

Abstract
Genomic alterations leading to aberrant activation of cyclin/cyclin-dependent kinase (cdk) complexes drive the pathogenesis of many common human tumor types. In the case of glioblastoma multiforme (GBM), these alterations are most commonly due to homozygous deletion of p16INK4a and less commonly due to genomic amplifications of individual genes encoding cyclins or cdks. Here, we describe deletion of the p18INK4c cdk inhibitor as a novel genetic alteration driving the pathogenesis of GBM. Deletions of p18INK4c often occurred in tumors also harboring homozygous deletions of p16INK4a. Expression of p18INK4c was completely absent in 43% of GBM primary tumors studied by immunohistochemistry. Lentiviral reconstitution of p18INK4c expression at physiologic levels in p18INK4c-deficient but not p18INK4c-proficient GBM cells led to senescence-like G1 cell cycle arrest. These studies identify p18INK4c as a GBM tumor suppressor gene, revealing an additional mechanism leading to aberrant activation of cyclin/cdk complexes in this terrible malignancy.
Introduction
Glioblastoma multiforme (GBM) is the most lethal primary brain tumor. Approximately 10,000 GBMs are diagnosed each year in the United States, with an average survival of ~1 year. Activation of the epidermal growth factor signaling pathway by amplification and/or mutation of the epidermal growth factor receptor is found in most GBM tumors (1). Additionally, activation of the phosphatidylinositol-3-OH kinase signaling pathway via mutational inactivation of the PTEN tumor suppressor or mutational activation of the PIK3CA oncogene is also very common in GBM (2, 3). Finally, virtually all GBMs harbor genomic alterations that lead to the constitutive activation of cyclin-dependent kinases (cdk).
Several different genomic alterations are present in GBM tumors that lead to activation of cdks. A remarkable 60% to 80% of GBMs harbor homozygous deletions of the p16INK4a tumor suppressor, which binds to and inhibits cdk4 and cdk6 (4, 5). Another 5% to 10% of GBMs have amplifications of individual cyclins and cdks, including cdk4, cdk6, and cyclin D1 (5–7). As such, it is by now clear that aberrant activation of cdks is a particularly important genetic event contributing to the pathogenesis of GBM.
We have used high resolution single nucleotide polymorphism (SNP) arrays to interrogate the genomes of GBM samples in an effort to identify recurrent copy number alterations that may drive the pathogenesis of GBM. Using this approach, we have identified inactivation of the p18INK4c cdk inhibitor as an additional genomic alteration that drives the pathogenesis of GBM.
Materials and Methods
Cell lines, xenografts, and primary tumors
Cell lines were obtained from the American Type Culture Collection (U87MG, U138MG, M059J, Hs683, H4, A172, LN18, LN229, CCF-STTG1, T98G, and DBTRG-05MG), DSMZ (8MGBA, 42MGBA, DKMG, GAMG, GMS10, LN405, and SNB19), and the Japan Health Sciences Foundation Health Science Research Resources Bank (AM38, NMC-G1, and KG-1-C). Normal human astrocytes (NHAs) were obtained from Clonetics and Allcells. All cell lines were grown in DMEM + 10% fetal bovine serum at 37° in 5% CO2.
S.c. xenografts in immunodeficient mice were obtained from the Duke University Brain Tumor Center or created in the Lombardi Comprehensive Cancer Center Animal Shared Resource from tissue taken from patients undergoing craniotomy at Georgetown University Hospital (IRB #2006-344).
Snap-frozen primary GBM tumors and paired blood samples were obtained from the Brain Tumour Tissue Bank (London Health Sciences Centre) funded by the Brain Tumour Foundation of Canada. All tumors were graded by a neuropathologist as good or moderate on a scale of good to poor depending on the amount of tumor cells present (as opposed to hemorrhagic, necrotic, or fibrous tissue). All tumor samples were further categorized as “tumor center.”
Microarrays and bioinformatics
Genomic DNA derived from GBM cell lines and xenografts was interrogated with Affymetrix 250K Nsp I Human Gene Chip microarrays using protocols described by the manufacturer. Data processing was performed using dCHIP (8, 9). Additional details regarding the protocols used for data acquisition and processing, as well as the complete raw and processed data sets will be presented elsewhere.
PCR
Conventional PCR was performed using Taq Platinum (Invitrogen) as described by the manufacturer. qPCR was performed in an iCycler (Bio-Rad) using the Platinum Taq SYBR Green PCR Supermix (Invitrogen) according to the manufacturer’s instructions. DNA copy number was calculated using the 2−Δ(ΔCt) method, normalizing to the copy number of an arbitrarily chosen region on chromosome 9 that was neither amplified nor deleted in the samples studied. All assays were performed at least in triplicate.
Western blot
Primary antibodies used were as follows: p18INK4c clone DCS118 (Cell Signaling), p16INK4a #554079 (BD-PharMingen), a-tubulin Ab-2 clone DM1A (Neomarkers), and cdk6 clone C-21 (Santa Cruz Biotechnology).
DNA sequencing
Individual exons of p18INK4c were PCR amplified using conditions and primer pairs recently described by Sjoblom et al. (10). PCR products were purified using the Exo/SAP method followed by a Sephadex spin column. Sequencing reactions were performed using Big Dye v3.1 (Applied Biosystems) using an M13F primer, and analyzed on an Applied Biosystems 3730XL capillary sequencer. Sequences were analyzed using Mutation Surveyor (Softgenetics). Traces with putative mutations were reamplified and sequenced from both tumor and matched normal DNA.
Immunohistochemistry
GBM tissue microarrays were obtained from US Biomax and contained 35 cases of GBM spotted in duplicate. Immunohistochemistry was performed using standard techniques using the same antibodies as for Western blot except using p16INK4a antibody #554070 (BD-PharMingen). Antigen-antibody complexes were detected with the avidin-biotin peroxidase method using 3,3′-diaminobenzidine as the chromogenic substrate (Vectastain ABC kit; Vector Laboratories), and sections were counterstained with hematoxylin.
p18INK4c lentivirus
A full-length, wild-type p18INK4c cDNA (MGC 3907917) was obtained from Open Biosystems and cloned into the pCDF1-MCS2-EF1-Puro lentiviral expression vector backbone (System Biosciences). To make virus, this construct was cotransfected into 293T cells with pVSV-G (Addgene) and pFIV-34N (System Biosciences) helper plasmids using Fugene 6 (Roche) as described by the manufacturer. Virus-containing conditioned medium was harvested 48 h after transfection, filtered, and used to infect recipient cells in the presence of 8 μg/mL polybrene.
Flow cytometry
Cells were pulsed with 10 μmol/L BrdUrd for 1 h, trypsinized, and centrifuged. Cells were fixed and stained using the BrdUrd Flow kit (PharMingen) and analyzed by flow cytometry in a BD FACSort instrument using FCS Express v.3 software (DeNovo Software).
β-Galactosidase staining
Cells grown on coverslips were stained with the Senescence β-Galactosidase Staining kit (Cell Signaling) as described by the manufacturer.
Microscopy
All imaging was performed on an Olympus BX61 light microscope with a ×40 Plan-Apochromat objective.
Results
Homozygous deletion of p18INK4c in GBM
In an effort to identify novel copy number alterations that drive the pathogenesis of GBM, we initially interrogated genomic DNA derived from 35 GBM cell lines and xenografts with Affymetrix SNP microarrays, as described in Materials and Methods. This analysis revealed focal deletions of chromosome 1p in 7 of 35 (20%) of samples (Fig. 1A).
Figure 1.

Homozygous deletions of p18INK4c in GBM. A, copy number analysis of Affymetrix 250K SNP microarray data shows focal homozygous deletions of chromosome 1p in four GBM cell lines (left) and three GBM xenografts (right) but not in NHAs. B, PCR on genomic DNA derived from GBM cell lines confirms the presence of homozygous deletions in all four GBM cell lines with putative deletions (SNB19 not shown). C, copy number analysis of Affymetrix SNP microarray data shows p18INK4c deletions in five uncultured primary GBM tumors. The raw SNP microarray data on the primary tumors was generated as part of The Cancer Genome Atlas.
This ~ 200- to 300-kb region of chromosome 1 contains two annotated genes—p18INK4c and FAF1. p18INK4c is a cdk inhibitor that binds to and inhibits cdk4 and cdk6, and is a known tumor suppressor in mice (11, 12). FAF1 binds to the intracellular domain of Fas and is a proapoptotic signal transduction molecule (13). To determine whether either of these genes was the likely target of the deletion, we examined the copy number information at individual probesets to identify the consensus region of deletion (Supplementary Fig. S1). This analysis revealed that three probesets were deleted in all samples studied, narrowing the consensus region of deletion to a 56- to 133-kb interval containing only p18INK4c. These deletions of p18INK4c were confirmed by PCR and qPCR analysis using primer pairs specific to p18INK4c exon 1 (Supplementary Fig. S2; Fig. 1B). Finally, Western blot analysis showed the absence of p18INK4c expression in GBM cells with confirmed homozygous deletions (Supplementary Fig. S3).
p18INK4c deletions in primary tumor samples from the Cancer Genome Atlas
The NIH has recently sponsored a large-scale project to perform genomic analysis on GBM and other tumor types. As part of this project, they have recently released raw Affymetrix SNP microarray data on 106 primary GBM tumors. We analyzed these data with dCHIP to determine the copy number status of p18INK4c in primary GBM tumors. As depicted in Fig. 1C, deletions of p18INK4c were present in five tumors (5%). These deletions were not present in constitutional DNA from these patients studied with the same Affymetrix SNP microarrays (data not shown), demonstrating that the deletions are somatic. The available clinical and pathologic details of these samples are presented in Supplementary Fig. S4. These data show that deletions of p18INK4c are identifiable in uncultured primary human GBM samples, albeit at a lower apparent frequency than in our cultured samples.
Admixed nonneoplastic cells complicate the identification of p18INK4c deletions in uncultured GBMs
We hypothesized that the lower apparent frequency of p18INK4c deletions in uncultured GBM samples was due, at least in part, to the presence of admixed nonneoplastic human cells that are eliminated during ex vivo growth. To directly test this, we performed SNP microarray analysis on a primary GBM tumor and a matched first-passage xenograft that we derived from the tumor. Deletion of p18INK4c was easily detectable in the first passage xenograft but was much less apparent (and would have been missed) in the primary tumor from which the xenograft was derived (Fig. 2A). This experiment clearly shows that the presence of admixed normal human cells is one factor that confounds the identification of p18INK4c deletions in primary tumors. Xenograft growth eliminates admixed nonneoplastic human cells, enabling more efficient detection of p18INK4c deletions with available technologies.
Figure 2.

Heterogeneity of p18INK4c deletions in GBM. A, copy number analysis of Affymetrix SNP microarray data shows deletion of p18INK4c in a first-passage xenograft that is largely obscured by admixed normal cells in the primary tumor from which it was derived. B, copy number analysis of the p16INK4a gene in cell lines (left), xenografts (middle), and primary tumors (right) harboring deletions of p18INK4c reveals striking homogeneity with regard to p16INK4a deletion in all sample types.
Heterogeneity of p18INK4c deletion in GBM
Despite eliminating the influence of admixed nonneoplastic human cells by ex vivo growth, we did not observe complete copy number reduction at the p18INK4c locus in either GBM xenografts or uncultured primary tumors (Fig. 1A and C). To determine whether this was an issue with sample quality or instead reflected a more fundamental underlying biology, we examined the copy number at the p16INK4a locus in the same GBM samples that harbored deletions of p18INK4c. Deletions of p16INK4a were present in 12 of these 14 samples (86%). As expected, in cell lines, the copy number for both p16INK4a and p18INK4c was zero, reflecting the homogeneity of cultured cell lines (compare Fig. 2B to identical samples in Fig. 1A). In contrast, in more genetically heterogeneous xenografts and uncultured primary tumors, the copy number for p16INK4a was near zero, whereas the copy number for p18INK4c was 0.5 to 1.0 (compare Fig. 2B to identical samples in Fig. 1A and C). This analysis clearly shows that GBMs are homogenous with regard to p16INK4a deletions and more heterogeneous with regard to p18INK4c deletions.
Loss of p18INK4c expression in GBM primary tumors
We next used immunohistochemistry to measure p18INK4c expression in individual cells in primary GBM tumors. In particular, we measured the expression of p18INK4c protein in 35 primary GBM specimens as part of a GBM tissue microarray (Fig. 3A). Remarkably, expression of p18INK4c was completely lost in 15 of the 35 (43%) samples studied by immunohistochemistry (examples in i and ii). In samples expressing p18INK4c, staining was primarily nuclear, with some cytoplasmic staining observable as has been reported by Bartkova et al. (14). By comparison, expression of p16INK4a (known to be commonly deleted in GBM) was lost in 21 of the 35 (60%) samples studied (examples in iii and iv). Nine of the fifteen samples (60%) lacking expression of p18INK4c also lacked expression of p16INK4a. Staining for a-tubulin was positive in all but one of the 70 tumor cores, demonstrating that all p18INK4c-negative tumors were accessible for staining with other antibodies (example in v). This experiment showed that p18INK4c expression is lost in ~40% to 50% of GBMs (Fig. 3B), and therefore, its inactivation is likely to play a major role in the pathogenesis of GBM.
Figure 3.
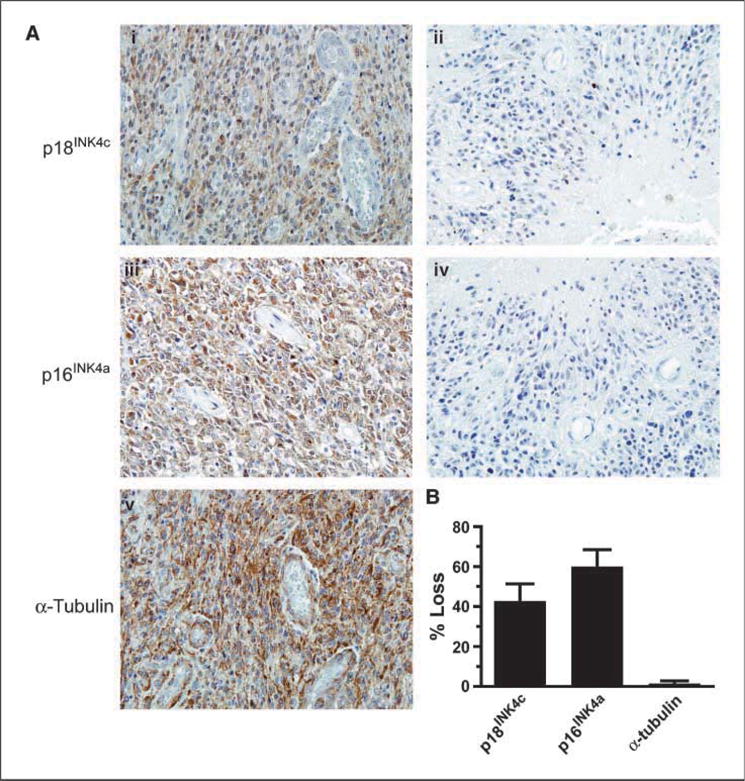
Loss of p18INK4c expression in GBM primary tumors. Immunohistochemistry to GBM tissue microarrays was performed as described in Materials and Methods. A, representative GBMs that (i) express p18INK4c, (ii) fail to express p18INK4c, (iii) express p16INK4a, and (iv) fail to express p16INK4a. v, representative staining for α-tubulin. B, bar graph depicting the percentage of samples lacking expression of p18INK4c, p16INK4a, and α-tubulin.
Absence of p18INK4c missense mutations in 82 GBM samples
We next sequenced p18INK4c in 82 GBM samples lacking homozygous deletions, including 51 primary tumors, 14 xenografts, and 17 cell lines as described in Materials and Methods. The p18INK4c coding region was wild-type in each of the 82 samples sequenced. Heterozygosity at a previously annotated synonymous SNP is depicted in Supplementary Fig. S5. This experiment showed that as for p16INK4a, homozygous deletion of p18INK4c is the predominant genetic mechanism leading to its inactivation during GBM pathogenesis.
Amplification of cdk6 in GBMs harboring intact p18INK4c genes
Cdk6 is thought to be a particularly important target of inhibition by p18INK4c, and Costello et al. (6) have previously described cdk6 amplifications in GBM samples. Therefore, we determined whether amplification of cdk6 might be present in our GBM samples with intact p18INK4c genes. To test this, we first analyzed SNP microarray data and found amplifications of cdk6 in both a GBM cell line and xenograft with wild-type p18INK4c (Supplementary Fig. S6A). Next, we expanded the analysis by using qPCR to measure cdk6 copy number in a panel of 51 GBM primary tumors and, in this way, identified an additional sample with cdk6 amplification (Supplementary Fig. S6B).
Lentiviral reconstitution of p18INK4c expression in GBM cells leads to G1 cell cycle arrest
To determine the phenotypic consequences of p18INK4c deletion in GBM cells, we created a p18INK4c-expressing lentivirus as described in Materials and Methods. Next, we infected five GBM cell lines-LN229, U87MG, T98G, SNB19 (each deleted for p18INK4c), and M059J (wild-type p18INK4c) with either empty vector or p18INK4c virus. Infected cells were studied by Western blot (Fig. 4A), flow cytometry/BrdUrd incorporation (Supplementary Fig. S7; Fig. 4B), phase-contrast microscopy (Fig. 4C), and staining for senescence-associated β-galactosidase activity (Fig. 4D).
Figure 4.

Reconstitution of p18INK4c leads to senescence in GBM cells. A, Western blot for p18INK4c 24 h postinfection shows lentiviral reconstitution of p18INK4c expression in p18INK4c-null LN229, U87MG, and T98G GBM cells (SNB19 not shown). The level of reconstituted expression is only slightly higher than the endogenous levels present in p18INK4c-proficient M059J cells. B, cell cycle distributions 60 h postinfection with control or p18INK4c-expressing lentiviruses. Infection with p18INK4c lentivirus causes G1 cell cycle arrest in p18INK4c-deleted LN229 and T98G cells but not in p18INK4c-proficient M059J cells. C, phase contrast microscopy of cells 7 d postinfection shows that reconstitution of p18INK4c expression in p18INK4c-deficient LN229 and T98G cells leads to morphologic changes resembling senescence, whereas expression of p18INK4c in SNB19 cells leads to frank cell death. D, reconstitution of p18INK4c expression in LN229 cells leads to induction of senescence-associated β-galactosidase activity. Similar results were observed in T98G cells but not in p18INK4c-proficient M059J cells (data not shown).
Infection with the p18INK4c lentivirus led to a physiologic level of expression, comparable with the endogenous levels of expression found in M059J GBM cells harboring an intact p18INK4c gene (Fig. 4A). Importantly, ectopic expression of p18INK4c led to a rapid and complete senescence-like G1 cell cycle arrest in cells with homozygous deletions of p18INK4C but not in cells with an intact p18INK4C gene (Supplementary Fig. S7; Fig. 4B–D; data not shown). Interestingly, expression of p18INK4c in p18INK4c-deficient SNB19 cells led to rapid and complete cell death (Fig. 4C).
Discussion
Here, we identify p18INK4c as a new GBM tumor suppressor gene by describing homozygous deletions in GBM cell lines, xenografts, and primary tumors, and complete loss of expression in 43% of GBMs studied by immunohistochemistry. Furthermore, we show that re-expression of p18INK4c at physiologic levels in GBM cells that lack it leads to immediate senescent-like arrest in the G1 phase of the cell cycle.
p18INK4c is a member of the INK4 family of cdk inhibitors, which includes p16INK4a, p15INK4b, p18INK4c, and p19INK4d. Members of this family bind to cdk4 and cdk6 and inhibit their ability to bind to D-type cyclins, thereby inhibiting the formation of an active cdk/cyclin complex and leading to cell cycle arrest. Deletions at the p18INK4c locus have previously been implicated in the pathogenesis of other tumor types, including those of the brain (12, 15–18). Intriguingly, loss of both p18INK4c and PTEN (both GBM tumor suppressors) has been shown to have synergistic effects on tumor formation in mice (19).
INK4 family members differ from each other in their patterns of expression and in the potency with which they bind individual cyclin/cdk complexes. p18INK4c is thought to bind most potently to cdk6-specific complexes, although there is conflicting data on this point (11). The phenotypic consequences of binding to and inhibiting cdk4 and cdk6 with differing affinities are not well-understood.
We have shown that the presence of admixed nonneoplastic cells and intratumoral heterogeneity complicates the efficient identification of p18INK4c deletions in uncultured primary tumors, using conventional technologies. However, it is also a formal possibility that the greater frequency of p18INK4c deletions in cell lines and xenografts are artifacts of ex vivo culture.
It is also notable that the same GBM samples harboring heterogeneous deletions of p18INK4c also often harbor remarkably homogeneous deletions of p16INK4a. This finding was important as it enabled us to rule out issues of sample quality as a trivial explanation for our finding of heterogeneity. Furthermore, it suggests that homozygous deletion of p16INK4a is an early event in the pathogenesis of GBM, whereas inactivation of p18INK4c seems to occur later in the neoplastic process.
p18INK4c seems to be inactivated in GBM predominantly by homozygous deletion. This is similar to the situation for p16INK4a, in which homozygous deletion is the major mechanism of inactivation in GBM (although point mutations in p16INK4a also occur, albeit at a lower frequency; ref. 20). In the case of p16INK4a, this has been rationalized by suggesting that there is selection pressure for loss of p14ARF as well. It is possible that there is similar selection pressure for simultaneous codeletion of p18INK4c and FAF1 (or an adjacent as yet uncharacterized gene or noncoding RNA) during the pathogenesis of GBM.
In summary, here we have identified p18INK4c as a tumor suppressor gene that is genetically inactivated by homozygous deletion during the pathogenesis of GBM. Additional detailed studies are warranted to identify the phenotypic consequences of p18INK4c deletion during the pathogenesis of GBM.
Supplementary Material
Acknowledgments
Grant support: Georgetown University School of Medicine (T. Waldman and W. Jean), a training grant T32-CA009686 from the National Cancer Institute, NIH (S. Jenkins), and the Lombardi Comprehensive Cancer Center is funded by P30-CA051008 from the National Cancer Institute, NIH.
We thank Michelle Lombard and Karen Cresswell for assistance with flow cytometry, Aaron Foxworth and Syid Abdullah for assistance with animal husbandry, Marcela White of the Brain Tumor Tissue Bank at the London Health Sciences Centre in Ontario, Canada for her assistance with the procurement of high quality GBM specimens, and Yardena Samuels for her comments on the manuscript.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
The results published here are, in part, based on data generated by The Cancer Genome Atlas pilot project established by the National Cancer Institute and National Human Genome Research Institute. Information about The Cancer Genome Atlas and the investigators and institutions who constitute The Cancer Genome Atlas research network can be found at http://cancergenome.nih.gov.
References
- 1.Libermann TA, Nusbaum HR, Razon N, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumors of glial origin. Nature. 1985;313:144–7. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- 2.Rasheed BK, Stenzel TT, Mclendon RE, et al. PTEN gene mutations are seen in high-grade but not in low-grade gliomas. Cancer Res. 1997;57:4187–90. [PubMed] [Google Scholar]
- 3.Broderick DK, Di C, Parrett TJ, et al. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004;64:5048–50. doi: 10.1158/0008-5472.CAN-04-1170. [DOI] [PubMed] [Google Scholar]
- 4.Jen J, Harper JW, Bigner SH, et al. Deletion of p16 and p15 genes in brain tumors. Cancer Res. 1994;54:6353–8. [PubMed] [Google Scholar]
- 5.Schmidt EE, Ichimura K, Reifenberger G, Collins VP. CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994;54:6321–4. [PubMed] [Google Scholar]
- 6.Costello JF, Plass C, Arap W, et al. Cyclin-dependent kinase 6 (CDK6) amplification in human gliomas identified using two-dimensional separation of genomic DNA. Cancer Res. 1997;57:1250–4. [PubMed] [Google Scholar]
- 7.Buschges R, Weber RG, Actor B, Lichter P, Collins VP, Reifenberger G. Amplification and expression of cyclin D genes (CCND1, CCND2, and CCND3) in human malignant gliomas. Brain Pathol. 1999;9:435–42. doi: 10.1111/j.1750-3639.1999.tb00532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98:31–6. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin M, Wei LJ, Sellers WR, Lieberfarb M, Wong WH, Li C. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics. 2004;20:1233–40. doi: 10.1093/bioinformatics/bth069. [DOI] [PubMed] [Google Scholar]
- 10.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 11.Guan KL, Jenkins CW, Li Y, et al. Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type Rb function. Genes Dev. 1994;8:2939–52. doi: 10.1101/gad.8.24.2939. [DOI] [PubMed] [Google Scholar]
- 12.Bai F, Pei XH, Godfrey VL, Xiong Y. Haploinsufficiency of p18(INK4c) sensitizes mice to carcinogen-induced tumorigenesis. Mol Cell Biol. 2003;23:1269–77. doi: 10.1128/MCB.23.4.1269-1277.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chu K, Niu X, Williams LT. A Fas-associated protein factor, FAF1, potentiates Fas-mediated apoptosis. Proc Natl Acad Sci U S A. 1995;92:11894–8. doi: 10.1073/pnas.92.25.11894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartkova J, Thullberg M, Rajpert-De Meyts E, Skakkebaek NE, Bartek J. Cell cycle regulators in testicular cancer: loss of p18INK4C marks progression from carcinoma in situ to invasive germ cell tumours. Int J Cancer. 2000;85:370–5. doi: 10.1002/(sici)1097-0215(20000201)85:3<370::aid-ijc13>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 15.Pohl U, Cairncross JG, Louis DN. Homozygous deletions of the CDKN2C/p18INK4C gene on the short arm of chromosome 1 in oligodendrogliomas. Brain Pathol. 1999;9:639–43. doi: 10.1111/j.1750-3639.1999.tb00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kulkarni MS, Daggett JL, Bender TP, Kuehl WM, Bergsagel PL, Williams ME. Frequent inactivation of the cyclin-dependent kinase inhibitor p18 by homozygous deletion in multiple myeloma cell lines: ectopic p18 expression inhibits growth and induces apoptosis. Leukemia. 2002;16:127–34. doi: 10.1038/sj.leu.2402328. [DOI] [PubMed] [Google Scholar]
- 17.Franklin DS, Godfrey VL, Lee H, et al. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigene-sis. Genes Dev. 1998;12:2899–911. doi: 10.1101/gad.12.18.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uziel T, Zindy F, Xie S, et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev. 2005;19:2656–67. doi: 10.1101/gad.1368605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai F, Pei XH, Pandolfi PP, Xiong Y. p18Ink4c and PTEN constrain a positive regulatory loop between cell growth and cell cycle control. Mol Cell Biol. 2006;26:4564–76. doi: 10.1128/MCB.00266-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kyritsis AP, Zhang B, Zhang W, et al. Mutations of the p16 gene in gliomas. Oncogene. 1996;12:63–7. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.