5′-AAGCTTTAATACCCGAGCTTCAAG-3′ |
| P4 | kan (R), fliG1 inactivation | 5′-AAGCTTTCAAGTCAGCGTAATGCT-3′ |
| P5 | flgBp (F), complementation | 5′-GGATCCTAATACCCGAGCTTCAAG-3′ |
| P6 | flgBp (R), complementation | 5′-CATATGACCTCCCTCATTTAAAATTGC-3′ |
| P7 | fliG1 (F), complementation | 5′-CATATGAAAGTTATGCAGGATCCTAGG-3′ |
| P8 | fliG1 (R), complementation | 5′-GCATGCGTGTCATTAAATAAATTCCTC-3′ |
| P9 | fliG2 (F), inactivation | 5′-ATTCTGGATGTTTCTGCT-3′ |
| P10 | fliG2 (R), inactivation | 5′-GACAAGCACATCTTCTTC-3′ |
| P11 | kan (F), fliG2 inactivation | 5′-TCTAGATAATACCCGAGCTTCAAG-3′ |
| P12 | kan (R), fliG2 inactivation | 5′-CAATTGTCAAGTCAGCGTAATGCT-3′ |
| P13 | fliG2 (F), complementation | 5′-CATATGGAAGAAAAAAAAGAAAAGC-3′ |
| P14 | fliG2 (R), complementation | 5′-GCATGCGACAAGCACATCTTCTTC-3′ |
| P15 | fliG1 (F), His-tagged FliG1 | 5′-GAGCTCGATCCTAGGCTTTCCAAGTAT-3′ |
| P16 | fliG1 (R), His-tagged FliG1 | 5′-CTGCAGTTTTTCTATATATTATTAAATCG-3′ |
| P17 | fliG2 (F), His-tagged FliG2 | 5′-GGATCCATTCTGGATGTTTCTGCT-3′ |
| P18 | fliG2 (R), His-tagged FliG2 | 5′-CTGCAGGACAAGCACATCTTCTTC-3′ |
| P19 | flgBp (F), expressing gfp | 5′-CTGCAGTAATACCCGAGCTTCAAG-3′ |
| P20 | flgBp (R), expressing gfp | 5′-GGATCCTTTAAAATTGCTTTTAAC-3′ |
| P21 | gfp (F) | 5′-GGATCCAAGAAGGAGATATACATATGAG-3′ |
| P22 | gfp (R), with 5 × glycin linker | 5′-GCATGCACCTCCACCTCCACCTTTGTATAGTTCATCCATGCCATGTG-3′ |
| P23 | T7 terminator (F) | 5′-GCATGCTAACAAAGCCCGAAAGGAAGC-3′ |
| P24 | T7 terminator (R) | 5′-AAGCTTGCAGATCCGGATATAGTTCCT-3′ |
| P25 | fliG1 (F), GFP fusion | 5′-GCATGCAAAGTTATGCAGGATCCTAGGCTT-3′ |
| P26 | fliG1 (R), GFP fusion | 5′-GCATGCGTGTCATTAAATAAATTCCTC-3′ |
Complementation of the fliG1− and fliG2− mutants
The similar methods described before were used to complement the fliG1− mutant (Bakker et al., 2006; Sal et al., 2008). As shown in Fig. 2, the flgB promoter was first amplified by PCR with an engineered BamHI site at the 5′ end and an NdeI site at the 3′ end (primers were listed in Table 4). The intact fliG1 gene was also amplified by PCR with engineered NdeI site at 5′ and SphI site at 3′ end. The resultant PCR fragments were cloned into the pGEM-T Easy vector. The fliG1 gene was fused to the 3′ end of flgB promoter at the NdeI site, and confirmed by DNA sequence analysis. The BamHI–SphI-digested flgB-fliG1 fragment was further cloned into pKFSS1 vector (Frank et al., 2003; Bakker et al., 2006), which resulted in the complementing plasmid FlgBG1/ pKFSS1. The resultant construct was transformed into the mutant by electroporation as previously described (Bakker et al., 2006; Sal et al., 2008). The obtained transformants were characterized by PCR and Western blot as previously described (Bakker et al., 2006; Motaleb et al., 2007; Sal et al., 2008). One complemented strain was selected for further studies and was named as fliG1−/+. The similar method was applied to complement fliG2−. Although the complementation recovered the expression of fliG2 (Fig. 3C), it could not restore the phenotype of the wild type. The obtained strain was referred to as fliG2−/+.
Construction of the fliG1 mutant and its complemented strain in B31-A3
To construct the fliG1 mutant in the virulent strain B31-A3, the same construct described above was transformed into B31-A3 by electroporation. Cell plating and mutant screening were the same as the above. The plasmid profile in the obtained mutant was detected by PCR as previously described (Elias et al., 2002; Stewart et al., 2005). Only the mutant containing the same plasmid content as its parental strain B31-A3 was selected for the further characterization and the complementation. To complement the obtained fliG1−mutant, the above construct flgB-fliG1 was cloned into the shuttle vector pBSV2G that contains gentamicin-resistant marker (Elias et al., 2003; Grimm et al., 2004). The resultant construct, FlgBG1/pBSV2G, was then transformed into the fliG1− mutant that was derived from B31-A3 as described before (Grimm et al., 2004). The plasmid profile in the complemented strain was further confirmed by PCR analysis (Elias et al., 2002). The obtained mutant and complemented strains on B31-A3 background were referred to as vfliG1−and vfliG1−/+ respectively.
Generation of polyclonal antiserum against FliG1 or FliG2
The fliG1 and fliG2 genes were amplified using primers P15 to P18 (Table 4), and the PCR products were first cloned into the pGME-T Easy vector and the resulted insert was further subcloned into the pQE30 expression vector (Qiagen, Valencia, CA), which encodes an N-terminal histidine tag. The expression of fliG1 and fliG2 was induced using 0.1 M isopropyl-β-D-thiogalactoside (IPTG), and the recombinant proteins were purified by a nickel agarose column, and concentrated in 10 kDa molecular weight cut-off Amicon Ultra centrifugal concentrators (Millipore, Billerica, MA). Rats were immunized with 1 mg of purified recombinant proteins during a 1-month period. Polyclonal antisera were further purified by an affinity chromatography with the AminoLink Plus Immobilization Kit (Thermo Scientific, Rockford, IL), and eluted as recommended by the manufacturer.
Electrophoresis and Western blotting analysis
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting using the enhanced chemiluminescent detection system (ECL) were performed as previously described (Li et al., 2002; Sal et al., 2008). All gels were 10% polyacrylamide unless otherwise noted. Whole cell lysates were prepared by first washing cells in PBS, followed by boiling for at least 5 min in Laemmli sample buffer. The antibodies against FliG1 or FliG2 were described above, and the antibodies against B. burgdorferi DnaK, FlaA and FlaB were described in our previous studies (Sal et al., 2008). Monoclonal antibody against GFP was purchased from Invitrogen (Carlsbad, California).
Swarm plate assay and bacterial tracking analysis
The motility of the above constructed mutants and complemented strains were analysed by a computer-based bacteria tracking system and swarm plate assay as previously described (Li et al., 2002; Bakker et al., 2006; Motaleb et al., 2007). For bacterial tracking analysis, the strains were tracked under both a low viscous medium (BSK-II) and a highly viscous medium (1% methylcellulose). The swarm plate assay was performed on 1:10 diluted BSK-II medium containing 0.35% argarose.
Construction of a plasmid that expresses N-terminal GFP-fusion proteins
The same methods described before (with slight modification) were used to construct a vector that expresses N-terminal GFP-fusion protein (Sourjik and Berg, 2000). Figure 6A is the map of the vector expressing N-terminal GFP. The gfp gene was kindly provided by Dr Radolf’s laboratory, which is driven by the flgB promoter (Eggers et al., 2002). In this vector, a five-glycine linker was added between GFP and a fused protein, which allows GFP and the fused protein to fold independently and correctly (Sourjik and Berg, 2000). A unique restriction enzyme (SphI) cut site was engineered for the insertion of a fused gene, and a T7 terminator was added at the end of a fused gene. The final construct was named as pFBgfp/T7t. To express fusion protein GFP–FliG1, the full-length fliG1 gene was PCR amplified with engineered SphI cut site at each end. The obtained PCR product was first cloned into pGEM-T easy vector and further subcloned into pFBgfp/T7t at the SphI site. The orientation of fliG1 in this vector was further confirmed by restriction digestion and DNA sequencing. To localize GFP–FliG1 protein, the obtained plasmid was transformed into the fliG1− mutant by electroporation. The transformants were selected and confirmed as described above. The primers (P19 to P26) for constructing this vector and GFP–FliG1 were enclosed in Table 4.
Light, fluorescence and electron microscopy
Cell morphology and motility of the wild type, fliG1 mutant cells, as well as the complemented strains were characterized using light microscopy as previously described (Li et al., 2002; Bakker et al., 2006; Sal et al., 2008). To visualize the motion of B31A and the fliG1− mutant, the cells in BSK-II medium were videotaped with dark-field illumination at 100× for at least 1 min. Video was digitized to allow for frame-by-frame observation of cell motion.
Immunofluorescence microscopic analysis was conducted to localize FliG2 as previously described (Hiraga et al., 1998). Briefly, 1.5 ml of B31A and the fliG2− mutant cultures were harvested, washed two times with PBS buffer (phosphate-buffered saline, pH 7.5), and then treated with methanol at −20°C for 1 h. The collected cells were treated with lysozyme (1 mg ml−1) in GTE buffer (50 mM glucose, 25 mM Tris, 1 mM EDTA, pH 7.5) for 1 h at room temperature, and then placed on a polysine-coated coverslip, allowed to fully dry in air. The obtained coverslips were first incubated in a blocking solution (2% BSA in PBS, pH 7.5) for 1 h, and then incubated in the blocking solution containing 1:500 diluted anti-FliG2 antibody for 1 h at room temperature. Finally, the coverslips were washed five times with PBS, incubated with secondary goat anti-rat Texas red antibody (Invitrogen) for 1 h at room temperature, washed with PBS again, and mounted in 40% glycerol for image process.
GFP images were taken using a Zeiss Axiostar plus microscope at a wave length of 480 nm. Texas red images were taken using a Zeiss Axioimager Z1 Axiophot wide-field microscope with an excitation filter (541–569) and an emission filter (581–654 nm). The images were captured and processed using the program Axiovision (Zeiss, Germany). To assay for the presence of PFs by electron microscopy, the wild-type and mutant cells were fixed, embedded, and observed by a JEOL JEM 1220 transmission electron microscopy as previously described (Li et al., 2008; Sal et al., 2008).
Infection studies in mice
Both BALB/c and BALB/c SCID mice at ages of 4–8 weeks (provided by the Division of Laboratory Animal Medicine at Louisiana State University) were used in the study as described previously (Xu et al., 2005; 2008). All animal procedures were performed in compliance with the guidelines and with the approval of the Institutional Animal Care and Use Committee (IACUC). Briefly, BALB/c mice were given a single subcutaneous injection of 104 spirochetes, and sacrificed 4 weeks post inoculation. Heart, tibiotarsal joint and skin specimens were aseptically collected for spirochete culture. SCID mice received two intradermal/subcutaneous injections of 104 spirochetes. The two inoculation sites were at least 2 cm apart. Animals were sacrificed 24, 48 or 72 h later; inoculation site skin tissues were harvested for spirochete isolation.
Supplementary Material
Acknowledgments
We thank J. Radolf for providing gfp construct, S. Samuels and P. Rosa for providing the shuttle vectors and B. burgdorferi strains, N. Charon for sharing the computer-based bacteria tracking system, and the Confocal Microscope and Flow Cytometry Facility in the School of Medicine and Biomedical Sciences, University at Buffalo for the assistance of fluorescent microscope. This research was supported by Public Service AI073354, AI078958, and American Heart Association grants to C. Li, and AI29743 to N. Charon.
Footnotes
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aldridge P, Hughes KT. Regulation of flagellar assembly. Curr Opin Microbiol. 2002;5:160–165. doi: 10.1016/s1369-5274(02)00302-8. [DOI] [PubMed] [Google Scholar]
- Antal GM, Lukehart SA, Meheus AZ. The endemic treponematoses. Microbes Infect. 2002;4:83–94. doi: 10.1016/s1286-4579(01)01513-1. [DOI] [PubMed] [Google Scholar]
- Bakker RG, Li C, Miller MR, Cunningham C, Charon NW. Identification of specific chemoattractants and genetic complementation of a Borrelia burgdorferi chemotaxis mutant: a flow cytometry-based capillary tube chemotaxis assay. Appl Environ Microbiol. 2006;73:1180–1188. doi: 10.1128/AEM.01913-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthold SW. Animal models for Lyme disease. Lab Invest. 1995;72:127–130. [PubMed] [Google Scholar]
- Berg HC, Turner L. Movement of microorganisms in viscous environments. Nature (London) 1979;278:349–351. doi: 10.1038/278349a0. [DOI] [PubMed] [Google Scholar]
- Blevins JS, Revel AT, Smith AH, Bachlani GN, Norgard MV. Adaptation of a luciferase gene reporter and lac expression system to Borrelia burgdorferi. Appl Environ Microbiol. 2007;73:1501–1513. doi: 10.1128/AEM.02454-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botkin DJ, Abbott A, Stewart PE, Rosa PA, Kawabata H, Watanabe H, Norris SJ. Identification of potential virulence determinants by Himar1 transposition of infectious. Infect Immun. 2006;74:6690–6699. doi: 10.1128/IAI.00993-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PN, Hill CP, Blair DF. Crystal structure of the middle and C-terminal domains of the flagellar rotor protein FliG. EMBO J. 2002;21:3225–3234. doi: 10.1093/emboj/cdf332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PD, Carrington DG, Gravekamp C, van de Kemp H, Edwards CN, Jones SR, et al. Direct detection of leptospiral material in human postmortem samples. Res Microbiol. 2003;154:581–586. doi: 10.1016/S0923-2508(03)00166-9. [DOI] [PubMed] [Google Scholar]
- Charon NW, Goldstein SF. Genetics of motility and chemotaxis of a fascinating group of bacteria: the spirochetes. Annu Rev Genet. 2002;36:47–73. doi: 10.1146/annurev.genet.36.041602.134359. [DOI] [PubMed] [Google Scholar]
- Charon NW, Goldstein SF, Marko M, Hsieh C, Gebhardt LL, Motaleb MA, et al. The flat-ribbon configuration of the periplasmic flagella of Borrelia burgdorferi and its relationship to motility and morphology. J Bacteriol. 2009;191:600–607. doi: 10.1128/JB.01288-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZQ, Zhang GC, Gong XD, Lin C, Gao X, Liang GJ, et al. Syphilis in China: results of a national surveillance programme. Lancet. 2007;369:132–138. doi: 10.1016/S0140-6736(07)60074-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevance FF, Hughes KT. Coordinating assembly of a bacterial macromolecular machine. Nat Rev Microbiol. 2008;6:455–465. doi: 10.1038/nrmicro1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa NE, Peng F, Klose KE. Roles of the regulatory proteins FlhF and FlhG in the Vibrio cholerae flagellar transcription hierarchy. J Bacteriol. 2005;187:6324–6332. doi: 10.1128/JB.187.18.6324-6332.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta N, Arora SK, Ramphal R. fleN, a gene that regulates flagellar number in Pseudomonas aeruginosa. J Bacteriol. 2000;182:357–364. doi: 10.1128/jb.182.2.357-364.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers CH, Caimano MJ, Clawson ML, Miller WG, Samuels DS, Radolf JD. Identification of loci critical for replication and compatibility of a Borrelia burgdorferi cp32 plasmid and use of a cp32-based shuttle vector for the expression of fluorescent reporters in the lyme disease spirochaete. Mol Microbiol. 2002;43:281–295. doi: 10.1046/j.1365-2958.2002.02758.x. [DOI] [PubMed] [Google Scholar]
- Elias AF, Stewart PE, Grimm D, Caimano MJ, Eggers CH, Tilly K, et al. Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect Immun. 2002;70:2139–2150. doi: 10.1128/IAI.70.4.2139-2150.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias AF, Bono JL, Kupko JJ, III, Stewart PE, Krum JG, Rosa PA. New antibiotic resistance cassettes suitable for genetic studies in Borrelia burgdorferi. J Mol Microbiol Biotechnol. 2003;6:29–40. doi: 10.1159/000073406. [DOI] [PubMed] [Google Scholar]
- Frank KL, Bundle SF, Kresge ME, Eggers CH, Samuels DS. aadA confers streptomycin resistance in Borrelia burgdorferi. J Bacteriol. 2003;185:6723–6727. doi: 10.1128/JB.185.22.6723-6727.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, Dodson R, et al. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science. 1998;281:375–388. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- Goldstein SF, Charon NW, Kreiling JA. Borrelia burgdorferi swims with a planar waveform similar to that of eukaryotic flagella. Proc Natl Acad Sci USA. 1994;91:3433–3437. doi: 10.1073/pnas.91.8.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg EP, Canale-Parola E. Motility of flagellated bacteria in viscous environments. J Bacteriol. 1977a;132:356–358. doi: 10.1128/jb.132.1.356-358.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg EP, Canale-Parola E. Relationship between cell coiling and motility of spirochetes in viscous environments. J Bacteriol. 1977b;131:960–969. doi: 10.1128/jb.131.3.960-969.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Tilly K, Byram R, Stewart PE, Krum JG, Bueschel DM, et al. Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc Natl Acad Sci USA. 2004;101:3142–3147. doi: 10.1073/pnas.0306845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraga S, Ichinose C, Niki H, Yamazoe M. Cell cycle-dependent duplication and bidirectional migration of SeqA-associated DNA–protein complexes in E. coli. Mol Cell. 1998;1:381–387. doi: 10.1016/s1097-2765(00)80038-6. [DOI] [PubMed] [Google Scholar]
- Holt SC. Anatomy and chemistry of spirochetes. Microbiol Rev. 1978;42:114–160. doi: 10.1128/mr.42.1.114-160.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irikura VM, Kihara M, Yamaguchi S, Sockett H, Macnab RM. Salmonella typhimurium fliG and fliN mutations causing defects in assembly, rotation, and switching of the flagellar motor. J Bacteriol. 1993;175:802–810. doi: 10.1128/jb.175.3.802-810.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimsey RB, Spielman A. Motility of Lyme disease spirochetes in fluids as viscous as the extracellular matrix. J Infect Dis. 1990;162:1205–1208. doi: 10.1093/infdis/162.5.1205. [DOI] [PubMed] [Google Scholar]
- Klose KE, Mekalanos JJ. Differential regulation of multiple flagellins in Vibrio cholerae. J Bacteriol. 1998;180:303–316. doi: 10.1128/jb.180.2.303-316.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper H, van Dam AP, Spanjaard L, de Jongh BM, Widjojokusumo A, Ramselaar TC, et al. Isolation of Borrelia burgdorferi from biopsy specimens taken from healthy-looking skin of patients with Lyme borreliosis. J Clin Microbiol. 1994;32:715–720. doi: 10.1128/jcm.32.3.715-720.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFond RE, Lukehart SA. Biological basis for syphilis. Clin Microbiol Rev. 2006;19:29–49. doi: 10.1128/CMR.19.1.29-49.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadbetter JR, Schmidt TM, Graber JR, Breznak JA. Acetogenesis from H2 plus CO2 by spirochetes from termite guts. Science. 1999;283:686–689. doi: 10.1126/science.283.5402.686. [DOI] [PubMed] [Google Scholar]
- Levett PN. Leptospirosis. Clin Microbiol Rev. 2001;14:296–326. doi: 10.1128/CMR.14.2.296-326.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Motaleb A, Sal M, Goldstein SF, Charon NW. Spirochete periplasmic flagella and motility. J Mol Microbiol Biotechnol. 2000;2:345–354. [PubMed] [Google Scholar]
- Li C, Bakker RG, Motaleb MA, Sartakova ML, Cabello FC, Charon NW. Asymmetrical flagellar rotation in Borrelia burgdorferi nonchemotactic mutants. Proc Natl Acad Sci USA. 2002;99:6169–6174. doi: 10.1073/pnas.092010499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Wolgemuth CW, Marko M, Morgan DG, Charon NW. Genetic analysis of spirochete flagellin proteins and their involvement in motility, filament assembly, and flagellar morphology. J Bacteriol. 2008;190:5607–5615. doi: 10.1128/JB.00319-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lin T, Botkin DJ, McCrum E, Winkler H, Norris SJ. Intact flagellar motor of Borrelia burgdorferi revealed by cryo-electron tomography: evidence for stator ring curvature and rotor/C ring assembly flexion. J Bacteriol. 2009;191:5026–5036. doi: 10.1128/JB.00340-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd SA, Tang H, Wang X, Billings S, Blair DF. Torque generation in the flagellar motor of Escherichia coli: evidence of a direct role for FliG but not for FliM or FliN. J Bacteriol. 1996;178:223–231. doi: 10.1128/jb.178.1.223-231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd SA, Whitby FG, Blair DF, Hill CP. Structure of the C-terminal domain of FliG, a component of the rotor in the bacterial flagellar motor. Nature. 1999;400:472–475. doi: 10.1038/22794. [DOI] [PubMed] [Google Scholar]
- Lux R, Moter A, Shi W. Chemotaxis in pathogenic spirochetes: directed movement toward targeting tissues? J Mol Microbiol Biotechnol. 2000;2:355–364. [PubMed] [Google Scholar]
- Lybarger SR, Maddock JR. Polarity in action: asymmetric protein localization in bacteria. J Bacteriol. 2001;183:3261–3267. doi: 10.1128/JB.183.11.3261-3267.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride AJ, Athanazio DA, Reis MG, Ko AI. Leptospirosis. Curr Opin Infect Dis. 2005;18:376–386. doi: 10.1097/01.qco.0000178824.05715.2c. [DOI] [PubMed] [Google Scholar]
- Masuzawa T, Beppu Y, Kawabata H, Yanagihara Y, Iwamoto Y, Shimizu T, Johnson RC. Experimental Borrelia burgdorferi infection of outbred mice. J Clin Microbiol. 1992;30:3016–3018. doi: 10.1128/jcm.30.11.3016-3018.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikosza AS, Hampson DJ. Human intestinal spirochetosis: Brachyspira aalborgi and/or Brachyspira pilosicoli? Anim Health Res Rev. 2001;2:101–110. [PubMed] [Google Scholar]
- Motaleb MA, Corum L, Bono JL, Elias AF, Rosa P, Samuels DS, Charon NW. Borrelia burgdorferi periplasmic flagella have both skeletal and motility functions. Proc Natl Acad Sci USA. 2000;97:10899–10904. doi: 10.1073/pnas.200221797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motaleb MA, Miller MR, Li C, Bakker RG, Goldstein SF, Silversmith RE, et al. CheX is a phosphorylated CheY phosphatase essential for Borrelia burgdorferi chemotaxis. J Bacteriol. 2005;187:7963–7969. doi: 10.1128/JB.187.23.7963-7969.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motaleb MA, Miller MR, Bakker RG, Li C, Charon NW. Isolation and characterization of chemotaxis mutants of the Lyme disease spirochete Borrelia burgdorferi using allelic exchange mutagenesis, flow cytometry, and cell tracking. Methods Enzymol. 2007;422:419–437. doi: 10.1016/S0076-6879(06)22021-4. [DOI] [PubMed] [Google Scholar]
- Murray TS, Kazmierczak BI. FlhF is required for swimming and swarming in Pseudomonas aeruginosa. J Bacteriol. 2006;188:6995–7004. doi: 10.1128/JB.00790-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocton JJ, Bloom BJ, Rutledge BJ, Persing DH, Logigian EL, Schmid CH, Steere AC. Detection of Borrelia burgdorferi DNA by polymerase chain reaction in cerebrospinal fluid in Lyme neuroborreliosis. J Infect Dis. 1996;174:623–627. doi: 10.1093/infdis/174.3.623. [DOI] [PubMed] [Google Scholar]
- Norris SJ, Cox DL, Weinstock GM. Biology of Treponema pallidum: correlation of functional activities with genome sequence data. J Mol Microbiol Biotechnol. 2001;3:37–62. [PubMed] [Google Scholar]
- Palaniappan RU, Ramanujam S, Chang YF. Leptospirosis: pathogenesis, immunity, and diagnosis. Curr Opin Infect Dis. 2007;20:284–292. doi: 10.1097/QCO.0b013e32814a5729. [DOI] [PubMed] [Google Scholar]
- Paster BJ, Dewhirst FE. Phylogenetic foundation of spirochetes. J Mol Microbiol Biotechnol. 2000;2:341–344. [PubMed] [Google Scholar]
- Ren SX, Fu G, Jiang XG, Zeng R, Miao YG, Xu H, et al. Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature. 2003;422:888–893. doi: 10.1038/nature01597. [DOI] [PubMed] [Google Scholar]
- Rosa PA, Tilly K, Stewart PE. The burgeoning molecular genetics of the Lyme disease spirochaete. Nat Rev Microbiol. 2005;3:129–143. doi: 10.1038/nrmicro1086. [DOI] [PubMed] [Google Scholar]
- Ruby JD, Charon NW. Effect of temperature and viscosity on the motility of the spirochete Treponema denticola. FEMS Microbiol Lett. 1998;169:251–254. doi: 10.1111/j.1574-6968.1998.tb13325.x. [DOI] [PubMed] [Google Scholar]
- Sadziene A, Thomas DD, Bundoc VG, Holt SC, Barbour AG. A flagella-less mutant of Borrelia burgdorferi. Structural, molecular, and in vitro functional characterization. J Clin Invest. 1991;88:82–92. doi: 10.1172/JCI115308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sal MS, Li C, Motalab MA, Shibata S, Aizawa S, Charon NW. Borrelia burgdorferi uniquely regulates its motility genes and has an intricate flagellar hook-basal body structure. J Bacteriol. 2008;190:1912–1921. doi: 10.1128/JB.01421-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels DS. Electrotransformation of the spirochete Borrelia burgdorferi. Methods Mol Biol. 1995;47:253–259. doi: 10.1385/0-89603-310-4:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels DS, Mach KE, Garon CF. Genetic transformation of the Lyme disease agent Borrelia burgdorferi with coumarin-resistant gyrB. J Bacteriol. 1994;176:6045–6049. doi: 10.1128/jb.176.19.6045-6049.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri R, Myers GS, Tettelin H, Eisen JA, Heidelberg JF, Dodson RJ, et al. Comparison of the genome of the oral pathogen Treponema denticola with other spirochete genomes. Proc Natl Acad Sci USA. 2004;101:5646–5651. doi: 10.1073/pnas.0307639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro L, Losick R. Dynamic spatial regulation in the bacterial cell. Cell. 2000;100:89–98. doi: 10.1016/s0092-8674(00)81686-4. [DOI] [PubMed] [Google Scholar]
- Sourjik V, Berg HC. Localization of components of the chemotaxis machinery of Escherichia coli using fluorescent protein fusions. Mol Microbiol. 2000;37:740–751. doi: 10.1046/j.1365-2958.2000.02044.x. [DOI] [PubMed] [Google Scholar]
- Stanek G, Klein J, Bittner R, Glogar D. Isolation of Borrelia burgdorferi from the myocardium of a patient with longstanding cardiomyopathy. N Engl J Med. 1990;322:249–252. doi: 10.1056/NEJM199001253220407. [DOI] [PubMed] [Google Scholar]
- Steere AC, Coburn J, Glickstein L. The emergence of Lyme disease. J Clin Invest. 2004;113:1093–1101. doi: 10.1172/JCI21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PE, Hoff J, Fischer E, Krum JG, Rosa PA. Genome-wide transposon mutagenesis of Borrelia burgdorferi for identification of phenotypic mutants. Appl Environ Microbiol. 2004;70:5973–5979. doi: 10.1128/AEM.70.10.5973-5979.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PE, Byram R, Grimm D, Tilly K, Rosa PA. The plasmids of Borrelia burgdorferi: essential genetic elements of a pathogen. Plasmid. 2005;53:1–13. doi: 10.1016/j.plasmid.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Szczepanski A, Furie MB, Benach JL, Lane BP, Fleit HB. Interaction between Borrelia burgdorferi and endothelium in vitro. J Clin Invest. 1990;85:1637–1647. doi: 10.1172/JCI114615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanbichler M, Shapiro L. Getting organized –how bacterial cells move proteins and DNA. Nat Rev Microbiol. 2008;6:28–40. doi: 10.1038/nrmicro1795. [DOI] [PubMed] [Google Scholar]
- Toker AS, Macnab RM. Distinct regions of bacterial flagellar switch protein FliM interact with FliG, FliN and CheY. J Mol Biol. 1997;273:623–634. doi: 10.1006/jmbi.1997.1335. [DOI] [PubMed] [Google Scholar]
- Vijayachari P, Sugunan AP, Shriram AN. Leptospirosis: an emerging global public health problem. J Biosci. 2008;33:557–569. doi: 10.1007/s12038-008-0074-z. [DOI] [PubMed] [Google Scholar]
- Warnecke F, Luginbuhl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560–565. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- Weinstock GM, Hardham JM, McLeod MP, Sodergren EJ, Norris SJ. The genome of Treponema pallidum: new light on the agent of syphilis. FEMS Microbiol Rev. 1998;22:323–332. doi: 10.1111/j.1574-6976.1998.tb00373.x. [DOI] [PubMed] [Google Scholar]
- Xu Q, Seemanapalli SV, Lomax L, McShan K, Li X, Fikrig E, Liang FT. Association of linear plasmid 28-1 with an arthritic phenotype of Borrelia burgdorferi. Infect Immun. 2005;73:7208–7215. doi: 10.1128/IAI.73.11.7208-7215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, McShan K, Liang FT. Essential protective role attributed to the surface lipoproteins of Borrelia burgdorferi against innate defences. Mol Microbiol. 2008;69:15–29. doi: 10.1111/j.1365-2958.2008.06264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Li C. Transcription and genetic analyses of a putative N-acetylmuramyl-L-alanine amidase in Borrelia burgdorferi. FEMS Microbiol Lett. 2009;290:164–173. doi: 10.1111/j.1574-6968.2008.01416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.