

Abstract
A new radiosynthetic protocol for the preparation of [11C]aryl nitriles has been developed. This process is based on the direct reaction of in situ prepared L•Pd(Ar)X complexes (L=biaryl phosphine) with [11C]HCN. The strategy is operationally simple, exhibits a remarkably wide substrate scope with short reaction times, and demonstrates superior reactivity compared to previously reported systems. With this procedure, a variety of [11C]nitrile-containing pharmaceuticals were prepared with high radiochemical efficiency.
Incorporation of short-lived radioisotopes such as 11C (t1/2 = 20.4 minutes) into organic molecules for application in positron emission tomography (PET)1 presents unique synthetic challenges.2 The short half-life of 11C necessitates reaction times on the order of seconds/minutes with clean reaction profiles to facilitate rapid purification. Additionally, the radiochemical reaction must outcompete all possible side reactions, which can be a challenge because there is a large stoichiometry bias between radionuclide and all other species present in the reaction mixture (including the carrier gas), and submicromolar concentrations of radionuclides are typically employed. Lastly, the reaction conditions should be mild (i.e., room temperature under neutral conditions) to minimize decomposition of the radiotracer that is formed. Due to these restrictions, many high-yielding catalytic and even stoichiometric reactions cannot be applied to the synthesis of radiolabeled compounds.
A promising method to introduce a radioactive carbon unit into organic molecules is the Pd-mediated cyanation of aryl (pseudo)halides (Scheme 1).3,4 This transformation is based on oxidative addition (OA) of the aryl (pseudo)halide to an “L•Pd(0)” intermediate to form complex A, transmetallation (TM) with cyanide, and reductive elimination (RE) to form the desired aryl nitrile product.
Scheme 1.
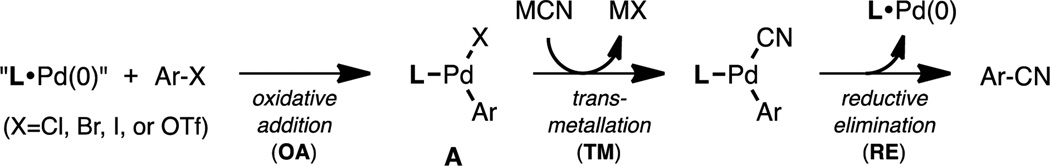
Mechanism of Pd-mediated cyanation of aryl (pseudo)halides.
However, extension of this strategy to the radiosynthetic setting does not necessarily meet the challenges discussed above.2,3 The use of traditional ligands (e.g., PPh3 or 1,1′-bis(diphenylphosphino)ferrocene (dppf)) requires harsh reaction conditions, such as high temperatures, relatively long reaction times, and the use of base (e.g., K2CO3) to capture [11C]HCN in the gas phase, which diminishes the substrate scope (Scheme 2, a).5 In addition, these reactions are typically carried out in DMF or DMSO, which can hamper purification or multi-step syntheses involving reactions incompatible with these solvents. On the other hand, we have recently demonstrated that biaryl phosphine ligands, specifically L1 (Figure 1), facilitate stoichiometric transmetallation with cyanide and reductive elimination of aryl nitriles at ambient temperature in under 5 minutes.6 Thus, we hypothesized that [11C]cyanation of aryl (pseudo)halides, even with an extremely low concentration of [11C]HCN, could be achieved under mild conditions using biaryl phosphines instead of traditional ligands (Scheme 2, b).
Scheme 2.

Synthetic strategies for [11C]aryl nitriles.
Figure 1.

Structure of ligands L1–L4 and complexes 1–4.
Even with this strategy, reagent activation (i.e., generation of “L•Pd(0)” from the Pd source) and oxidative addition, steps that do not involve the radionuclide, still take place after the end of bombardment (EOB), when [11C]HCN addition is complete, resulting in an unnecessary loss of radioactivity. Ideally, a preformed oxidative addition complex A, with L1, could be used directly (Scheme 2, c)7 to avoid this loss of radioactivity. However, the independent synthesis and purification of A, which can be a challenging task for non-experts, would limit the applicability of this strategy to radiotracer synthesis for PET imaging. Recently, we introduced a new class of biaryl phosphine (L1–L4)-bound Pd(0) complexes 1–4, which are functionally equivalent to "L•Pd(0)" at ambient temperature (Figure 1).8 We envisioned that utilizing complexes 1–4, along with the desired aryl (pseudo)halide, should allow for the convenient generation of A in situ to set the stage for the formation of the aryl-[11C]CN bond (Scheme 2, d). This setup should allow us to take full advantage of the established system to prepare [11C]aryl nitriles in a mild and expeditious manner.
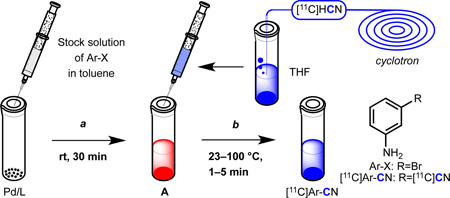
To test this hypothesis, a synthesis of [11C]3-aminobenzonitrile was attempted using 3-bromoaniline as a substrate (Table 1). In these experiments, the Pd source and the aryl halide were mixed and allowed to stand for 30 minutes to generate A in situ (Table 1, a). Then, [11C]HCN in THF9 was introduced into the reaction vial, allowed to react for one minute (Table 1, b), and the reaction mixture was analyzed by radio TLC. The desired product was successfully formed with 81% radiochemical conversion (RCC) in the presence of 1 (entry 1).10 In fact, nearly the same RCC was observed after only 10 seconds of reaction time (entry 2), though subsequent reactions were conducted for 1 minute for consistency. In contrast, conventionally used Pd(0) sources, including [Pd(PPh3)4] (entry 3) and a combination of Pd2(dba)3/dppf (entry 4) did not produce any product at either room temperature or after heating the reaction mixture at 50 °C for 5 minutes. Only when the reaction with [Pd(PPh3)4 was carried out under much harsher conditions (DMF, 100 °C, 5 minutes) was the desired product formed, and then only in low yield (entry 5). Notably, Pd2(dba)3/L1 (entry 6) and L1-bound palladacycle 5,6 alone (entry 7) or with activating base (entry 8), could also be used to furnish the product, though both were less efficient than 1. Therefore, 1 is uniquely effective at forming A at room temperature in high yield, allowing for the effective [11C]cyanation of 3-bromoaniline under extremely mild conditions.
Table 1.
Pd source and ligand effect in the synthesis of [11C]3-aminobenzonitrile.a
 | |||||
|---|---|---|---|---|---|
| Entry | Pd/L | Temp. (°C) | Time | RCC (%) | |
| 1 | 1 | 23 | 1 min | 81 |  |
| 2 | 1 | 23 | 10 s | 73 | |
| 3 | [Pd(PPh3)4] | 50 | 5 min | 0 | |
| 4 | Pd2(dba)3/dppf | 50 | 5 min | 0 | |
| 5 | [Pd(PPh3)4]b | 100 | 5 min | 30 | |
| 6 | Pd2(dba)3/L1 | 23 | 1 min | 57 | |
| 7 | 5 | 23 | 1 min | 27 | |
| 8 | 5, K2CO3, K3PO4 or KOt-Bu | 23 | 1 min | 18–25 | |
Reaction conditions: Pd/L (0.004 mmol), 3-bromoaniline (0.008 mmol), toluene (0.1 mL), rt, 30 min; then [11C]HCN (1–5 mCi), THF (0.1 mL) rt, 1 min.
[Pd(PPh3)4] (0.004 mmol), 3-bromoaniline (0.008 mmol), K2CO3 (0.008 mmol), DMF (0.1 mL); then [11C]HCN (1–5 mCi), DMF (0.1 mL), 100 °C, 5 min.
Having demonstrated the viability of this strategy, we next applied it to the syntheses of a variety of functionalized [11C](hetero)aryl nitriles (Table 2). Although 1 showed the most general reactivity, the ease of setting up these reactions with a stock solution of [11C]HCN in THF allowed for the quick determination of the ideal solvent and reagent (1–4) for each substrate. The reaction tolerates nucleophilic amine (6) and hydroxyl (7) groups, as well as a formyl group (8), which is susceptible to benzoin condensation at elevated temperatures.6 Both aryl iodides and bromides can be used to furnish [11C]aryl nitriles in high yield, with aryl chlorides providing lower but still radiosynthetically useful yields (7).11 The electronic nature of the arene did not bias the outcome of the reaction: highly electron-rich (7, 9) and electron-deficient (8, 10) [11C]nitriles were prepared with comparable efficiency. Heterocyclic compounds, such as quinoxaline (11), quinoline (12), thiazole (13) and pyrazole (14), were also viable substrates for this reaction. To our knowledge, these are the first examples of the [11C]-cyanation of 5-membered heteroaryl halides. Finally, a [11C]CN-labeled derivative of estrone was successfully synthesized from the corresponding triflate with high radiochemical yield (15). In all cases, the same straightforward setup as described above was used, and the results proved to be highly reproducible.
Table 2.
Radiosyntheses of [11C]aryl nitriles.a
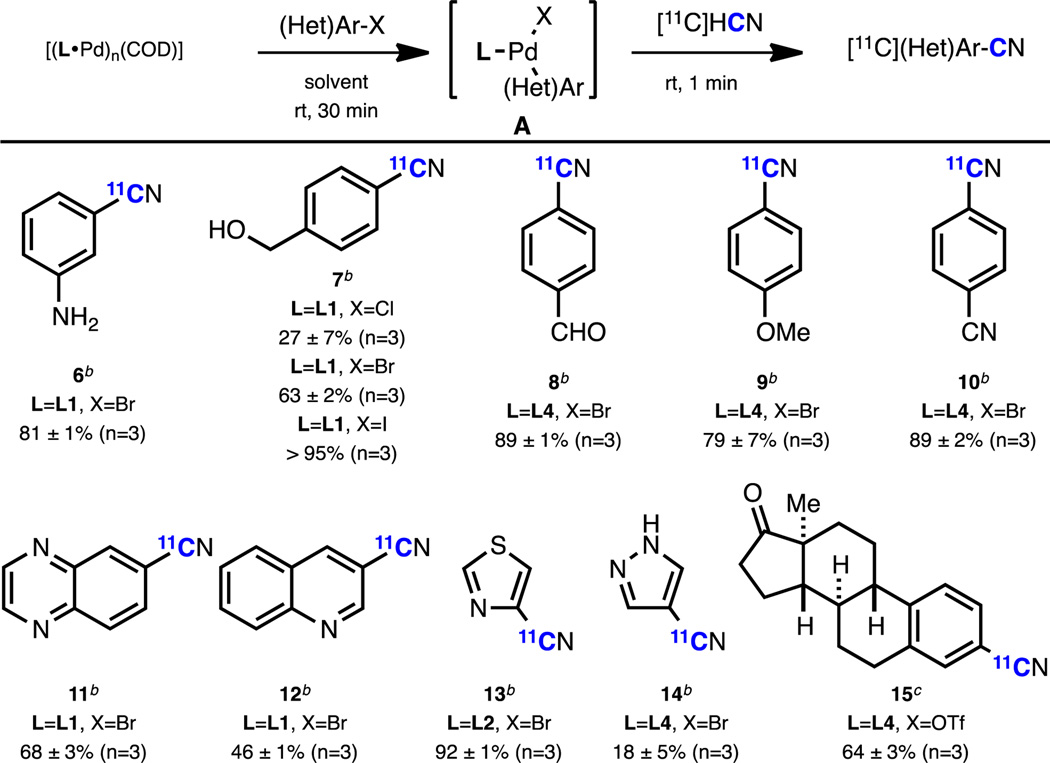 |
Reported values indicate radiochemical conversion. Reaction conditions: [(L•Pd)n(COD)] (0.004 mmol in Pd), ArX (0.008 mmol), solvent (0.1 mL), rt, 30 min; then [11C]HCN (1–5 mCi), THF (0.1 mL) rt, 1 min.
Toluene as solvent.
THF as solvent.
The susceptibility of the [11C]-cyanation of 3-bromoaniline with 1 or [Pd(PPh3)4] to inhibition by common contaminants and functional groups was also evaluated (Table 3).12 Potential inhibitors were placed in the reaction vessel together with the aryl halide before carrying out oxidative addition to form A. The presence of common acidic compounds, such as water, phenol, or an amide did not significantly affect the outcome of the reaction with 1 (entries 2–4a). Nitrogen-containing heterocycles that can impede metal-catalyzed cross-couplings, such as pyridine (entry 5a), imidazole (entry 6a), and indole (entry 7a), also did not show any inhibitory effects when using 1. In contrast, while the reaction with [Pd(PPh3)4] could tolerate phenol (entry 3b), the presence of a free amide (entry 4b) or pyridine (5b) diminished the yield, and imidazole completely inhibited the desired reaction (entry 6b). Thus, reactions carried out with 1 at room temperature are more robust than those carried out with [Pd(PPh3)4] at 100 °C.
Table 3.
Robustness screen of the radiosynthesis of [11C]3-aminobenzonitrile with 1 (a) and [Pd(PPh3)4] (b).a
 | |||
|---|---|---|---|
| Entry | Additive | RCC (%) w/ 1a (a) | RCC (%) w/ [Pd(PPh3)4]b (b) |
| 1 | None | 81 | 30 |
| 2 | water | 80 | n.d. |
| 3 | phenol | 94 | 25 |
| 4 | 4-methoxybenzamide | 88 | 9 |
| 5 | pyridine | 90 | 14 |
| 6 | imidazole | 92 | 0 |
| 7 | 5-fluoroindole | 80 | n.d. |
Reaction conditions: 1 (0.002 mmol), additive (0.008 mmol), 3-bromoaniline (0.008 mmol), toluene (0.1 mL), rt, 30 min; then [11C]HCN (1–2 mCi), THF (0.1 mL) rt, 1 min.
[Pd(PPh3)4] (0.004 mmol), additive (0.008 mmol), 3-bromoaniline (0.008 mmol), K2CO3 (0.008 mmol), DMF (0.1 mL); then [11C]HCN (1–5 mCi), DMF (0.1 mL), 100 °C, 5 min.
n.d. =not determined.
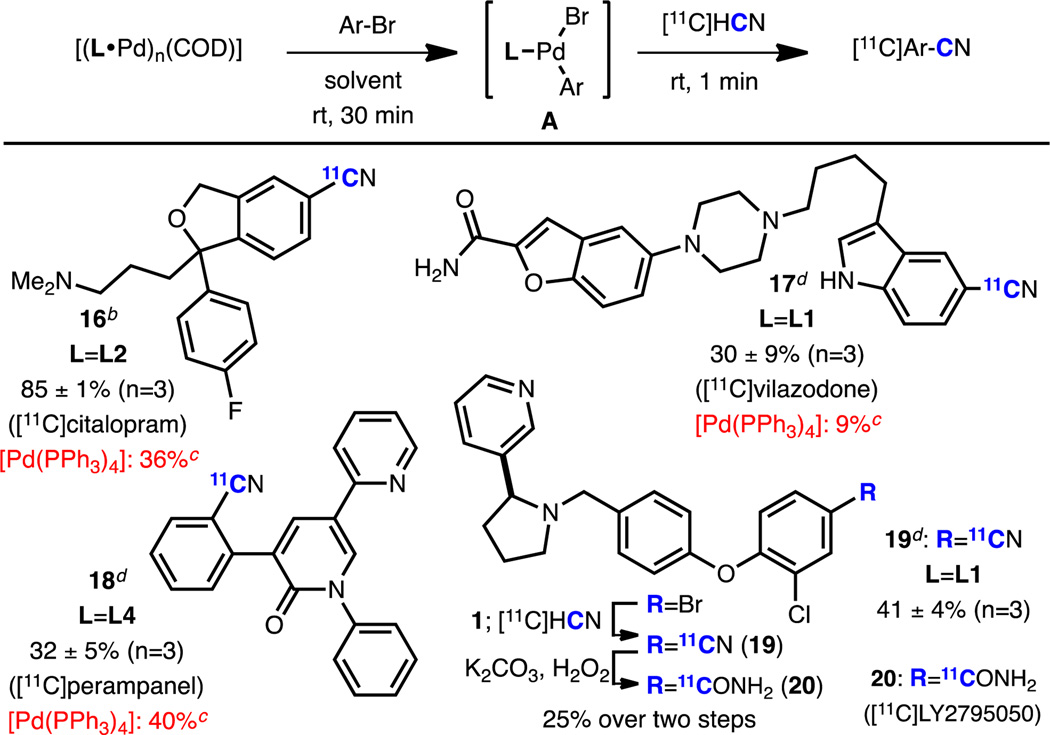
Another motivation in the development of our [11C]cyanation protocol is the abundance of cyano groups in pharmaceutically important small molecules,13 as well as their ability to be readily transformed into other common functional groups such carboxylic acids, amides,14 amidines,15 and tetrazoles.16 Therefore, the syntheses of complex drug-based radiotracers using the corresponding aryl bromides as precursors were carried out (Table 4). All compounds were prepared using the same simple procedure presented above with no need to isolate the oxidative addition complexes. [11C]Citalopram (16, antidepressant),17 [11C]vilazodone (17, antidepressant),18 and [11C]perampanel (18, antiepileptic)19 could be formed with high efficiency. The nitrile precursor to [11C]LY279505020 (19) was also easily prepared. Notably, [11C]LY2795050 itself (20, κ-opioid receptor antagonist) was also effectively prepared with 25% RCC from the corresponding bromide, demonstrating the potential utility of our protocol in multistep radiosyntheses. It is important to note that, to the best of our knowledge, this is the first preparation of [11C]vilazodone and [11C]perampanel. Although 18 could be prepared using [Pd(PPh3)4] with comparable yield as with 1 (albeit with a less clean reaction profile), both 16 and 17 were formed in significantly higher yields with 1 at room temperature than with [Pd(PPh3)4] at 100 °C (Table 4).
Table 4.
Syntheses of [11C]-labeled drugs.a
 |
Reported values indicate radiochemical conversion. Reaction conditions: [(L•Pd)n(COD)] (0.004 mmol in Pd), ArBr (0.008 mmol), solvent (0.1 mL), rt, 30 min; then [11C]HCN (1–5 mCi), THF (0.1 mL) rt, 1 min.
Toluene/acetone (5:1) was used as solvent.
[Pd(PPh3)4] (0.004 mmol), ArBr (0.008 mmol), K2CO3 (0.008 mmol), DMF (0.1 mL); then [11C]HCN (1–5 mCi), DMF (0.1 mL), 100 °C, 5 min.
DMF as solvent.
Finally, high-radioactivity syntheses of compounds 17–19 were carried out to assess applicability of this method for in vivo imaging (Table 5). To avoid loss of activity during transfer, [11C]HCN was captured from the carrier gas into the reaction mixture (vide supra). After achieving maximum radioactivity in the reaction mixture, HPLC eluent was added and the product was purified by reverse-phase HPLC. The absolute (decay uncorrected) radioactivity of the purified product ranged from 15–35 mCi21 and the whole process including chromatography took only 12–23 minutes after the solution capture of [11C]HCN, which is only 0.61–1.1 half lives of 11C. In addition, the specific activities of the products were all sufficiently high and reproducible. This is especially important in the preparation of high specific activity [11C]radiotracers. For comparison, the previous report of the synthesis of [11C]LY2795050 provided the final product with a specific activity of 0.64 Ci/µmol.20 It should also be mentioned that the protocol is suitable for automation and remote control due to the ease of setup and purification of the product. This permits translation to human PET studies, which require strict validation.22
Table 5.
High-radioactivity synthesis of [11C]-labeled drugs.
 | |||
|---|---|---|---|
| [11C]Ar-CN | 17 | 18 | 19 |
| starting / final activity (mCi) | 188 / 35.3 | 156 / 15.1 | 70.0 / 14.5 |
| RCY (%)a | 18.8 | 9.7 | 20.7 |
| total synthesis time (min)b | 16.5 | 22.5 | 12.4 |
| specific activity (Ci/µmol) | 3.97 | 1.34 | 2.69 |
Decay-uncorrected isolated radiochemical yield at end of synthesis.
Calculated after [11C]HCN delivery peaked to product isolation after HPLC purification.
In conclusion, we have demonstrated the capability of new Pd complexes 1–4 to promote the nearly instantaneous [11C]cyanation of (hetero)aryl halides and triflates at room temperature, an otherwise difficult transformation. The remarkable reactivity stems from the capability of 1–4 to produce oxidative addition complex A in situ. As a consequence of the mild reaction conditions and robustness of this methodology, it exhibits wide functional group tolerance and is highly amenable for high-activity synthesis, even of densely functionalized molecules.
Supplementary Material
ACKNOWLEDGMENT
Technical assistance from Nathaniel Schauer, Judit Sore, and Dr. Ramesh Neelamegam (MGH/HST) is appreciated. We are grateful to Dr. Todd Senecal (MIT) for performing preliminary experiments.
Funding Sources
Research reported in this publication was supported by the National Institutes of Health (GM46059). A portion of this research was carried out at the Athinoula A. Martinos Center for Biomedical Imaging at the Massachusetts General Hospital, using resources provided by the Center for Functional Neuroimaging Technologies, P41EB015896, a P41 Regional Resource supported by the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health. This work also involved the use of instrumentation supported by the National Institutes of Health Shared Instrumentation Grant Program and/or High-End Instrumentation Grant Program (S10RR017208 and S10RR026666). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. P.J.M. thanks the National Science Foundation for a predoctoral fellowship (2010094243). We also thank Amgen for an educational donation. M.S.P. thanks NIH-NIDA for a postdoctoral fellowship (2T32DA015036).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental details and compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest: MIT has obtained or has filed patents on some of the ligands/precatalysts used in this work from which S.L.B. and former/current coworkers receive royalty payments.
REFERENCES
- 1.Ametamey SM, Honer M, Schübiger PA. Chem. Rev. 2008;108:1501. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- 2.Miller PW, Long NJ, Vilar R, Gee AD. Angew. Chem. Int. Ed. 2008;47:8998. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- 3.Alternatively, the less widely employed Rosenmund-von Braun reaction can be used. See reference 2.
- 4.Anbarasan P, Schareina T, Beller M. Chem. Soc. Rev. 2011;40:5049. doi: 10.1039/c1cs15004a. [DOI] [PubMed] [Google Scholar]
- 5.For example, a base induced loss of stereochemical integrity during the radiosynthesis of a [11C]aryl nitrile has been reported: Donohue SR, Pike VW, Finnema SJ, Truong P, Andersson J, Gulyás B, Halldin C. J. Med. Chem. 2008;51:5608. doi: 10.1021/jm800329z.
- 6.Senecal TD, Shu W, Buchwald SL. Angew. Chem. Int. Ed. 2013;52:10035. doi: 10.1002/anie.201304188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.A related strategy for the preparation of [18F]aryl fluorides has been documented. See: Lee E, Kamlet AS, Powers DC, Neumann CN, Boursalian GB, Furuya T, Choi DC, Hooker JM, Ritter T. Science. 2011;334:639. doi: 10.1126/science.1212625.
- 8.Lee HG, Milner PJ, Colvin MT, Andreas L, Buchwald SL. Inorg. Chim. Acta. 2014;422:188. doi: 10.1016/j.ica.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.To set up multiple experiments at a time, a stock solution of [11C]HCN was used, although it could be directly introduced in a gaseous form (vide infra).
- 10.A variety of solvents, including Et2O, DMSO, acetone, and t-amyl alcohol could be used to furnish the product in good RCC (see Supporting Information Table S1).
- 11.Andersson Y, Långström B. J. Chem. Soc., Perkin Trans. 1. 1994:1395. [Google Scholar]
- 12.Collins KD, Glorius F. Nat. Chem. 2013;5:597. doi: 10.1038/nchem.1669. [DOI] [PubMed] [Google Scholar]
- 13.Fleming FF, Yao L, Ravikumar PC, Funk L, Shook BC. J. Med. Chem. 2010;53:7902. doi: 10.1021/jm100762r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andersson Y, Bergstrom M, Långström B. Appl. Radiat. Isot. 1994;45:707. doi: 10.1016/0969-8043(94)90251-8. [DOI] [PubMed] [Google Scholar]
- 15.Siméon F, Sobrio F, Gourand F, Barré L. J. Chem. Soc. Perkin Trans. 1. 2001:690. [Google Scholar]
- 16.Ponchant M, Hinnen F, Demphel S, Crouzel C. Appl. Radiat. Isot. 1997;48:755. [Google Scholar]
- 17.Dorell K, Cohen MA, Huprikar SS, Gorman JM, Jones M. Psychosomatics. 2005;46:91. doi: 10.1176/appi.psy.46.1.91. [DOI] [PubMed] [Google Scholar]
- 18.Robinson DS, Kajdasz DK, Gallipoli S, Whalen H, Wamil A, Reed CR. J. Clin. Psychopharmacol. 2011;31:643. doi: 10.1097/JCP.0b013e31822c6741. [DOI] [PubMed] [Google Scholar]
- 19.Hanada T, Hashizume Y, Tokuhara N, Takenaka O, Kohmura N, Ogasawara A, Hatakeyama S, Ohgoh M, Ueno M, Nishizawa Y. Epilepsia. 2011;52:1331. doi: 10.1111/j.1528-1167.2011.03109.x. [DOI] [PubMed] [Google Scholar]
- 20.Zheng M-Q, Nabulsi N, Kim SJ, Tomasi G, Lin S-F, Mitch C, Quimby S, Barth V, Rash K, Masters J, Navarro A, Seest E, Morris ED, Carson RE, Huang Y. J. Nucl. Med. 2013;54:455. doi: 10.2967/jnumed.112.109512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.PET radiotracer administered doses vary but are generally 0.1–0.3 mCi/kg (human), 0.1–0.5 mCi/kg (non-human primates), 1–5 mCi/kg (rodent) for 11C.
- 22.<823> Positron Emission Tomography Drugs for Compounding, Investigational, and Research Uses. United States Pharmacopeia and National Formulary (USP 35-NF 30) Rockville, MD: United States Pharmacopeia Convention; 2012.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.