

Abstract
Epidermal growth factor receptor (EGFR) is expressed, albeit at low or intermediate levels, in human melanomas at the different stages of tumor progression. Coexpression of EGFR with its ligand TGFα indicates their role in paracrine and autocrine growth regulation of melanomas. As it was previously observed for several types of cancer, specific inhibitors of EGFR-mediated signaling may reduce antiapoptotic properties of cancer cells and sensitize them to cytotoxic drugs. We recently reported that arsenite, particularly in combination with inhibitors of the PI3K-AKTand mitogen-activated protein kinase (MAPK) kinase (MEK)-extracellular signal-regulated kinase (ERK) pathways, induces high levels of apoptosis in different melanomas. Since EGFR signaling operates via activation of the PI3K-AKTand MEK-ERK pathways, we suggested that the combination of arsenite and EGFR inhibitors might also effectively induce apoptosis in melanoma. Here, we demonstrate that a moderate concentration of arsenite (5–10 μM) indeed upregulates apoptosis induced by EGFR inhibitors in EGFR-positive melanomas. In contrast, induction of apoptosis in melanomas with negligible surface expression of EGFR or with defective EGFR signaling requires direct suppression of the PI3K-AKTand MAPK pathways by specific pharmacological inhibitors in the presence of arsenite. Under these conditions, metastatic melanoma cell lines undergo TNF-related apoptosis-inducing ligand (TRAIL)- and tumor necrosis factor alpha (TNFα)-mediated apoptosis. Taken together, these data provide additional approaches in sensitizing melanomas to the cytotoxic effects of specific inhibitors of survival pathways.
Keywords: melanoma, EGFR, arsenite, ERK, AKT, TRAIL
Introduction
Epidermal growth factor receptor signaling has become an important target in anticancer drug development due to its ability to control tumor cell proliferation and to suppress apoptosis (Yarden and Sliwkowski, 2001). Extensive efforts to develop epidermal growth factor receptor (EGFR) inhibitorsfor anticancer therapy (Levitzki and Gazit, 1995; Wakeling et al., 1996; Dancey and Sausville, 2003; Gschwind et al., 2004) finally led to approval of one of the small reversible inhibitors, gefitinib (Iressa, AstraZeneca), for non-small-cell lung cancer treatment and of an inhibitory anti-EGFR monoclonal antibody, IMC-C225 (Erbitux, ImClone Systems), for metastatic colorectal cancer treatment. Activation of the EGFR pathway in cancer cells is based on several distinct mechanisms, such as amplification and/or overexpression of the EGFR gene, increased production of ligands, EGF or TGFα, decreased receptor turnover, and the presence of altered forms of receptors due to specific activating mutations or profound gene rearrangement. Effects of EGFR signaling on cell proliferation and survival are mediated by the mitogen-activated protein kinase (MAPK), PI3K-AKT and STAT pathways (Schlessinger, 2000; Sibilia et al., 2000; Yarden and Sliwkowski, 2001; Gschwind et al., 2004). Despite a ubiquitous low-level expression of EGFR in normal cells, inactivation of the EGFR gene in mice has minimal effects on development and survival (Threadgill et al., 1995), indicating that pharmacological inhibition of EGFR signaling should have relatively minor effects on normal cells. The results of clinical trialsfor non-small-cell lung cancer with the EGFR inhibitor gefitinib were relatively modest, showing objective tumor response rates of 10–18% (Blackledge and Averbuch, 2004; Dancey, 2004). However, the identification of genetic changes in the EGFR kinase domain that can predict tumor response to gefitinib was described in two recent publications (Lynch et al., 2004; Paez et al., 2004), resolving the problem of when gefitinib-based monotherapy will be effective for non-small-cell lung cancer treatment. Combined chemotherapy to treat different forms of cancer, as a rule, is much more effective than monotherapy. However, the addition of gefitinib to traditional chemotherapy failed to induce significant responses (Giaccone et al., 2004), indicating the importance of finding proper combinations of EGFR inhibitor with modulators of other signaling pathways in the cell.
Melanoma is largely curable at early stages of cancer development; however, there are no adequate treatments for locally advanced or metastatic stages of this disease (Perlisand Herlyn, 2004). Therefore, it isvery important to develop novel agents, especially in different combinations, that are both effective and low in toxicity for melanoma treatment. In contrast to some other tumors, melanomas express EGFR only at low or intermediate levels while other types of growth factor receptors, such as FGF R1 and IGF-1R, and their correspondent growth factors are also involved in the paracrine and autocrine control of melanoma cell proliferation (Halaban, 1996; Lazar-Molnar et al., 2000). In the present study, we have addressed the question with regard to effects of suppression of EGFR signaling on melanoma survival. Previously, we reported that arsenite in combination with inhibitors of the PI3K-AKT and mitogen-activated protein kinase (MEK)-extracellular signal-regulated kinase (ERK) pathways induces high levels of apoptosisin different melanomas(Ivanov and Hei, 2004). Since EGFR signaling operates via activation of the PI3K-AKT and MEK-ERK pathways (Yarden and Sliwkowski, 2001), we suggested that the combination of arsenite and EGFR inhibitors would also be an effective inducer of apoptosis in melanoma. Here, we demonstrate that a moderate concentration of arsenite (5–10 μM) induces an additive or synergistic upregulation of apoptosis stimulated by EGFR inhibitors in EGFR-positive melanomas. In contrast, apoptosis induction in melanomas with negligible surface expression of EGFR requires direct suppression of the PI3K-AKT and MAPK pathways by specific pharmacological inhibitors in the presence of arsenite. Under these conditions, metastatic melanoma cells may undergo TNF-related apoptosis-inducing ligand (TRAIL)- or tumor necrosis factor alpha (TNFα)-mediated apoptosis. These data provide additional approaches to increase apoptotic responses of melanomas to cytotoxic effects of specific inhibitors of survival pathways.
Results
EGFR surface expression in human melanomas and sensitivity of cancer cells to EGFR inhibitors
The EGFR surface expression has been determined using monoclonal anti-EGFR antibody and flow cytometry in both human melanocytes and melanoma cell lines at different stages of tumor progression (Figure 1a). Melanocytes possess low, almost negligible surface levels of EGFR (data not shown). Radial growth phase WM35 and metastatic HHMSX, FEMX, LOX and OM431 melanoma cells were also characterized by negligible surface expression of EGFR. On the other hand, two early melanoma cell lines, SBcl2 (radial growth phase), WM793 (vertical growth phase), and WM9 metastastic melanoma demonstrated average levels of surface expression of EGFR; LU1205 metastatic melanoma cells possess a low surface expression of EGFR (Figure 1a). These data indicate an absence of a direct correlation between surface EGFR expression and the stage of melanoma development. Furthermore, high levels of surface EGFR expression may not directly correlate to the efficiency of EGFR signaling due to the possibility of inactivating mutations in the EGFR gene (Lynch et al., 2004). The WM793 and WM9 melanoma cells were chosen for treatment with increased doses of AG1478, a highly specific inhibitor of EGFR signaling (Levitzki and Gazit, 1995). AG1478 (20 μM) induced reasonably high levels of apoptosis 48 h after treatment of these cells (Figure 1b); PD153035, another inhibitor of EGFR signaling, demonstrated the similar effects (data not shown). Two EGFR inhibitors (20 μM) have been applied for treatment of human melanocytesan d additional melanoma cell lines. These concentrations of inhibitors are equivalent to doses, which have been used recently in clinical trails of gefitinib (Iressa, AstraZeneca) for non-small-cell lung cancer treatment (Levitzki and Gazit, 1995; Wakeling et al., 1996; Dancey and Sausville, 2003; Gschwind et al., 2004). Besides WM793 and WM9, only SBcl2 melanoma cells with intermediate levels of surface expression of EGFR developed high levels of apoptosis following treatment with inhibitors (Figure 1c).
Figure 1.

EGFR inhibitors induce apoptosis in human melanomas. (a) Cell surface EGFR expression on melanocytes and human melanomas was determined by staining with PE-conjugated anti-human EGFR monoclonal antibody and flow cytometry. Filled histograms represent nonspecific staining with mouse Ig-PE. Mean fluorescent intensity (MFI) is indicated. WM35 cells, as well as HHMSX, FEMX, LOX and OM431, are EGFR negative. (b) Dose–response induction of apoptosis by AG1478 in two melanoma lines, WM793 and WM9. (c) Cell-cycle apoptosis analysis of human melanocytes and melanoma cells treated with AG1478 or PD153035 (20 μM) for 24 h. Cells were stained with PI and percentage of cells with hypodiploid content of DNA was determined by flow cytometry
Arsenite increases apoptosis induced by EGFR inhibitors
We previously observed that a moderate concentration of arsenite (5–10 μM) may effectively induce apoptosis in melanomas, particularly in combination with inhibitors of the MEK-ERK and PI3K-AKT pathways(Ivanov and Hei, 2004). We suggested that the combined treatment of arsenite and EGFR inhibitor should also be an effective inducer of apoptosis in melanoma, because EGFR-mediated signaling operates via MEKERK and PI3K-AKT pathways(Yarden and Sliwkowski, 2001). Indeed, AG1478 (20 μM) in concert with arsenite (10 μM) demonstrated additive effects in the induction of apoptosis of WM793 cells 24 and 48 h after treatment (Figure 2a). It is interesting to note that the effects of AG1478 on apoptosis were quite similar to the effects of LY294002, an inhibitor of the PI3K-AKT pathway (Figure 2b). Indeed, Western blot analysis of the total protein of WM793 cells indicated a suppression of EGFR phosphorylation and a substantial inhibition of phospho-AKT levels following treatment with AG1478 or combination of AG1478 and arsenite. Simultaneously, EGFR inhibitor AG1478 did not affect basal or arsenite-induced phospho-ERK levels, revealing that the MEK-ERK pathway was not activated by EGFR signaling in WM793 melanoma cells. Heme oxygenase-1 (HO-1) induction was chosen as a hallmark of oxidative stress stimulated by arsenite; AG1478 effectively blocked HO-1 induction by arsenite, indicating possible involvement of the PI3K-AKT pathway in this regulation (Figure 2c). So, from two main signaling pathways, MEK-ERK and PI3K-AKT usually activated by EGFR in different cell systems, only the PI3K-AKT pathway was under the control of EGFR signaling in early melanoma WM793 cells. It correlates with the presence of BRAF activating mutation T1796A (V599E), which makes the downstream MEK-ERK cascade partially independent of GFR stimulation, in many melanoma lines including WM793 (Dong et al., 2003; Satyamoorthy et al., 2003). Our data also indicate that alternative FGFR1-, IGF-1R- or c-Met-mediated signaling may be involved in the regulation of the MEK-ERK cascade in melanomas (Satyamoorthy et al., 2003).
Figure 2.

Arsenite increases the apoptotic response of WM793 melanoma (vertical growth phase) cells to the EGFR inhibitor. (a) Additive effects of AG1478 (20 μM) and arsenite (10 μM) for the induction of apoptosis of WM793 melanoma cells 24 and 48 h after treatment. (b) Effects of LY294002 (50 μM), rapamycin (100 nM) or herbimycin (10 and 20 μM) on arsenite-induced apoptosis. Cell-cycle apoptosis analysis was 24 h after treatment performed using flow cytometry. (c) Western blot analysis of total and phospho-EGFR, phospho-ERK and phospho-AKT levels following treatment with AG1478 (20 μM), arsenite (10 μM) or their combination. Induction of HO-1 was used as a demonstration of arsenite-induced oxidative stress. β-Actin served as an internal standard. (d) PARP cleavage was determined by Western blot analysis 18 h after treatment with AG1478 (20 μM), PD153035 (20 μM), arsenite (10 μM) and indicated combinations of inhibitors with arsenite. (e) Effects of caspase inhibitors, Ac-IETD-CHO (50 μM), Ac-LEHD-CHO (50 μM) and zVAD-fmk (50 μM), on induction of apoptosis of WM793 cells by AG1478 or PD153035 (20 μM) treatment
The PI3K-AKT pathway affects general cell survival and proliferation via control of protein levels and activities of several transcription factors and correspondent target genes, as well asvia regulation of the mTOR kinase that is the central controller of general protein synthesis (Bjornsti and Houghton, 2004; Hanada et al., 2004). Furthermore, the PI3K-AKT pathway has some specific antiapoptotic functions, such as an inactivation of caspase-9 and proapoptotic protein Bad (Kandel and Hay, 1999). We used rapamycin (100 nM) for specific inhibition of mTOR (Figure 2b). Rapamycin did not promote the effects of arsenite-induced apoptosis, indicating that the AKT-mTOR pathway was not critically involved in the regulation of this type of apoptosis in WM793 cells. On the other hand, herbimycin, a general inhibitor of protein tyrosine kinases, induced apoptosis of WM793 melanoma cells and increased arsenite-induced apoptosis of these cells highlighting the role of protein tyrosine kinases in general cell survival (Figure 2b). One of the prominent antiapoptotic actions of arsenite, suppression of nuclear factor kappa B (NF-κB) activity and NF-κB-dependent transcription (Kapahi et al., 2000; Ivanov and Hei, 2004), which was observed after treatment WM793 cells by arsenite alone or in concert with AG1478 (data not shown), may be responsible for enhancing AG1478-induced apoptosis.
Caspase-3-mediated cleavage of PARP, an important feature of apoptosis, was evident 18 h after treatment with AG1478, PD153035, arsenite and combinations of inhibitors with arsenite (Figure 2d). To further investigate the role of caspases in apoptosis induced by EGFR inhibitors, we used N-acetyl-Ile-Glu-Thr-Asp-CHO (aldehyde) (Ac-IETD-CHO) (an inhibitor of caspase-8 and -6), N-acetyl-Leu-Glu-His-Asp-CHO (aldehyde) (Ac-LEHD-CHO) (an inhibitor of caspase-9) and zVAD-fmk (a general caspase inhibitor). Both caspase-8 and -9 inhibitors notably suppressed apoptosis induced by AG1478, but were less effective for suppression of PD153035-induced cell death; zVAD-fmk treatment was an effective suppressor for death induction by AG1478 or PD153035 (Figure 2e).
We further elucidated the effects of arsenite on AG1478-induced apoptosis using two additional EGFR-positive melanomas, SBcl2 and WM9 (Figure 3). For both lines, EGFR inhibitors AG1478 (20 μM) or PD153035 (20 μM) and PI3K-AKT inhibitor LY294002 (50 μM) induced apoptosis and dramatically enhanced apoptotic effects of arsenite (Figure 3a and b). It was well correlated with the downregulation of EGFR phosphorylation and a decrease in basal phospho-AKT activity following AG1478 (20 μM) treatment (Figure 4a). No negative effects of AG1478 on basal or arsenite-induced phospho-ERK levels have been observed. Arsenite-induced HO-1 activity was substantially suppressed in the presence of AG1478 (Figure 4a). Asexpected, arsenite alone or in the combination with AG1478 downregulated NF-κB activity and NF-κB-dependent transcription (data not shown) providing conditions favorable to enhancement of apoptosis. Apoptotic abilities of AG1478, PD153035 and arsenite in WM9 metastatic melanoma that finally target caspase-3 were additionally confirmed by the specific caspase-3-dependent cleavage of PARP 18 h after treatment (Figure 4b). To further verify the role of AKT as a target of suppression by EGFR inhibitors, we used previously established WM9-AKTmyr cells(Ivanov et al., 2002) with high levels of permanently active phospho-AKT and control WM9-puro cells (Figure 4c). WM9-AKTmyr cells were substantially more resistant to apoptotic effects of LY294002 and AG1478 than control cells(Figure 4d).
Figure 3.
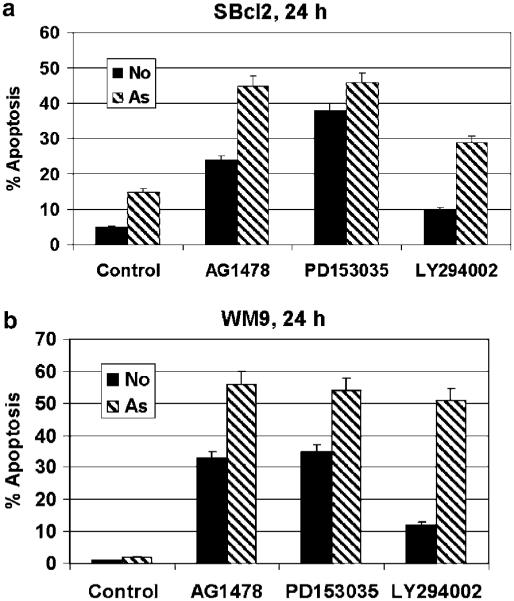
Arsenite synergizes with EGFR inhibitors for the induction of apoptosis in the sBCL2 melanoma (horizontal growth phase) and WM9 metastatic melanoma cells. (a and b) Synergistic effects of AG1478 (20 μM), PD153035 (20 μM), LY294002 (50 μM) and arsenite (10 μM) for the induction of apoptosis of sBCL2 and WM9 melanoma cells24 h after treatment. Cell-cycle apoptosis analysis was performed using flow cytometry
Figure 4.

Regulation of EGFR-mediated signaling in WM9 melanoma cells. (a) Western blot analysis of total and phospho-EGFR, phospho-ERK, phospho-p38 and phospho-AKT levels following AG1478 (20 μM), arsenite (10 μM) or combined treatment. Induction of HO-1 was used as an indication of arsenite-induced oxidative stress. β-Actin levelswere used asan internal control. (b) PARP cleavage wasdetermined by Western blot analysis 18 h after treatment with AG1478 (20 μM), PD153035 (20 μM) or arsenite (10 μM). (c and d) Overexpression of AKTmyr protectsWM9 melanoma cells from apoptosis induced by LY294002 (50 μM) or AG1478 (20 μM)
Suppression of PI3K-AKT and MEK-ERK pathways partially reproduces the effects of GFR inhibitors
LU1205 metastatic melanoma cells have low levels of surface EGFR (Figure 1a) and are relatively resistant to arsenite treatment. As expected in this case, EGFR inhibitors, AG1478 (Figure 5a) and PD153035 (not shown), induced only slight apoptotic effects and did not upregulate arsenite-induced apoptosis in LU1205 melanoma cells. By contrast, herbimycin, a general protein tyrosine kinase inhibitor, was very effective for increasing arsenite-induced apoptosis in LU1205 cells (Figure 5a), demonstrating a probable role of alternative growth factor receptor pathwaysin the regulation of cell survival in case of deficiency of EGFR signaling. Since inhibition of both PI3K-AKT and MEK-ERK pathwaysmay partially mimic the downstream effects of growth factor receptor inhibition, we treated LU1205 cells with LY294002 (50 μM) and PD98059 (50 μM) with or without arsenite (Figure 5a). We previously observed (Ivanov and Hei, 2004) that such treatments induced high levels of apoptosis in LU1205 cells. As expected, the combination of LY294002 with arsenite was less effective than the combination of LY294002, PD98059 and arsenite (data not shown).
Figure 5.

Combined treatment with LY294002, PD98059 and arsenite induces TRAIL-mediated apoptosis in LU1205 melanoma cells. (a) Herbimycin (20 μM), but not AG1478 (20 μM), enhances arsenite-induced apoptosis in LU1205 melanoma cells; combination of LY294002 (50 μM) and PD98059 (50 μM) also upregulates arsenite-induced apoptosis. Cell-cycle apoptosis analysis was performed using flow cytometry of PI-stained cells. (b) Western blot analysis of total and phospho-AKT, phospho-ERK and TRAIL levelsfollowing LY294002 (50 μM) and PD98059 (50 μM) treatment in the presence or absence of arsenite (10 μM). NF-κB DNA-binding activity of nuclear proteinswas determined by EMSA. Two DNA-binding complexesare indicated NF-κB p65-p50 and (p50)2. (c) Apoptosis analysis of LU1205 cells treated by LY294002 (50 μM) þ PD98059 (50 μM) with or without arsenite (10 μM) in the presence of nonspecific IgG, inhibitory anti-TRAIL or anti-TNFα mAb (5 mg/ml). FACS analysis was performed with PI-stained melanoma cells. A percentage of apoptotic cells 24 h after treatment is indicated
Staining LU1205 cells by Annexin-V–FITC and propidium iodide (PI) with subsequent flow cytometry revealed initial apoptotic and secondary necrotic events 6 h after arsenite treatment in the presence of inhibitors (data not shown). Western blot analysis indicated suppression of phospho-AKT and phospho-ERK2 activities following treatment with the combination of specific inhibitors, while electrophoretic mobility shift assay (EMSA) demonstrated a downregulation of NF-κB DNA-binding activity (for both bandsp65-p50 and p50-p50) (Figure 5b) and NF-κB-dependent transcription (data not shown) in the presence of arsenite. Since LU1205 cellsproduce TRAIL and TNFα and have surface expression of tumor necrosis factor receptor (TNFR)1, DR4 and DR5 death receptors(Griffith et al., 1998; Ivanov and Ronai, 1999), a decrease in the basal NF-κB activity by arsenite, a decrease in AKT activity by LY294002 and a decrease in ERK activity by PD98059 may sensitize these cells to TRAIL- or TNFα-mediated cell suicide (Chen et al., 2001; Franco et al., 2001; Zhang et al., 2003b). Indeed, pretreatment with anti-TRAIL or, to a lesser extent, with anti-TNFα antibodies partially suppressed apoptosis that was induced by arsenite in combination with LY294002 and PD98059 (Figure 5c). Hence, for melanoma cells with low or negligible levels of surface EGFR, it was possible to induce efficient levels of apoptosis by direct inhibition of downstream PI3K-AKT and MEK-ERK signaling pathways in the presence of arsenite.
HO-1 activity as a target of EGFR signaling
What are some of the target genes, whose expressions are dependent on both the EGFR- and arsenite-mediated signaling? Determination of arsenite-affected gene expression in several cell systems using microarrays revealed a dramatic upregulation of HO-1 following arsenite treatment (Liu et al., 2001a; Yih et al., 2002; Rea et al., 2003; Zheng et al., 2003). Induction of HO-1 is the hallmark feature of oxidative stress (Durante, 2003). Arsenite efficiently induced HO-1 in melanomas 6 h after treatment with maximal levels at a dose 10– 20 μM (Figure 6a). The role of AP-1/activating transcription factor 2 (ATF2) transcription factors in the regulation of HO-1 gene expression has been well established (Alam and Den, 1992; Lee et al., 2000; Gong et al., 2002). AP-1 exists in multiple homo or hetero combinations of Jun–Jun, Jun–Fos and Jun– ATF2 proteins, which may be targets of different signaling pathways (Shaulian and Karin, 2002). A probable function of HIF-1 in the transcriptional activation of HO-1 was also described (Lee et al., 1997). SB203580 (an MAPK p38-ATF2 inhibitor) and most effectively LY294002 (a PI3K-AKT inhibitor) downregulate the HO-1 induction in both WM793 and LU1205 melanoma cells. PD98059 (an MEK-ERK-Elk1-cFosinhibitor) was partially effective only in LU1205 cells. SP600125 (Jun N-terminal kinase (JNK)’s inhibitor) did not notably affect HO-1 protein levels in indicated melanoma lines(Figure 6a). Taken together, these data supported the role of AP-1/ATF2-dependent regulation of HO-1 expression (via MAPK p38 and ERK) in melanomas. Targets of PI3K-AKT signaling for the HO-1 expression are not completely identified. Probable targets are transcription factors NRF2 and HIF-1, which controlsthe HO-1 gene transcription. The stability of NRF2 and HIF-1 proteins is dependent on PI3K-AKT signaling (Jiang et al., 2001; Alam and Cook, 2003; Martin et al., 2004). If inhibitors of EGFR signaling downregulate the PI3K-AKT pathway, they also should suppress HO-1 induction by arsenite. Indeed, we observed an effective suppression of arsenite-induced HO-1 expression in the EGFR-positive WM793 melanoma cells by AG1478 and the absence of suppression for the EGFR-negative FEMX cells (Figure 6b and c).
Figure 6.
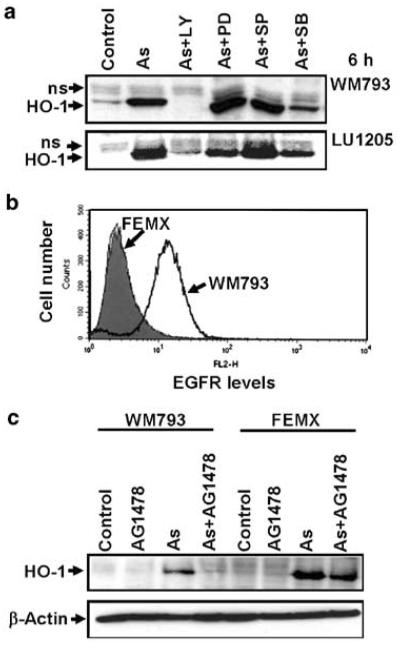
Regulation of HO-1 activity by arsenite in melanoma cells: effects of inhibition of signal transduction pathways. (a) Effects of LY294002 (50 μM), PD98059 (50 μM), SP600125 (10 μM) and SB203580 (10 μM) on arsenite (10 μM)-induced HO-1 protein levelsin melanoma cells. (b) Surface EGFR levelswere determined by flow cytometry of stained FEMX and WM793 cells. Filled histogram represents nonspecific staining with mouse Ig-PE. (c) Western blot analysis of HO-1 levels from WM793 and FEMX cellsfollowing treatment with AG1478 (20 μM), arsenite (10 μM) or their combination
HO-1 is known to possess numerous antioxidative stress functions, including interference and suppression of TNF- and FasL-mediated apoptosis (Brouard et al., 2002; Zhang et al., 2003a). Direct suppression of HO-1 by introduction of its pharmacological inhibitor, Zn-containing protoporphyrin IX (PP IX (Zn2+)) (50 μM), notably accelerated arsenite-induced apoptosis of WM793 melanoma cellsand had a certain, but relatively mild increase in apoptosis for LU1205 cells (data not shown). Positive regulation of expression of antiapoptotic protein HO-1 by PI3K-AKT and MEK-ERK pathways or by EGFR signaling allows connecting the upregulation of arsenite-induced apoptosis after LY294002, PD98059 or AG1478 cotreatment (see Figures 2 and 3), to a certain extent, with suppression of the HO-1 expression.
It is interesting to note that downregulation of arsenite-induced HO-1 protein levels in cancer cells by EGFR inhibitors may serve as an easily performed additional test in the determination of the physiologically active state of EGFR signaling in these cells (as an alternative to the determination of EGFR phosphorylation). The absence of the surface EGFR expression or mutations of EGFR that disturb EGFR-mediated signaling may suppress the effects of EGFR inhibitors on HO-1 activity, as we observed for FEMX melanoma cells(Figure 6b and c).
Discussion
Apoptosisisone of the key regulatory mechanisms in the development of multicellular organism and in the regulation of its general homeostasis, and also in the initiation and progression of degenerative diseases. Apoptosis serves as a defensive mechanism to remove potentially dangerouscells such asprimary tumor cells and virus-infected cells. However, most if not all metastatic cancer cells have serious defects in the control of apoptosis providing cancer cells the ability to resist death (Evan and Vousden, 2001; Johnstone et al., 2002; Kitada et al., 2002). Two fundamental signaling pathways, Ras-Raf-MEK-ERK-(Elk1, activator protein-1 (AP-1)) and PI3K-AKT-(FKHD, mTOR), which are initiated by the interactions of growth factor receptors with cognate growth factors, are involved in the regulation of cell proliferation and general cell survival (Figure 7). Components of these two pathwaysare frequent targetsfor gene mutations, which may change their inducible status to a permanently active state. For example, RAS and BRAF genestogether have activating mutations in almost 80% of all melanomas(Davies et al., 2002; Tuveson et al., 2003; Reifenberger et al., 2004). The EGFR gene by itself is also very often mutated in different tumors(Lynch et al., 2004). The NF-κB signaling pathway TNFR-TRAF2-IKK-IkB*NF-κB-(NF-κB) (Figure 7), which balances cell survival and proapoptotic functions, is also often overactivated in different tumors as a result of epigenetic or genetic alterations(Karin et al., 2002, 2004; Orlowski and Baldwin, 2002; Lin and Karin, 2003; Chen and Greene, 2004).
Figure 7.

Kinase targets and inhibitors. A schematic illustration of the principal signaling pathways that are affected by the inhibitors, which are described in this paper. Protein tyrosine kinase activity of EGFR is inhibited with AG1478 or PD153035. Downstream events of EGFR-mediated signaling include a formation of multisubunit scaffold complex with the activation of the Ras-Raf-MEK-ERK pathway. PD98059 and U0126 are specific inhibitors of MEK1/2. The PI3K-AKT pathway that is initiated at the active EGFR can be suppressed with pharmacological inhibitor LY294002 or with the endogenous inhibitor PTEN. The MEK-ERK and PI3K-AKT pathways activate the specific sets of transcription factors, which are involved in the antiapoptotic and cell-cycle gene expression. AKT also regulates general protein translation via mTOR kinase, which is sensitive to rapamycin. The TNFR-mediated signaling induces via adaptor proteins IKK activity followed by phosphorylation and proteasome-dependent degradation of IκB, and release and nuclear translocation of NF-κB, which regulates the expression of many different genes, including antiapoptotic genes. Simultaneously, the TNFR-mediated signaling may activate caspase-8/caspase-3 proapoptotic pathway; at normal conditions, however, this pathway is suppressed by the NF-κB-dependent antiapoptotic proteins. Arsenite upregulates the MAPK pathways, but eventually suppresses IKK-NF-κB activation affecting a balance between cell survival and apoptosis
We recently investigated apoptotic activity of arsenite in human melanomas(Ivanov and Hei, 2004), which is based on two main aspects of arsenite function in the cell: (1) arsenite acts as a sulfhydryl reagent, which binds to free thiol (–SH) group of enzymes and inhibits their functions (Snow, 1992) and (2) arsenite induces strong production of reactive oxygen species (ROS) with subsequent development of oxidative stress, which affectsmultiple targetsin the cell (Hei et al., 1998; Liu et al., 2001b; Li et al., 2002). There are two important targets of enzymatic inhibition by arsenite: (1) inhibitor nuclear factor kappa B-kinase β (IKKβ) (Kapahi et al., 2000; Roussel and Barchowsky, 2000; Hershko et al., 2002), which results in the suppression of NF-κB transcription factor activation followed by the dramatic change of the anti- and proapoptotic functions of the cells(Karin and Lin, 2002; Amit and Ben-Neriah, 2003; Mathas et al., 2003) and (2) JAK2 tyrosine kinase, which results in the inhibition of tyrosine phosphorylation and functional activation of Stat3, which controls numerous survival functions of cells (Darnell, 2002; Levy and Darnell, 2002; Hayashi et al., 2003; Cheng et al., 2004). As a result of oxidative stress, stress-induced kinase pathways (MKK6-MAPK p38, MKK4/7-JNK and MEK-ERK1/2) simultaneously activate and upregulate the AP-1 (Jun–Fos, Jun–ATF2) transcription factors and induce AP-1-dependent gene expression (Cavigelli et al., 1996; Chen et al., 1998; Ludwig et al., 1998; Bode and Dong, 2002). On the one hand, arsenite suppresses general cell survival via inhibition of NF-κB and Stat3, and on the other, it stimulates MAP kinases and some antiapoptotic functions. To enhance proapoptotic events, it would be necessary to suppress the MAPK and PI3K-AKT pathways in concert with arsenite treatment (Figure 7). We present here data that show the additive or synergistic effects of EGFR inhibitors and arsenite on apoptosis in EGFR-positive melanomas. In these melanomas, EGFR inhibitors suppressed the PI3K-AKT pathway, while the MEK-ERK pathway was independent of their actions. This obviously indicates that other GFRs (such asFGFR1, IGF-1R and c-Met) are involved in the regulation of MEK-ERK signaling in melanomas (Satyamoorthy et al., 2003; von Willebrand et al., 2003). Furthermore, BRAF T1796A (V599E) activating mutation, which has been found in WM793, WM9, LU1205, OM431 and LOX melanomas(but not in SBcl2, WM35 or FEMX) upregulates basal levels of ERK activity (Dong et al., 2003; Krasilnikov et al., 2003; Satyamoorthy et al., 2003). The combination of LY294002 (an inhibitor of PI3K-AKT), U0126 (an inhibitor of MEK1/2-ERK) and arsenite (an inhibitor of IKKb and JAK2), which affectsthe downstream targets of GFR and NF-κB signaling, was extremely effective for the induction of apoptosis in all tested melanomas, as well as in the prostate adenocarcinomas(VN Ivanov and TK Hei, unpublished observations).
There is an obvious mechanistic analogy in the general arrangement of pro- and antiapoptotic signaling between melanomas and prostate adenocarcinomas. In this respect, androgen-dependent LNCaP cells with low basal NF-κB activity and high sensitivity to TNFα- or arsenite-induced apoptosis are similar to early WM793 melanoma cells. On the other hand, highly aggressive androgen-independent DU145 prostate cancer cells with a high basal NF-κB activity and resistance to both TNFα- or arsenite-induced apoptosis are quite similar to metastatic melanoma LU1205. For both cell systems, downregulation of the basal NF-κB activities by different agents may induce TNFα- and TRAIL-mediated apoptosis (Huerta-Yepez et al., 2004). Surprisingly, in spite of the high surface expression of EGFR in PC3 and DU145 prostate cancer cells, inhibitors of EGFR (alone or in concert with arsenite) induced only modest levels of apoptosis (VN Ivanov and TK Hei, unpublished observations). This was opposite to EGFR-positive melanomas, which are highly responsive to the combined treatment of EGFR inhibitorsand arsenite, as was shown in this study. This discrepancy may reflect potential mutations and alterations of the EGFR gene in prostate cancer cell lines, as has been recently reported in non-small-cell lung cancer cells(Lynch et al., 2004; Paez et al., 2004).
Materials and methods
Materials
Sodium arsenite, herbimycin and rapamycin were obtained from Sigma (St Louis, MO, USA), pharmacological inhibitors AG1478, LY294002, PD98059, PD153035, SB203580 and U0126 were obtained from Calbiochem (La Jolla, CA, USA) and SP600125 waspurchased from Biomol (Plymouth Meeting, PA, USA). Caspase inhibitors zVAD-fmk, Ac-IETD-CHO (an inhibitor of caspase-8 and -6) and Ac-LEHD-CHO (an inhibitor of caspase-9) were purchased from Calbiochem (La Jolla, CA, USA). Precast SDS–polyacrylamide gels were purchased from BioRad (Hercules, CA, USA).
Cell lines
Human melanoma cell lines, WM35, SBcl2, LU1205 (also known as1205lu), WM9, WM793 (Satyamoorthy et al., 1997; Berking et al., 2001; Li et al., 2001, 2003) and OM431 were maintained in DMEM medium supplemented with 10% fetal bovine serum, l-glutamine and antibiotics. FEMX, HHMSX and LOX, human melanoma lines(Myklebust et al., 1994), were maintained in RPMI1640 medium supplemented with 10% fetal bovine serum, and antibiotics. Normal human melanocytes were maintained in TICVA medium.
Treatment, apoptosis studies and fluorescence-activated cell sorter (FACS) analysis
Cells were exposed to arsenite (1–10 μM) in the medium for 6– 48 h. Specific inhibitors of EGFR AG1478 (20 μM) and PD153035 (20 μM), PI3K-AKT LY294002 (50 μM), of MEK-ERK PD98059 (50 μM) or U0126 (10 μM), of JNK’sSP600125 (10–20 μM) and of MAPK p38 SB203580 (5–10 μM) were used with or without 5–10 μM arsenite. Inhibitors were added to media 30 min before arsenite treatment. Antibodies against TNFα (BD Pharmingen, San Diego, CA, USA) and TRAIL (Alexis, San Diego, CA, USA) were added (1–5 mg/ml) 1 h before arsenite treatment. Apoptosis was assessed by quantifying the percentage of hypodiploid nuclei undergoing DNA fragmentation (Nicoletti et al., 1991) or by quantifying the percentage of Annexin-V–FITC-positive cells (BD Pharmingen, San Diego, CA, USA). Flow cytometric analysis was performed on a FACS Calibur flow cytometer (Becton Dickinson) using the CellQuest program. Surface expression of EGFR on cancer cells was determined by staining them with phycoerythrin (PE)-anti-EGFR monoclonal antibody (BD Pharmingen) and by flow cytometry.
Western blot analysis
Cell lysates (50–100 mg protein) were resolved on 10% SDS– PAGE, and processed according to the standard protocols. The antibodies used were polyclonal anti-phospho-p44/42 MAP kinase (Thr202/Tyr204), anti-p44/42 MAP kinase, anti-phospho-AKT (Ser473), anti-AKT, anti-phospho-p38 MAP kinase (Thr180/Tyr182), anti-p38 MAP kinase, anti-PARP (Cell Signaling, Beverly, MA, USA), polyclonal anti-HO-1 (Stressgen, Victoria, Canada) and monoclonal anti-β-actin (Sigma) (optimal dilutions of Abswere 1 : 1000 to 1 : 10 000). The secondary Abs (anti-rabbit or anti-mouse) were conjugated with horseradish peroxidase (dilution 1 : 5000 to 1 : 10000). Signals were detected using the ECL system (Amersham, Piscataway, NJ, USA).
EMSA
EMSA was performed for detection of NF-κB DNA-binding activity, as previously described (Ivanov et al., 1994) using the labeled double-strand oligonucleotide AGCTTGGGGAC TTTCCAGCCG (binding sites are underlined).
Transfection and luciferase assay
The NF-κB luciferase reporter containing two kB binding sites wasused for the determination NF-κB transactivation. Transient transfection of NF-κB reporter construct (0.5 mg) and pCMV-β-gal (0.25 mg) into 5 × 105 melanoma cells was performed using Lipofectamine (Life Technologies-Invitrogen). Proteinswere prepared for β-gal and Luciferase analysis 16 h after transfection. Luciferase activity was determined using the Luciferase assay system (Promega, Madison, WI, USA) and was normalized based on β-galactosidase levels.
Acknowledgements
This work was supported by NIH Grant ES 11804, ES 05786 Superfund Grant P42 ES 10349 and Environmental Center Grant P30 ES 09089. We thank Drs M Herlyn, O Fodstad, R Halaban and Z Ronai for the cell lines; Dr A Chan, Dr S Fuchs, Ms S Baker and Mr JA Gillespie for critical reading of the manuscript.
Abbreviations
- Ac-IETD-CHO
N-acetyl-Ile-Glu-Thr-Asp-CHO (aldehyde)
- Ac-LEHD-CHO
N-acetyl-Leu-Glu-His-Asp-CHO (aldehyde)
- AP-1
activator protein-1
- ATF2
activating transcription factor 2
- EGFR
epidermal growth factor receptor
- EMSA
electrophoretic mobility shift assay
- ERK
extracellular signal-regulated kinase
- FACS
fluorescence-activated cell sorter
- HO-1
heme oxygenase-1
- JNK
Jun N-terminal kinase
- IkB
inhibitor of NF-κB
- IKK
inhibitor nuclear factor kappa B kinase
- MAPK
mitogen-activated protein kinase
- MEK
MAPK kinase
- MFI
medium fluorescence intensity
- mTOR
mammalian target of rapamycin
- NF-κB
nuclear factor kappa B
- PARP
poly (ADP-ribose) polymerase
- PI
propidium iodide
- PP IX (Zn2+)
Zn-containing protoporphyrin IX
- ROS
reactive oxygen species
- TNFα
tumor necrosis factor alpha
- TNFR
tumor necrosis factor receptor
- TRAIL
TNF-related apoptosis-inducing ligand
References
- Alam J, Cook JL. Curr. Pharm. Des. 2003;9:2499–2511. doi: 10.2174/1381612033453730. [DOI] [PubMed] [Google Scholar]
- Alam J, Den Z. J. Biol. Chem. 1992;267:21894–21900. [PubMed] [Google Scholar]
- Amit S, Ben-Neriah Y. Semin. Cancer Biol. 2003;13:15–28. doi: 10.1016/s1044-579x(02)00096-2. [DOI] [PubMed] [Google Scholar]
- Berking C, Takemoto R, Schaider H, Showe L, Satyamoorthy K, Robbins P, Herlyn M. Cancer Res. 2001;61:8306–8316. [PubMed] [Google Scholar]
- Bjornsti MA, Houghton PJ. Nat. Rev. Cancer. 2004;4:335–348. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- Blackledge G, Averbuch S. Br. J. Cancer. 2004;90:566–572. doi: 10.1038/sj.bjc.6601550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode AM, Dong Z. Crit. Rev. Oncol. Hematol. 2002;42:5–24. doi: 10.1016/s1040-8428(01)00215-3. [DOI] [PubMed] [Google Scholar]
- Brouard S, Berberat PO, Tobiasch E, Seldon MP, Bach FH, Soares MP. J. Biol. Chem. 2002;277:17950–17961. doi: 10.1074/jbc.M108317200. [DOI] [PubMed] [Google Scholar]
- Cavigelli M, Li WW, Lin A, Su B, Yoshioka K, Karin M. EMBO J. 1996;15:6269–6279. [PMC free article] [PubMed] [Google Scholar]
- Chen LF, Greene WC. Nat. Rev. Mol. Cell. Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- Chen W, Martindale JL, Holbrook NJ, Liu Y. Mol. Cell. Biol. 1998;18:5178–5188. doi: 10.1128/mcb.18.9.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Thakkar H, Tyan F, Gim S, Robinson H, Lee C, Pandey SK, Nwokorie C, Onwudiwe N, Srivastava RK. Oncogene. 2001;20:6073–6083. doi: 10.1038/sj.onc.1204736. [DOI] [PubMed] [Google Scholar]
- Cheng HY, Li P, David M, Smithgall TE, Feng L, Lieberman MW. Oncogene. 2004;23:3603–3612. doi: 10.1038/sj.onc.1207466. [DOI] [PubMed] [Google Scholar]
- Dancey J, Sausville EA. Nat. Rev. Drug Discov. 2003;2:296–313. doi: 10.1038/nrd1066. [DOI] [PubMed] [Google Scholar]
- Dancey JE. Cancer Cell. 2004;5:411–415. doi: 10.1016/s1535-6108(04)00122-9. [DOI] [PubMed] [Google Scholar]
- Darnell JE., Jr Nat. Rev. Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Dong J, Phelps RG, Qiao R, Yao S, Benard O, Ronai Z, Aaronson SA. Cancer Res. 2003;63:3883–3885. [PubMed] [Google Scholar]
- Durante W. J. Cell. Physiol. 2003;195:373–382. doi: 10.1002/jcp.10274. [DOI] [PubMed] [Google Scholar]
- Evan GI, Vousden KH. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- Franco AV, Zhang XD, Van Berkel E, Sanders JE, Zhang XY, Thomas WD, Nguyen T, Hersey P. J. Immunol. 2001;166:5337–5345. doi: 10.4049/jimmunol.166.9.5337. [DOI] [PubMed] [Google Scholar]
- Giaccone G, Herbst RS, Manegold C, Scagliotti G, Rosell R, Miller V, Natale RB, Schiller JH, Von Pawel J, Pluzanska A, Gatzemeier U, Grous J, Ochs JS, Averbuch SD, Wolf MK, Rennie P, Fandi A, Johnson DH. J. Clin. Oncol. 2004;22:777–784. doi: 10.1200/JCO.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Gong P, Stewart D, Hu B, Vinson C, Alam J. Arch. Biochem. Biophys. 2002;405:265–274. doi: 10.1016/s0003-9861(02)00404-6. [DOI] [PubMed] [Google Scholar]
- Griffith TS, Chin WA, Jackson GC, Lynch DH, Kubin MZ. J. Immunol. 1998;161:2833–2840. [PubMed] [Google Scholar]
- Gschwind A, Fischer OM, Ullrich A. Nat. Rev. Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- Halaban R. Semin. Oncol. 1996;23:673–681. [PubMed] [Google Scholar]
- Hanada M, Feng J, Hemmings BA. Biochim. Biophys. Acta. 2004;1697:3–16. doi: 10.1016/j.bbapap.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Hideshima T, Anderson KC. Br. J. Haematol. 2003;120:10–17. doi: 10.1046/j.1365-2141.2003.03929.x. [DOI] [PubMed] [Google Scholar]
- Hei TK, Liu SX, Waldren C. Proc. Natl. Acad. Sci. USA. 1998;95:8103–8107. doi: 10.1073/pnas.95.14.8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko DD, Robb BW, Hungness ES, Luo G, Hasselgren PO. J. Cell. Biochem. 2002;84:687–698. doi: 10.1002/jcb.10083. [DOI] [PubMed] [Google Scholar]
- Huerta-Yepez S, Vega M, Jazirehi A, Garban H, Hongo F, Cheng G, Bonavida B. Oncogene. 2004;23:4993–5003. doi: 10.1038/sj.onc.1207655. [DOI] [PubMed] [Google Scholar]
- Ivanov V, Fleming TJ, Malek TR. J. Immunol. 1994;153:2394–2406. [PubMed] [Google Scholar]
- Ivanov VN, Hei TK. J. Biol. Chem. 2004;279:22747–22758. doi: 10.1074/jbc.M314131200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov VN, Ronai Z. J. Biol. Chem. 1999;274:14079–14089. doi: 10.1074/jbc.274.20.14079. [DOI] [PubMed] [Google Scholar]
- Ivanov VN, Krasilnikov M, Ronai Z. J. Biol. Chem. 2002;277:4932–4944. doi: 10.1074/jbc.M108233200. [DOI] [PubMed] [Google Scholar]
- Jiang BH, Jiang G, Zheng JZ, Lu Z, Hunter T, Vogt PK. Cell Growth Differ. 2001;12:363–369. [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, Lowe SW. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Kandel ES, Hay N. Exp. Cell Res. 1999;253:210–229. doi: 10.1006/excr.1999.4690. [DOI] [PubMed] [Google Scholar]
- Kapahi P, Takahashi T, Natoli G, Adams SR, Chen Y, Tsien RY, Karin M. J. Biol. Chem. 2000;275:36062–36066. doi: 10.1074/jbc.M007204200. [DOI] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. Nat. Rev. Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A. Nat. Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- Karin M, Yamamoto Y, Wang QM. Nat. Rev. Drug Discov. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- Kitada S, Pedersen IM, Schimmer AD, Reed JC. Oncogene. 2002;21:3459–3474. doi: 10.1038/sj.onc.1205327. [DOI] [PubMed] [Google Scholar]
- Krasilnikov M, Ivanov VN, Dong J, Ronai Z. Oncogene. 2003;22:4092–4101. doi: 10.1038/sj.onc.1206598. [DOI] [PubMed] [Google Scholar]
- Lazar-Molnar E, Hegyesi H, Toth S, Falus A. Cytokine. 2000;12:547–554. doi: 10.1006/cyto.1999.0614. [DOI] [PubMed] [Google Scholar]
- Lee PJ, Camhi SL, Chin BY, Alam J, Choi AM. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000;279:L175–82. doi: 10.1152/ajplung.2000.279.1.L175. [DOI] [PubMed] [Google Scholar]
- Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, Choi AM. J. Biol. Chem. 1997;272:5375–5381. [PubMed] [Google Scholar]
- Levitzki A, Gazit A. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr Nat. Rev. Mol. Cell. Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Li G, Kalabis J, Xu X, Meier F, Oka M, Bogenrieder T, Herlyn M. Oncogene. 2003;22:6891–6899. doi: 10.1038/sj.onc.1206819. [DOI] [PubMed] [Google Scholar]
- Li G, Satyamoorthy K, Herlyn M. Cancer Res. 2001;61:3819–3825. [PubMed] [Google Scholar]
- Li M, Cai JF, Chiu JF. J. Cell. Biochem. 2002;87:29–38. doi: 10.1002/jcb.10269. [DOI] [PubMed] [Google Scholar]
- Lin A, Karin M. Semin. Cancer Biol. 2003;13:107–114. doi: 10.1016/s1044-579x(02)00128-1. [DOI] [PubMed] [Google Scholar]
- Liu J, Kadiiska MB, Liu Y, Lu T, Qu W, Waalkes MP. Toxicol. Sci. 2001a;61:314–320. doi: 10.1093/toxsci/61.2.314. [DOI] [PubMed] [Google Scholar]
- Liu SX, Athar M, Lippai I, Waldren C, Hei TK. Proc. Natl. Acad. Sci. USA. 2001b;98:1643–1648. doi: 10.1073/pnas.031482998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig S, Hoffmeyer A, Goebeler M, Kilian K, Hafner H, Neufeld B, Han J, Rapp UR. J. Biol. Chem. 1998;273:1917–1922. doi: 10.1074/jbc.273.4.1917. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. N. Engl. J. Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Martin D, Rojo AI, Salinas M, Diaz R, Gallardo G, Alam J, De Galarreta CM, Cuadrado A. J. Biol. Chem. 2004;279:8919–8929. doi: 10.1074/jbc.M309660200. [DOI] [PubMed] [Google Scholar]
- Mathas S, Lietz A, Janz M, Hinz M, Jundt F, Scheidereit C, Bommert K, Dorken B. Blood. 2003;102:1028–1034. doi: 10.1182/blood-2002-04-1154. [DOI] [PubMed] [Google Scholar]
- Myklebust AT, Helseth A, Breistol K, Hall WA, Fodstad O. J. Neurooncol. 1994;21:215–224. doi: 10.1007/BF01063770. [DOI] [PubMed] [Google Scholar]
- Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. J. Immunol. Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- Orlowski RZ, Baldwin AS., Jr Trends Mol. Med. 2002;8:385–389. doi: 10.1016/s1471-4914(02)02375-4. [DOI] [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Perlis C, Herlyn M. Oncologist. 2004;9:182–187. doi: 10.1634/theoncologist.9-2-182. [DOI] [PubMed] [Google Scholar]
- Rea MA, Gregg JP, Qin Q, Phillips MA, Rice RH. Carcinogenesis. 2003;24:747–756. doi: 10.1093/carcin/bgg010. [DOI] [PubMed] [Google Scholar]
- Reifenberger J, Knobbe CB, Sterzinger AA, Blaschke B, Schulte KW, Ruzicka T, Reifenberger G. Int. J. Cancer. 2004;109:377–384. doi: 10.1002/ijc.11722. [DOI] [PubMed] [Google Scholar]
- Roussel RR, Barchowsky A. Arch. Biochem. Biophys. 2000;377:204–212. doi: 10.1006/abbi.2000.1770. [DOI] [PubMed] [Google Scholar]
- Satyamoorthy K, De Jesus E, Linnenbach AJ, Kraj B, Kornreich DL, Rendle S, Elder DE, Herlyn M. Melanoma Res. 1997;7(Suppl 2):S35–42. [PubMed] [Google Scholar]
- Satyamoorthy K, Li G, Gerrero MR, Brose MS, Volpe P, Weber BL, Van Belle P, Elder DE, Herlyn M. Cancer Res. 2003;63:756–759. [PubMed] [Google Scholar]
- Schlessinger J. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M. Nat. Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- Sibilia M, Fleischmann A, Behrens A, Stingl L, Carroll J, Watt FM, Schlessinger J, Wagner EF. Cell. 2000;102:211–220. doi: 10.1016/s0092-8674(00)00026-x. [DOI] [PubMed] [Google Scholar]
- Snow ET. Pharmacol. Ther. 1992;53:31–65. doi: 10.1016/0163-7258(92)90043-y. [DOI] [PubMed] [Google Scholar]
- Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, La Mantia C, Mourton T, Herrup K, Harris RC, Barnard JA, Yuspa SH, Coffey RJ, Magnuson T. Science. 1995;269:230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- Tuveson DA, Weber BL, Herlyn M. Cancer Cell. 2003;4:95–98. doi: 10.1016/s1535-6108(03)00189-2. [DOI] [PubMed] [Google Scholar]
- von Willebrand M, Zacksenhaus E, Cheng E, Glazer P, Halaban R. Cancer Res. 2003;63:1420–1429. [PubMed] [Google Scholar]
- Wakeling AE, Barker AJ, Davies DH, Brown DS, Green LR, Cartlidge SA, Woodburn JR. Breast Cancer Res. Treat. 1996;38:67–73. doi: 10.1007/BF01803785. [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX. Nat. Rev. Mol. Cell. Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Yih LH, Peck K, Lee TC. Carcinogenesis. 2002;23:867–876. doi: 10.1093/carcin/23.5.867. [DOI] [PubMed] [Google Scholar]
- Zhang X, Shan P, Alam J, Davis RJ, Flavell RA, Lee PJ. J. Biol. Chem. 2003a;278:22061–22070. doi: 10.1074/jbc.M301858200. [DOI] [PubMed] [Google Scholar]
- Zhang XD, Borrow JM, Zhang XY, Nguyen T, Hersey P. Oncogene. 2003b;22:2869–2881. doi: 10.1038/sj.onc.1206427. [DOI] [PubMed] [Google Scholar]
- Zheng XH, Watts GS, Vaught S, Gandolfi AJ. Toxicology. 2003;187:39–48. doi: 10.1016/s0300-483x(03)00025-8. [DOI] [PubMed] [Google Scholar]