

Abstract
Human papillomavirus (HPV) is the most prevalent sexually transmitted disease in the United States, and can cause cancer with persistent infection. The most common cancer caused by HPV is cervical carcinoma with an average of 12,000 cases reported every year in the U.S. Worldwide, over 500,000 cases of cervical cancer are reported yearly with over 250,000 deaths attributed to the disease. Although much is known about the serious health risks associated with HPV infection, there is still much to be discovered about how HPV binds and enters target cells. Understanding how HPV infects will lead to strategies and therapies for reducing the number of infections and HPV-related diseases, including cancers.
The HPV viral particle is composed of two viral proteins, L1 and L2. Data suggest that binding of the viral capsid to cells is dependent on the L1 protein. We hypothesize that this initial binding to a heparan sulfate is composed of two independent events: the first results in a structural change that exposes a hidden portion of the L1 protein leading to a second binding event on the heparan sulfate.
Our experiments tested if this “hidden” portion of L1 is necessary for infection and explored the nature of this binding. We generated a peptide with the sequence of the “hidden” portion of L1. Infection of HaCaT cells in the presence of this peptide is highly reduced. Our results suggest that the binding of the L1 C-terminal domain is dependent on amino acid sequence, and is necessary for infection.
Keywords: HPV, L1 C-terminus, HSPG binding
INTRODUCTION
Human papillomavirus type 16 (HPV16) is a small non-enveloped virus, about 55nm in diameter with a genome of 8kb in size [1, 2]. The virus infects squamous epithelial cells in the cervix, glans of the penis, penile shaft, scrotum and anal verge [2, 3]. To initiate a successful infection, HPV has been shown to bind to a heparan sulfate proteoglycan (HSPG) and subsequently to an endocytic complex that may include cell-surface HSPG, integrins, tetraspanins, and growth factor receptors [4, 5]. This complex internalizes the virus and is responsible for the movement of the viral particle through an endosome, and possibly to the trans-Golgi network [5]. Infection will then be established when the viral genome reaches the nuclei, and viral gene transcription occurs. Mechanism of how the viral genome travels from the endosome to the nucleus is not fully defined.
The HPV capsid is composed of two virally encoded proteins, L1 and L2, the major and minor capsid proteins respectively. Five L1 molecules associate to form a capsomere, in the center of which the minor capsid L2 possibly associates [6]. Seventy-two capsomeres of L1 make up the capsid, creating a total of 360 L1 molecules [7]. Structural analysis has been helpful in describing the overall arrangement of the capsid proteins, and it is proposed that the L1 C-Terminal region in each capsomere invades a neighboring L1 and together form a disulfide bond that stabilizes interactions [7, 8]. The model postulates that the interacting arm of the L1 (the C-Terminus) lies in the vertex between two capsomeres, i.e. an intercapsomeric region that is not exposed on the outside of a mature capsid.
Data suggest that the initial interaction of the L1 capsid protein to the heparan sulfate proteoglycan (HSPG) can occur on the extracellular matrix or on the surface of the cells [9, 10]. This initial binding is comparable to the binding of chemokines. The chemokines are a family of proteins that bind to HSPGs by interacting with cell-surface heparan sulfate in an electrostatic-dependent manner, i.e., not sequence dependent [11]. Much like the chemokines, electrostatic binding interactions between basic amino acids on L1 and negatively charged sulfate and carboxyl groups on glycosaminoglycan (GAG) side chains on cell surfaces have been demonstrated. Single, double, and triple replacement of basic residues in the L-1 protein revealed that this initial interaction by the capsid is charge-dependent [10].
It has been demonstrated that the initial attachment of viral particles to a HSPG results in a conformational change of the viral capsid. This change in capsid conformation possibly exposes the intercapsomeric C-terminus region of the L1 protein [12, 13]. In this current manuscript, we corroborate this finding by describing that the exposed region of the L1 C-terminus plays a role in infection. We showed that a peptide created with L1 C-terminus sequence of HPV-16 was able to successfully reduce viral infection. This observed decrease in infection is sequence-specific and not charge-dependent. Mechanistically we showed that this second heparan-binding event is a separate heparan-binding step that we now term “intermediate,” and we showed that the binding influences viral endocytosis.
EXPERIMENTAL PROCEDURES
Cells
Immortalized epithelial cells derived from normal adult skin (HaCaT) were originally derived in the lab of Norbert Fusenig at DKFZ, Heidelberg, Germany, and obtained as a gift from Dr. Ozbun (The University of New Mexico School of Medicine, Albuquerque, NM). HaCaTs were cultured in Dulbecco’s Modified Eagle’s media (DMEM) supplemented with 10% Fetal Bovine Serum (FBS, DMEM-10).
HPV16 sequence alignment
The sequences of the HPV16 genotypes 16, 31, 11, 6 and 18 were extracted from the NCBI database, formatted into FASTA and aligned with the ClustalW alignment software tool version 1.83.
HPV16 PsV production and purification
HPV16 Pseudovirions (PsV) were generated in 293TT as described by Buck et al [14]. 293TT cells, bicistronic HPV16 L1 and L2 plasmid HPV16 pShell, and the GFP plasmid 8fwb were a generous gift from Drs. Day and Schiller (NCI/NIH Bethesda, MD). In brief, after high salt extraction, PsV were purified on an optiprep gradient (27–39%). Purity and integrity of the viral fractions were confirmed by negative staining electron-microscopy analysis, coomassie, and western blot techniques to detect the levels of L1 and L2 protein. Viral titers were determined by measuring the percentage of GFP positive cells by flow cytometry analysis after 48 hours infection of HaCaT cells.
Reagents
Wild Type (WT) peptide -TSSTSTTAKRKKRKL- and scrambled (SC) peptide -SKTTKARLRTSKTKS- were synthesized and fluorescently labeled with cy5 at Alpha Diagnostics Intl. (ADI), San Antonio, TX.
Peptide competition assay
HaCaT cells were pre-incubated for 1 hour at 4°C with indicated concentrations of WT, SC, or FLAG peptide (Sigma) prior to the addition of pseudovirus. Pseudovirus was allowed to bind to HaCaTs for two hours on ice. Soluble heparan from porcine intestinal mucosa was purchased from Sigma-Aldrich, St. Louis, MO and incubated with pseudovirions 30 minutes before infection to allow it to coat viral particles.
HPV16 infection assay
HaCaT cells plated at 50,000 cells/well in a 24 well plate were grown to 100,000 and were then incubated with peptide for 1 hour on ice. Cells were then washed in PBS to remove any unbound peptide and HPV16 PsVs were and allowed to bind for 2 hours on ice; incubation on ice minimized endocytosis. Cells were then incubated at 37°C for 48 hours. Infection was measured as the percentage of GFP-transduced cells, as the encapsidated plasmid 8fwb encodes the GFP cDNA (8fwb is the pseudogenome). Infections were analyzed by flow cytometry at Fordham University (BD Accuri C6 flow cytometer).
Time of peptide competition
Peptide was incubated for 2 hours with cells at 0, +30, and +60 minutes in relation to the addition of pseudovirus. Pseudovirus incubation with cells was for 2 hours on ice. Excess pseudovirus and peptide were removed after the 2 hour pseudovirion incubation. Level of infection was analyzed by flow cytometry after 48 hours incubation as before.
Adenovirus infection in the presence of peptide
Adenovirus type 5 carrying the GFP transgene was a kind gift of Dr. Imperiale (Univ. of Michigan, Ann Arbor, MI). HaCaT cells were pre-incubated for 1 hour at 4°C with indicated concentrations of WT, SC peptide prior to the addition of Ad-5 GFP. Virus was allowed to bind to HaCaTs for two hours on ice. Level of infection was analyzed 48 hours post infection using flow cytometry. Ad-5 GFP infection in absence of peptides was normalized to 100% and compared to infections in the presence of 100uM peptide.
Laser Scanning Immunofluorescence and Colocalization Studies
HaCaT cells were plated on a 6 well plate containing four glass coverslips per well and 200,000 cells/well (Fisher Scientific, Piscataway, NJ. Catalog #12-545-80). Cells were incubated overnight in DMEM-10 at 37°C. Cells were fixed for 10 minutes in 4% paraformaldehyde in PBS at 4°C and washed 3x in 1X PBS. Cells were permeabilized in blocking buffer (0.2% fish skin gelatin (Sigma) and 0.2% Triton X-100 in PBS) for 5 minutes. Coverslips were washed 3x in 1X PBS and incubated with mouse anti-HPV16.V5 antibody (generously provided by Neil Christensen, Penn State University, Hershey, PA) and the following antibodies: early endosome marker goat anti-EEA1 (Santa Cruz, California), or anti-LAMP-2 antibody produced in goat (Santa Cruz, California), or goat anti-syndecan-1 (Santa Cruz), or goat anti-α6 integrin (Santa Cruz, California). Primary antibodies were used at a 1:100 dilution for 1 hour. The following fluorescently labeled Alexa-Fluor secondary antibodies were used at a 1:2,000 dilution: donkey anti-goat 647 and donkey anti-mouse 488. All dilutions were made in blocking buffer. Coverslips were mounted using Prolong Gold Anti-Fade with DAPI for nuclear staining (Invitrogen). Fluorescence confocal microscopy was performed. All images were acquired with Leica TCS SP5 confocal microscope (Fordham University). Z-stacked images were taken at 0.75 micron sections (8 sections) of the cell in an x, y, and z plane.
Colocalization quantification
Colocalization between proteins was quantified using the program, ImageJ (National Institutes of Health, 1.46r). Images used were an average of all 8 sections, 0.75 micron Z-stack images taken within each treatment condition. ImageJ was used to count overlapping pixels of either membrane protein signal and HPV-16 signal, or cell organelle signal and HPV-16 signal. Overlapping of signal suggest co-localization of pseudovirions and organelle/protein and is observed as yellow. Percentage of colocalization was determined by number of “overlapping/yellow” pixels signal divided by pixels of pseudovirus. Pseudovirions were localized in relation to a6 integrin, tetraspanin 151, EEA1, and LAMP-2.
RESULTS
C-terminus sequences of major HPV types are highly conserved
The amino acid sequence of the L1 C-terminus of several HPV genotypes, including types 16, 31, 11, 6, and 18 was compared (Figure 1A). All genotypes analyzed contain a highly conserved sequence rich in lysine (K) and arginine (R) residues. These residues have been demonstrated to be essential in the formation of the L1 major capsid and in viral interactions with the cell surface [4, 15]. To determine the biological significance of the L1 C-terminus region, synthetic peptides were designed (Figure 1B). The C-terminus of oncogenic HPV type 16 includes the suggested heparan binding residues KRKKRK (shown in bold in Figure 1A). The full synthetic sequence contains fifteen amino acids: TSSTSTTAKRKKRKL (Figure 1B, Wildtype, WT). A scrambled version of the WT peptide was created containing the same amino acids but in different order SKTTKARLRTSKTKS (Figure 1B, Scrambled, SC). The SC peptide maintained the same overall charge as the WT peptide. We also used the FLAG epitope peptide as an additional control for non-specific effects (Figure 1B, FLAG).
Figure 1.
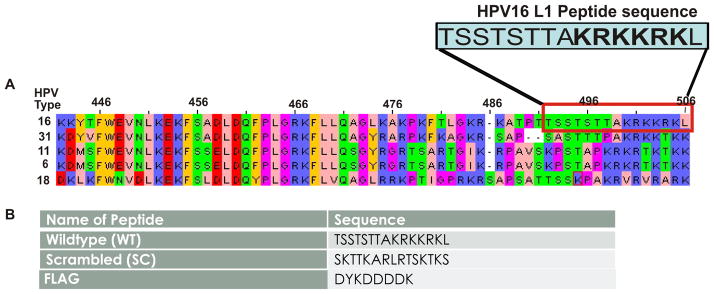
(A) Sequence alignment of major HPV types 16, 31, 11, 6, and 18. (B) Sequences of our synthetic peptides, WT and SC, and of the FLAG epitope peptide used as a non-specific control.
WT peptide effectively blocks infection by HPV-16, in comparison to SC peptide
Our analyses of the L1-C terminus sequences suggest that it may bind to heparan sulfate at the cell surface and contribute to infection. To examine if the L1 C-terminus synthetic peptide would competitively bind to HaCaT and interfere with infection, we tested whether cells treated with peptide experience a decrease in infection. Infection was measured by flow cytometry count of GFP positive cells 48 hours after pseudovirus addition. Addition of WT peptide at increasing concentrations of 1μM, 10μM and 100μM, led to a decrease in infection by 20%, 25% and 72%, respectively (Figure 2A, bars 3, 4, 5). These infection levels were compared to untreated cells and use of FLAG peptide (Figure 2A, bars 2 and 6 respectively). A 1-tailed T-test, comparing viral infection of untreated cells to those treated with WT, determined that our findings were significant with p = 0.0002. Control infections in the absence of peptides were normalized to 100%.
Figure 2.

(A) A comparison of HPV-16 infection in cells treated with FLAG peptide and increasing concentrations of WT peptide. The FLAG peptide reduces infection minimally. A 100 uM concentration of WT peptide reduces infection by 72%. (B) A comparison of HPV-16 infection in cells treated with increasing concentrations of SC peptide.
To address if the drop in infection was a result of the WT peptide’s sequence or its charge, we performed a similar experiment with the SC peptide. Pre-incubating cells with SC peptide at increasing concentrations of 1μM, 10μM and 100μM led to a decrease in infectivity of 5%, 11% and 21%, respectively (Figure 2B). These data suggested that the synthetic WT peptide competitively reduced infection in a sequence-dependent manner and not based solely on charge.
WT Peptide has no effect on Adenovirus infection
Our synthetic peptide is highly amphipathic in nature. Nonpolar, hydrophobic serine and threonine residues are concentrated towards one end of the peptide while polar, hydrophilic arginine and lysine residues are concentrated at the other end. It is the tendency of short, amphipathic peptides to bind into the plasma membrane, disrupting cell structure and leading to cell death. To ensure the drop in infection observed in the presence of the peptide in Figure 2 was not just an artifact of cell death, we performed the same peptide competition assay described in Figure 2 with Adenovirus (Ad-5 GFP) used as the infectious agent rather than HPV-16. Infection levels in the absence of peptide was normalized to 100% and compared to infection in the presence WT and SC peptide (Figure 3).
Figure 3.

Adenovirus infection in cells treated with the synthetic WT and SC peptide.
Cells pre-treated with WT peptide prior to Ad-5 GFP showed no drop in infection when compared to cells treated with Ad-5GFP alone (Figure 3, compare bar Adenovirus and bar Adenovirus, WT pep). There was also no change in infection in cells pre-treated with SC peptide. These data supports that the mechanism by which the synthetic peptide interferes with HPV-16 infection is not a result of its intercalation with the plasma membrane and causing cell death. During all the flow cytometry, Propidium Iodide (PI) staining was used to look for dead cells. No significant cell death was observed with addition of any of the peptides used.
L1 C-terminus peptide Early endosome trafficking is blocked by presence of interfering peptide
After observing decreased infection in cells treated with the WT peptide, we were interested in understanding whether the peptide was interfering with viral infection at the level of pseudovirus binding or during endocytosis. We observed no significant change in the amount of pseudovirus bound when comparing infection of cells treated with WT or SC peptide: at 60 minutes, 746,678 pixels of viral signal were counted in confocal images taken of control infections as compared to 657,483 the presence of WT peptide and 616,747 pixels in presence of SC peptide. Having observed that initial binding of the pseudovirus to the cell was not significantly affected by peptide treatment, we hypothesized that perhaps we would find decreased internalization or trafficking. An immunofluorescence assay was performed to visualize the location of HPV-16 in relation to early endosome marker EEA-1 or the late lysosomal marker LAMP-2. Figure 4 panels A–C show pseudovirus location four hours post-internalization and panels D–F shows pseudovirus location six hours post-internalization. EEA-1 (A–C) and LAMP-2 (D–F) are shown in red, pseudovirus in green and DNA in blue (A–F). Yellow color demonstrates co-localization of HPV-16 signals with EEA-1 or LAMP-2. Co-localization of signals was analyzed quantitatively using ImageJ, as discussed in Experimental Procedures. Pixels of overlapping red and green signal (yellow) were divided by pixels of pseudovirus signal to determine percentage of pseudovirus co-localized shown in graph (Figure 4G and H). At four hours post-internalization, we observed 3% co-localizations of EEA-1 and pseudovirus in cells without peptide (Figure 4G bar HPV16) as compared to 0% in cells treated with WT peptide (Figure 4G bar HPV16, WT) and 14% in SC peptide (Figure 4G bar HPV16, SC). At six hours, co-localization of LAMP-2 and pseudovirus was at 1% in absence of peptide (Fig 4H bar HPV16), 6% with WT peptide (Fig 4H bar HPV16, WT), and 1% with SC peptide (Fig 3H bar 4 HPV16, SC).
Figure 4.

(A–C) EEA-1 is shown in red, HPV-16 in green and DNA in blue. Cells shown were observed at four hours post viral internalization. (A) No peptide was added. (B) WT peptide was added. (C) SC peptide was added. (D–F) LAMP-2 is shown in red, HPV-16 in green, and DNA in blue. Cells shown were observed at six hours post viral internalization. (D) No peptide was added. (E) WT peptide was added. (F) SC peptide was added. (G) Overlap of HPV-16 and EEA-1 signal (yellow) was quantified using ImageJ. (H) Overlap of HPV-16 and LAMP-2 signal (yellow) was quantified using ImageJ.
These data suggest that the presence of WT peptide interferes with pseudovirus entry into endosomes and reroutes intracellular trafficking of HPV-16 to the lysosomes. It has been previously suggested that the non-infectious viral particles end up in the lysosome [16].
WT peptide binds to heparan sulfate
In order to determine if the WT peptide was binding to surface or ECM exposed heparan sulfate, we treated cells with heparanase before addition of peptide. Using confocal images and ImageJ we counted pixels of peptide signal present heparanase treated and untreated cells (Figure 5). Heparanase treated cells showed a dramatic decrease of bound WT peptide from 1,713,395 pixels (Figure 5 bar 2), to 307,008 pixels in heparanase treated cells (Figure 5 bar 5). Level of bound SC peptide was much lower than that of WT peptide, but experienced a similar decrease, with heparanase treated cells containing 265,210 pixels (Figure 5 bar 3) versus 26,267 pixels in heparanase treated cells (Figure 5 bar 6).
Figure 5.
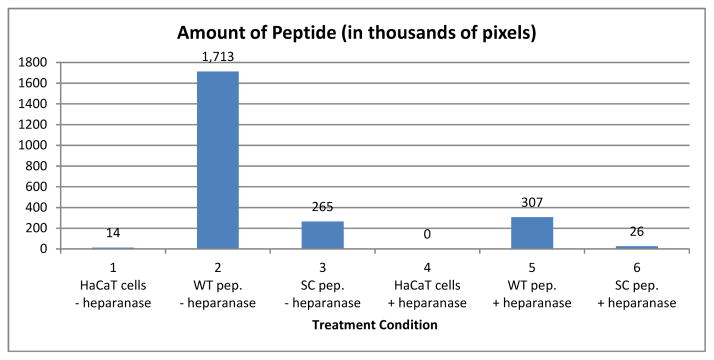
Amount of peptide bound to control cells compared to cells treated with heparanase was quantified from confocal images using ImageJ.
These data show the peptide binds to sites of sulfated heparan on the host cell or on the ECM. Furthermore the finding that less SC peptide binds to heparan sulfate than WT peptide (compare 5C to 5E and 5D to 5F) supports the sequence-dependent nature of peptide binding. These data also demonstrate that peptide binding is occurring at the cell membrane, and that peptide is not intercalating at plasma membrane; this reinforces previous data that the peptide is not decreasing HPV-16 infection through cell death, i.e., amphipathic effect.
L1 C-terminus peptide interacts with syndecan-1 and α6 integrin
All our data suggested that the peptide is preventing infection by competitively binding to heparan sulfate. To further explore this, we examined peptide location in relation to two surface proteins that have been implicated to play a role in the initial endocytosis of the virus. Syndecan-1 is a proteoglycan known to have heparan sulfate chains on the cell surface exposed regions, and α6 integrin is a cell surface protein involved in in-out or out-in signaling. Syndecan-1 and α6 integrin have been suggested to be HPV receptors and to be involved in the formation of high molecular weight complexes during endocytosis [4, 16].
Figure 6 shows peptide interaction with syndecan-1 after two hour incubation on ice (i.e., binding event) and interaction with α6 integrin under the same conditions. ImageJ was used to quantify the amount of colocalization between peptide present and membrane proteins using images acquired through confocal microscopy. Data showed that there was significant overlap of WT peptide with syndecan-1 as compared to WT peptide and α6 integrin (compare Fig 6 bar B with bar E). Specifically, colocalization of WT peptide with syndecan-1 was 20% (Fig 6 bar B) and colocalization of the WT peptide with α6 integrin was 1% (Fig 6 bar E). The amount of SC peptide colocalized with syndecan-1 or integrin was less than 2% (Fig 6 bars C and F). These data showing that the peptide was bound preferentially to syndecan-1, and our data showing loss of binding of peptide on cells treated with heparanase support that the block of infection is occurring at a HSPG.
Figure 6.

Colocalization of peptide with syndecan-1 or peptide with α6 integrin was quantified from confocal images using ImageJ.
WT Peptide Interferes with HPV-16 transition to the secondary receptor
To examine if our synthetic peptide interferes with viral entry at a described secondary binding step, we performed colocalization studies of the peptides with α6 integrin and tetraspanin CD151. α6 integrin and tetraspanin CD151 are two cell-surface receptors, which have been shown to engage in complexes with HPV-16 at this secondary binding step. A fluorescence assay was performed to visualize the location of HPV16 in relation to α6 integrin or tetraspanin CD151 in the presence or absence of peptide. Figure 7A–C shows confocal images taken one hour post viral internalization. α6 integrin is shown in red, pseudovirus in green, and DNA in blue. Figure 7D–F show confocal images taken two hours post viral internalization. Tetraspanin151 is shown in red, pseudovirus in green, and DNA in blue. Colocalization between pseudovirus and integrin or tetraspanin signal was quantified and compared using ImageJ (Figure 7G and H).
Figure 7.

(A–C) Colocalization of pseudovirus with α6 integrin was compared in the presence and absence of peptide. α6 integrin is shown in red, HPV16 in green and DNA in blue. Yellow signal represents overlap of alpha6 and HPV16 signals. Cells shown were fixed 1 hour post viral addition and binding. (A) No peptide was added. (B) WT peptide was added. (C) SC peptide was added. (D–F) Colocalization of pseudovirus with Tetraspanin CD151 was compared in the presence and absence of peptide. Tetraspanin CD151 is shown in red, HPV16 in green and DNA in blue. (D) No peptide was added. (E) WT peptide was added. (F) SC Peptide was added. (G) Overlap between HPV16 and alpha6 integrin signal (yellow) immediately after and 1 hour after viral addition was quantified using ImageJ. (H) Overlap between HPV16 and tetraspanin signal (yellow) 2 hours after pseudovirus addition was quantified using ImageJ.
One hour into infection, 9% of pseudovirus colocalized with integrin in the absence of peptide but only 2% in the presence of WT peptide (Figure 7G, compare bar 60′ HPV16 to bar 60′ HPV16, WT). We observed 6% of pseudovirus colocalized with integrin in the presence of SC peptide (Figure 7G, bar 60′ HPV16, SC).
Two hours into infection, 46% of pseudovirus colocalized with tetraspanin CD151 in the absence of peptide but only 12% in the presence of WT peptide (Figure 7H, compare bar HPV16 to bar HPV16, WT). In the presence of the SC peptide, 21% of pseudovirus colocalized with tetraspanin (Figure 7H bar HPV16, SC).
These data suggest that the WT peptide is preventing the binding of the virus with the secondary receptor. The observation that the SC peptide also decreases viral binding to integrin (though to a lesser extent), supports evidence that the SC peptide also partially blocks infection. We hypothesize that the SC peptide binds competitively to the putative intermediary step due to its electrostatic properties but that this interference is limited because of the scrambled arrangement of its amino acids.
Peptide interferes at the early steps of binding
To better understand when the peptide was blocking infection, we performed a peptide competition assay on ice. The addition of pseudovirus and peptide was entirely done on ice to prevent viral internalization. The goal was to determine the timeframe when the peptide acts to block infection. In figure 9, lanes A–C show that pseudovirus infection (lane B) is still reduced when WT peptide is added simultaneously with pseudovirus. In our initial experiments shown in figure 2, the cells were pre-incubated with peptide for 45 minutes (−45 minutes) prior to pseudovirus addition. Addition of peptide 30 minutes after pseudovirus (bar F) reduced infection by 52%, and addition of WT peptide 1hour after infection (bar H) reduced infection by only 28%. These cells experienced the same amount of peptide incubation although the length of pseudovirus incubation was extended by 30 minutes or 1 hour respectively. We kept the peptide/cell incubation times constant in order to ensure that results were not due to peptide cytotoxicity.
Figure 9.

(A) Our proposed model of HPV-16 binding. The initial viral interaction occurs electrostatically between the basic amino acids of the L1 capsid protein and cell-surface HSPGs. The viral capsid then undergoes a conformational change, exposing a previously internal and inaccessible region. Potentially, a complex forms between syndecan-1 and the integrin complex and the virus proceeds to the secondary binding step. The virus internalizes and is trafficked to the early endosomes. (B) Soluble heparan coats the virus surface preventing initial binding of the viral particle to the host cell. This prevents viral binding and internalization. (C) The virus binds electrostatically to the HSPG receptor but our synthetic WT peptide prevents the virus from a second intermediary HSPG binding step.
DISCUSSION
HPV-16 has been identified as the most commonly found oncogenic HPV type. Many questions remain regarding how HPV binds to host cells prior to entry. By gaining a better understanding of the interaction between the host cell and the virus at the cell surface, we hope to contribute to novel therapeutic strategies for reducing HPV infections.
It has been demonstrated that initial binding of HPV occurs between the virus’s L1 capsid protein and attachment receptors on the cell surface. In normal HPV binding, a secondary binding event has been demonstrated to allow the virus to engage cell membrane receptors in a high molecular weight (HMW) complex prior to intracellular trafficking perhaps via a retromer to the Golgi complex as recently described [4, 12, 17]. This HMW complex at the cell surface may encompass a HSPG, growth factor receptor, tetraspanin, and an integrin.
While the details of the initial and secondary binding events have been studied extensively, it is still contended how the virus transitions between the two. Initial binding of HPV types 16 and 18 to heparan sulfate has been found to be strongly exothermic and results in tight binding between the membrane receptor and viral protein [18]. It remains unclear why the virus would suddenly lose affinity for the primary binding site and transition to the secondary receptor complex. Of particular importance to this binding mechanism may be the amino acids of the L1 C-terminus.
During the preparation of this manuscript, a manuscript from Dr. Sapp’s laboratory was published proposing that following initial binding of HPV to the cell surface or ECM, a conformational change occurs in the L2 and L1 capsid [19]. This paper found that the conformational change L1 was required for viral internalization. This paper also suggested that a different set of residues than those required for initial binding engage in attachment to cell-surface receptors after this conformational change.
We hypothesized that there exists an intermediary step mediated by the region exposed after the L1 conformational change that results in the transfer, or movement, of the viral particle to the HMW complex. Our data suggest that the viral capsid undergoes a conformational change following primary binding that exposes this previously inaccessible L1 C-terminus intercapsomeric region. Our data also suggest that this L1 C-terminus sequence binds in a sequence-specific manner to the heparan sulfate chain of the HSPG prior to the viral particle binding to the HMW complex. In this manuscript, we present evidence that suggests the existence of this intermediary binding step in HPV 16 pseudovirion infection of HaCaT cells.
We performed infection competition experiments using a short peptide that contains the same residues of the highly conserved sequence found in the L1 C-terminus. As a control for sequence vs. charge specificity, we designed a randomly scrambled version of this sequence (SC peptide). Our hypothesis was that a secondary sequence specific binding to heparan sulfate occurs prior to the binding of HPV to an endocytic complex. The SC peptide maintained the same overall charge as the WT peptide, but its scrambled order allowed us to use it as a control to test the specific nature of the putative intermediary binding step, in particularly the role of the KRKKRK region. In our experiments, the addition of the WT peptide does not result in the loss of binding (see virus staining on Figures 4 and 5) but results in greater than 70% loss in infection. The data show that the sequence of the peptide is primarily responsible for the loss of infection as the drop of infection with the scrambled sequence peptide is minimal. The fact that cells with SC peptide did experience some drop in infection compared to cells untreated with peptide suggested that the peptide may experience either electrostatic binding or that there still remains some sequence similarity allowing SC to have proper binding. Indeed, we do see low binding of SC peptide in our experiments. Further mutations variations of amino acid sequence of the L1-C terminus peptides are needed to address the loss of infection observed by SC peptide.
We removed heparan sulfate (HS) from the culture cells using heparanase and observed a loss of WT and SC peptide binding. The HS is found on the cell surface receptors and on the extracellular matrix. Loss of peptide binding showed that the peptides are not intercalating into the membrane as some amphipathic peptide can. These data showing that the WT peptide was preferentially binding to HSPG was supported by experiments that showed that one likely protein to which the peptides bind is the HS-containing syndecan-1. We found large percentage of bound WT peptide colocalizing with syndecan-1 as compared to SC peptide. The data suggest that syndecan-1 may provide the primary binding sugar prior to the binding to the secondary binding complex. Because α6 integrin protein has been often identified in this putative secondary HPV receptor complex, we performed colocalization experiments with the peptide and α6 integrin. Unlike the overlapping with syndecan-1, we observed an insignificant amount of both SC and WT peptide bound to integrin, i.e, less than 3% of peptide was binding to the integrin, a level we believe to be due to non-specific sticking. These data support our hypothesis that the WT peptide is binding to HS and is preventing the transfer, or movement, of viral particles (or HSPG-virus complex) to the HMW endocytic complex.
It is of interest to note that in Adenovirus infection, a similar sequence KKTK has been suggested as being involved in HSPG binding or relevant to intracellular trafficking or sorting [20, 21]. In those experiments, infection was blocked in the presence of soluble heparan sulfate when using Adenovirus serotypes containing KKTK and the intracellular trafficking (including to an endosome) of KKTK mutant serotypes was altered. Our data showed that the WT peptide was not interfering with virus binding to the cell and suggested that peptide was binding to heparan sulfate on a HSPG, thus we investigated if the trafficking of the viral particles to the early endosome in the presence of the interfering peptide was altered. Indeed our data showed a decrease in virus trafficking to endosomes in the presence of WT peptide as compared to viral trafficking in the presence of the SC peptide, or in the absence of peptide. In addition, the data showed that in the presence of the WT peptide more virions were colocalized/trafficked to the lysosome (using marker LAMP-2) as compared to infection in the presence of the SC peptide or in the absence of peptide.
Our data combined with published literature, lead us to propose that under normal conditions HPV binds electrostatically to a HSPG (e.g., syndecan-1). Subsequently, after a conformational change occurs in the capsid, the virus has a secondary, “intermediary” binding event to HS prior to the formation of a complex that includes an integrin complex (see our model in Fig 9A). This complex would then internalize the virus into an EEA1 positive endosome. This is based on the following data: 1) addition of soluble heparan sulfate to infection experiments results in the coating of the viral particles, thus reducing infection by preventing the binding of the viral particle to the HSPG (Figure 9B), and 2) our finding that the presence of a peptide corresponding to the L1 C-terminus sequence during infection does not prevent the initial electrostatic binding of the viral particle to the HSPG, but does prevent a secondary binding step on the HS. During the preparation of this manuscript, a manuscript from Dr. Sapp’s laboratory described how the initial binding even prepares the virus for subsequent event by forcing a conformational change. The conformation is sufficient to expose previously hidden portions of L2 and L1 and changes in affinity [19]. We propose that this secondary step is an intermediate step between initial binding and the formation of a virus/HMW complex necessary for endocytosis into an endosome. Our data suggest that in the presence of WT peptide the virus does not traffic to the endosome and is diverted to the lysosome, and these two events contribute to the loss of infection.
Figure 8.
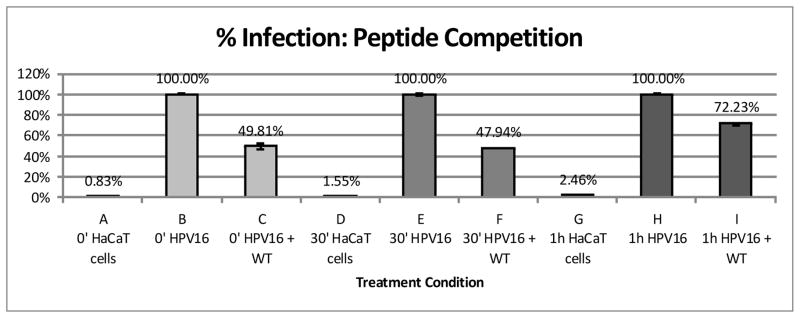
WT Peptide was added to infections at various timepoints (in relation to the addition of pseudovirus). Percent infection was compared between different time points of peptide addition.
Acknowledgments
The research was supported by the National Cancer Institute/NIH R21CA153096 to PIM, and by the American Cancer Society RSG 121721 to PIM. Thanks to Drs. Ozbun, Christensen, and Buck for reagents. We thank J Alexander and D Argyle for technical assistance, and Dr. Everly, C. Awad, X. Simon, and A Gonzalez for the reading of the manuscript.
References
- 1.Furumoto H, Irahara M. Human papilloma virus (HPV) and cervical cancer. The journal of medical investigation: JMI. 2002;49(3–4):124–33. [PubMed] [Google Scholar]
- 2.Knipe DM, HP, editors. Fundamental virology. 4. Lippincott Williams &Wilkins; Philadelphia, PA: 2001. p. 1395. [Google Scholar]
- 3.Cutts FT, et al. Human papillomavirus and HPV vaccines: a review. Bulletin of the World Health Organization. 2007;85(9):719–26. doi: 10.2471/BLT.06.038414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Surviladze Z, Dziduszko A, Ozbun MA. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS pathogens. 2012;8(2):e1002519. doi: 10.1371/journal.ppat.1002519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lipovsky A, et al. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(18):7452–7. doi: 10.1073/pnas.1302164110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buck CB, et al. Arrangement of L2 within the papillomavirus capsid. Journal of virology. 2008;82(11):5190–7. doi: 10.1128/JVI.02726-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Modis Y, Trus BL, Harrison SC. Atomic model of the papillomavirus capsid. Embo J. 2002;21(18):4754–62. doi: 10.1093/emboj/cdf494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buck CB, et al. Maturation of papillomavirus capsids. J Virol. 2005;79(5):2839–46. doi: 10.1128/JVI.79.5.2839-2846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schiller JT, Day PM, Kines RC. Current understanding of the mechanism of HPV infection. Gynecologic oncology. 2010;118(1 Suppl):S12–7. doi: 10.1016/j.ygyno.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knappe M, et al. Surface-exposed amino acid residues of HPV16 L1 protein mediating interaction with cell surface heparan sulfate. The Journal of biological chemistry. 2007;282(38):27913–22. doi: 10.1074/jbc.M705127200. [DOI] [PubMed] [Google Scholar]
- 11.Lortat-Jacob H, Grosdidier A, Imberty A. Structural diversity of heparan sulfate binding domains in chemokines. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(3):1229–34. doi: 10.1073/pnas.032497699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selinka HC, et al. Further evidence that papillomavirus capsids exist in two distinct conformations. Journal of virology. 2003;77(24):12961–7. doi: 10.1128/JVI.77.24.12961-12967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerqueira C, et al. Heparin increases the infectivity of Human Papillomavirus Type 16 independent of cell surface proteoglycans and induces L1 epitope exposure. Cellular microbiology. 2013 doi: 10.1111/cmi.12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buck CB, et al. Efficient intracellular assembly of papillomaviral vectors. J Virol. 2004;78(2):751–7. doi: 10.1128/JVI.78.2.751-757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deschuyteneer M, et al. Molecular and structural characterization of the L1 virus-like particles that are used as vaccine antigens in Cervarix, the AS04-adjuvanted HPV-16 and -18 cervical cancer vaccine. Human vaccines. 2010;6(5):407–19. doi: 10.4161/hv.6.5.11023. [DOI] [PubMed] [Google Scholar]
- 16.Dabydeen SA, Meneses PI. Smurf2 alters BPV1 trafficking and decreases infection. Archives of virology. 2011;156(5):827–38. doi: 10.1007/s00705-011-0924-0. [DOI] [PubMed] [Google Scholar]
- 17.Evander M, et al. Identification of the alpha6 integrin as a candidate receptor for papillomaviruses. Journal of virology. 1997;71(3):2449–56. doi: 10.1128/jvi.71.3.2449-2456.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jian Sun J-SY, Zhiwu Yu, Xiao Zha, Yuqing Wu. The differences in heparin binding for the C-terminal basic-sequence-rich peptides of HPV-16 and HPV-18 capsid protein L1. J Chem Thermodynamics. 2012;47:130–137. [Google Scholar]
- 19.Richards KF, et al. Multiple heparan sulfate binding site engagements are required for the infectious entry of human papillomavirus type 16. Journal of virology. 2013;87(21):11426–37. doi: 10.1128/JVI.01721-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Paolo NC, Kalyuzhniy O, Shayakhmetov DM. Fiber shaft-chimeric adenovirus vectors lacking the KKTK motif efficiently infect liver cells in vivo. Journal of virology. 2007;81(22):12249–59. doi: 10.1128/JVI.01584-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicklin SA, et al. The influence of adenovirus fiber structure and function on vector development for gene therapy. Molecular therapy: the journal of the American Society of Gene Therapy. 2005;12(3):384–93. doi: 10.1016/j.ymthe.2005.05.008. [DOI] [PubMed] [Google Scholar]