

Abstract
GNE myopathy is an autosomal recessive muscle disease due to biallelic mutations in GNE, a gene encoding for a single protein with key enzymatic activities in sialic acid biosynthetic pathway. The diagnosis should be considered primarily in patients presenting with distal weakness (foot drop) in early adulthood (other onset symptoms are possible too). The disease slowly progresses to involve other lower and upper extremities’ muscles, with marked sparing of the quadriceps. Characteristic findings found in biopsies of affected muscles include “rimmed” (autophagic) vacuoles, aggregation of various proteins, and fiber size variation. The diagnosis is confirmed by sequencing of the GNE gene. Note that we use a new mutation nomenclature based upon the longest transcript (GenBank: NM_001128227), which encodes a 31-amino-acid longer protein than the originally described one (GenBank: NM_005476), which has been used previously in most papers. Given the pathophysiology of the disease, recent clinical trials have evaluated the use of sialic acid or ManNAc (a precursor of sialic acid) in patients with GNE myopathy as well as early gene therapy trials. Now that therapies are under investigation, it is critical that a timely and accurate diagnosis is made in patients with GNE myopathy.
Keywords: GNE myopathy, hereditary inclusion body myopathy, distal myopathy with rimmed vacuoles, sialic acid, N-acetylmannosamine (ManNAc)
INTRODUCTION
GNE myopathy is a progressive muscle disease caused by mutations in the GNE gene, which encodes for key enzyme in the sialic acid biosynthesis pathway (Figure 1). In 2001 the gene defect associated with hereditary inclusion body myopathy (HIBM), was identified in Iranian Jews and other ethnicities [1]. Several mutations in the gene encoding sialic acid synthesis, called GNE, were identified. Soon afterwards it became clear that the distal myopathy with rimmed vacuoles (DMRV), first described in Japan by Nonaka is also caused by defects in the same gene [2]. In more than a decade afterwards numerous patients with GNE defects were described worldwide. Other names such as inclusion body myopathy type 2 and quadriceps-sparing myopathy have been used to describe this disease. To avoid confusion a group of international experts working in the field of GNE myopathy has recently met and later decided to unify the nomenclature to GNE myopathy (name of disease and its mutations) [3].
Figure 1. Sialic acid biosynthesis pathway.

The biosynthesis of sialic acid (Neu5Ac) occurs in the cytoplasm. The initial substrate for this pathway (UDP-GlcNAc) derives from glucose. In the rate-limiting step of the pathway, UDP-GlcNAc is epimerized into ManNAc by GlcNAc 2-epimerase, encoded by the epimerase domain of GNE. ManNAc is phosphorylated by ManNAc kinase encoded by “kinase” domain of GNE. Once Neu5Ac acid is synthesized, it becomes “activated” by the effect of CMP-sialic acid synthetase in the nucleus. CMP-sialic acid, the active form of Neu5Ac is used a donor of sialic acid to nascent proteins in the Golgi for the generation of glycoproteins. CMP-sialic acid also acts as a feedback inhibitor of the UDP-GlcNAc 2-epimerase enzyme by binding to its allosteric site.
In the passing decade much progress was achieved in clarifying some biochemical, genetic and phenotypic variations of this myopathy, but enigmas still persist about its pathogenesis [4]. Importantly, formal therapeutic trials have been initiated in the last two years. This timely review of the current knowledge about this unique myopathy also contains information presented at the recent 3rd meeting of the GNE Consortium (San Francisco, September 2013).
CLINICAL FEATURES
GNE myopathy is a relatively rare muscle disease with some typical clinical and pathological characteristics that may be very important for its correct identification. This is especially true in regions where the disease is probably less prevalent or under recognized (see Demographics). GNE myopathy is an adult onset muscle disorder with signs typically appearing in the third decade of life. However, onset at teenage has been reported, the earliest probably around 12 years of age. The commonest presentation is weakness of distal muscle of the leg (foot drop) thus GNE myopathy is still classified in the group of distal myopathies. Less common presentation include asymmetric foot drop or manifestations initially appearing in upper extremities and onset in the proximal leg musculature. The disease does not remain limited to the distal musculature but slowly progresses to involve more proximal leg muscles and the upper limbs. A very unique feature of this myopathy is the relative or full sparing of the quadriceps, even in advanced stages of the disease. This pattern when recognized in the patient is probably diagnostic and can be visualized by muscle imaging, which will also help differential diagnosis and selecting the biopsy site (see Diagnosis). However, the unique pattern of involvement becomes evident only after the proximal leg musculature becomes affected. It is of note that about 5% [5] of patients may have marked and early quadriceps involvement making the diagnosis more difficult. The pattern of muscle weakness in the upper limbs is more variable and can include scapular weakness (mimicking scapuloperoneal syndrome), or distal weakness of the hands with various degrees of involvement. There are patients with onset in proximal leg muscles only mimicking an unusual pattern of limb girdle muscular dystrophy [6] such onset may delay diagnosis but in retrospect both clinical and imaging features show that the posterior thigh muscles become markedly affected while the quadriceps is spared.
Cardiac involvement is not a classical feature of GNE myopathy. However, some patients with histologic or electrophysiologic evidence for heart disease have been reported [7]. Although its association with GNE myopathy needs to be further defined, ECG may need to be performed every several years. Respiratory muscle are usually not clinically affected during the course of the disease until the later stages when a proportion of wheelchair bound patients have reduced respiratory function [8]. It is very rare to have a patient with a need of respiratory support even in the final stage of the disease, but this may occur [9].
The course is slowly progressive with variable pace. In many patients, especially those of Persian Jewish ancestry, walking is still maintained (at least on flat ground) for 15–20 years (and even more) after the onset of the disease[5]. However, a study from a large cohort of patients in Japan noted an average 10 years till the need to use wheelchair. In this cohort there was a suggestion that patients with a homozygous kinase mutation do better than those with compound heterozygous for such GNE mutation[10]. The progression of GNE myopathy and the contribution of genetic and environmental factors to its variability need to be further delineated.
PATHOLOGICAL FEATURES
Pathological features of GNE myopathy include ‘rimmed’ vacuoles, aggregation of various proteins, and fiber size variation. ‘Rimmed’ vacuoles are recognized as small empty spaces surrounded by tiny red granules in the cytoplasm of muscle fibers typically on modified Gomori trichrome (mGT) staining. Although this empty space is called “vacuole”, this is a space artificially produced during staining procedures. The area was originally occupied mostly by red-colored granules, but they become detached from slide glass. On electron microscopy (EM), cluster of autophagic vacuoles are seen and each autophagic vacuole corresponds to red-colored granule on mGT.
Rimmed vacuoles are probably the most prominent finding on routine muscle histochemistry as protein aggregates are often hardly visualized without immunohistochemical staining. Aggregated proteins include β-amyloid, phosphorylated Tau, TDP-43 and α-synuclein. β-amyloid is supposed be detected on Congo red stain but in reality often need immunostaining for visualization.
Most of aggregated proteins are ubiquitinated and are believed to be targeted for autophagy clearance through p62-dependent aggresome formation, which is sometimes termed “aggrephagy” [11]. However, these proteins cannot be digested, thereby autophagy buildup occurs, which is detected as rimmed vacuoles on histochemistry. Therefore, protein aggregation should be in the upstream of the pathological cascade that produces rimmed vacuoles. In support of this notion, aggregation of β-amyloid is observed prior to the development of rimmed vacuoles in GNE myopathy model mouse. On EM, autophagic vacuoles are often present next to filamentous inclusions on EM, also suggesting close relationship between autophagy and protein aggregation. In nucleus, as well as cytoplasm, tubulofilamentous inclusions of 18–21 nm in diameter are observed. Of note, this protein aggregation-rimmed vacuole pathology is not an exclusively specific feature of GNE myopathy but is rather commonly seen in other hereditary and acquired myopathies, including sporadic inclusion body myositis (IBM).
Fiber size variation is mainly due to the presence of atrophied fibers, which are often angular in shape. For unknown reason, atrophic fibers tend to cluster in GNE myopathy, sometimes giving a false impression of neurogenic atrophy. In the mouse model, muscle fiber atrophy starts earlier than protein aggregation and rimmed vacuoles formation, indicating that, at least in part, mechanism of muscle fiber atrophy is independent from that of aggrephagy-related degenerative pathway.
Although inflammatory change is usually not a feature of GNE myopathy, there are reports of rare cases with lymphocyte infiltration into the endomysium [5 12 13] that could potentially mislead to a diagnosis of sporadic IBM. Nevertheless, the pattern of muscle involvement and the age of disease onset are different. Of note, a recent study showed upregulation of pro-inflammatory cell stress response with overexpression of αB-crystalin and iNOS, which seems to precede muscle degeneration with accumulation of β-amyloid, suggesting that inflammation may play a role in the early stages in the pathologic cascade of GNE myopathy even though cellular response is absent [14].
Another pitfall is the selection of biopsy site. As mentioned earlier, one of the most characteristic clinical features is quadriceps sparing. Therefore, biopsy of quadriceps muscle, which is one of the most frequently biopsied muscles, often gives a minimal or even completely normal histology. A significant number of cases may thus be undiagnosed or misdiagnosed because of quadriceps biopsy. When available muscle imaging is highly recommended for choosing an appropriate biopsy site.
GENETIC CAUSE AND POSSIBLE MOLECULAR MECHANISM
GNE myopathy is an autosomal recessive disease due to biallelic GNE gene mutations [1] [2][15] (Figure 2). Missense mutations account for the majority of alleles and no patient with biallelic null mutations has ever been found, suggesting that probably only ‘mildly deleterious’ mutations that are not associated with complete loss of GNE protein are necessary to cause this adult-onset myopathy. In fact, knocking out the Gne gene in mouse results in embryonic lethality [16]. It is possible that in humans biallelic null mutations are either lethal too or associated with a different, currently unrecognized disorder.
Figure 2. Schematic illustration of GNE gene structure.
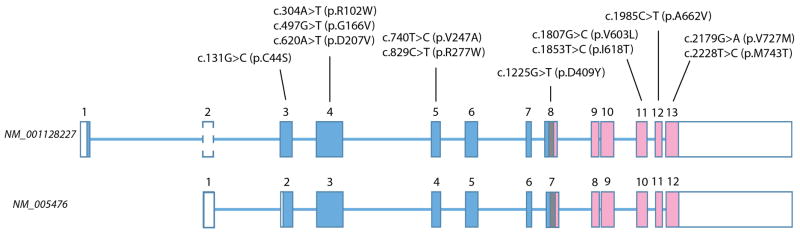
Gene structure for the two most representative transcripts is shown. The longest transcript (NM_001128227) encodes 753 amino acids, including 17 amino acid encoded by exon 1. The originally described transcript shown at the bottom (GenBank: NM_005476), uses an alternative first exon which is non-coding and the initial codon resides in the 43rd–45th nucleotides in the second exon, which makes the protein shorter by 31 amino acids. Note exon eight encodes the last part of epimerase domain, junctional region, and initial part of kinase domain.
Size of exons is to scale but that of introns is not. Boxes indicate exons. Open box means non-coding region. Blue and pick respectively indicate epimerase and kinase encoding regions.
Mutations mentioned in the text are included for reference.
In human, at least 6 different GNE transcripts have been described [3][17]. The originally described transcript (GenBank: NM_005476) (Ensembl: ENST00000377902) (UCSC: uc010mlh.3) encodes 722 amino acids while the longest transcript (GenBank: NM_001128227) (Ensembl: ENST00000396594) (UCSC: uc010mli.3), encodes 753 amino acids. Both transcripts are encoded in 12 exons and the difference between the two transcripts is in alternative first exons. NM_005476 has noncoding first exon and initial codon starts in the 43rd nucleotide in the second exon. In contrast, longer NM_001128227 uses a different, 17 amino-acid coding exon 1. The second exon is the same as NM_005476 but the first 42 nucleotides before NM_005476’s initial codon are also transcribed in NM_001128227, making the NM_001128227 transcript 31 amino acid longer than NM_005476. As this 31-amino acid coding sequence is added in the 5′ part of the NM_005476, description of the mutation position will be changed depending on which transcript is used as standard sequence. Since so far no pathogenic mutation has been found in NM_001128227 specific region, it is still unknown which transcript is crucial for causing GNE myopathy. We adopt the mutation nomenclature based upon NM_001128227 throughout this manuscript, following the guidelines of the Human Genome Variation Society (http://www.hgvs.org). Furthermore, as the NM_001128227’s first exon resides before the NM_005476’s first exon, now the former is named exon 1 and the latter exon 2, and the remaining exons are labeled exons 3–13 (Figure 2).
GNE encodes a single protein with two enzymatic activities in the biosynthetic pathway of 5-N-acetylneuraminic acid (Neu5Ac): UDP-N-acetylglucosamine 2-epimerase (GlcNAc 2-epimerase) and N-acetylmannosamine kinase (ManNAc) (Figure 1). Sialic acids are monosaccharides and 5-N-acetylneuraminic acid (Neu5Ac) is the most abundant sialic acid in mammals. Neu5Ac is usually present in the terminal portion of sugar chains in glycoproteins and glycolipids where they mediate several biological processes [18].
Due to recessive mutations in the GNE gene, sialic acid production is decreased and consequently, sialylation, i.e., incorporation of sialic acid to glycoproteins and glycolipids, is also decreased [19 20]. Hyposialylation appears to be a major cause of this myopathy as administration of sialic acid or its precursor ManNAc, prevents or arrests the development of disease in the mouse models of GNE myopathy [21]. This is the rationale behind current therapeutic trials (see below). However, the exact mechanism by which GNE defects lead to the human disease is still not fully understood and additional processes may contribute to it.
DIAGNOSIS
Currently, the diagnosis of GNE myopathy relies on identifying characteristic clinical manifestations and histopathologic findings on muscle biopsy and is confirmed by the identification of biallelic GNE mutations [22].
The diagnosis should be considered in patients presenting in young adulthood with foot drop, although the identification of the disease may be done at more advanced stages of the disease, when more proximal lower extremity or upper extremity muscles are affected. Clinically, the diagnosis may be confused with other conditions, such as other distal myopathies, limb girdle muscular dystrophy [23], spinal muscular atrophy, or Charcot-Marie-Tooth disease. The reliability of muscle biopsy for the diagnosis of GNE myopathy appears to depend on the technical skill and diagnostic expertise of those handling and evaluating the specimen (see above). GNE protein is present in the diseased muscle thus immunohistology may not identify the defect and furthermore no specific GNE antibody that could be used for diagnostics has yet been synthesized.
The use of muscle imaging can guide the choice of muscle to biopsy and can help establish disease severity. Muscle magnetic resonance imaging (MRI) of affected muscles initially shows increased hyperintensity on T2 STIR sequences followed by fatty-fibrous replacement evident on T1-weighted images [24].
The identification of biallelic mutations in GNE is the only definite diagnostic tool. Because there are 147 known GNE mutations associated with GNE myopathy to date (based upon HGMD Professional 2013.4 version), sequencing of GNE is necessary when considering the diagnosis. In regions where one mutation is very prevalent (e.g. p.M743T in the Middle East) testing for it may suffice. Patients with typical clinical and histological manifestations and only one heterozygous GNE mutation identified by sequencing have been encountered. Such patients may have deletions [25] not identified by sequencing, or mutations in noncoding regions of GNE on the other allele. Alternatively, they may have a genetically different disorder. In such cases, next generation sequencing could be considered in the further diagnostic effort. Heterozygous carriers have no phenotype, although heterozygous mice have decreased sialylation [19].
Because of the rarity of this disease and the diagnostic difficulties mentioned above, patients may remain undiagnosed for a long period of time. In one cohort of patients followed at the NIH the diagnosis was delayed by an average of 10 years (NCC, unpublished).
DEMOGRAPHICS
GNE myopathy is a disorder found worldwide, however, until recently it was mostly recognized in patients of Japanese and Persian Jewish ethnicity, where founder mutations are prevalent and different names, namely DMRV and HIBM, are used. However, after the identification of the genetic defect [1], it is now clear that this is a worldwide disorder with an estimated prevalence of about 1/1,000,000 (higher prevalence is seen in Middle Eastern Jews and Japanese) (Figure 3). In the last decade there have been a plethora of reports from Europe, many Asian countries and North America. Interestingly no patients were reported from South America, apart from two families of Persian Jewish ancestry residing in Argentina (ZA personal observations). The lack of report from South America may be due to a decreased recognition of the condition.
Figure 3.

The worldwide prevalence of GNE myopathy is estimated at 1/1,000,000.
Japan and Asian Oceanian region
Among all patients whose muscle biopsy was examined at the National Center of Neurology and Psychiatry (NCNP) in Tokyo between 1978 and 2005, 42 had GNE myopathy. During the same period of time, 502 had Duchenne muscular dystrophy (DMD), suggesting that the prevalence of GNE myopathy is roughly one log lower than that of DMD. In Japan, the prevalence of DMD ranges roughly from 1500 to 4000, indicating that 150–400 patients may be present in Japan. The cumulative number of Japanese patients who have been diagnosed to have biallelic GNE mutations at NCNP since 1978 is 237 at the time of writing. Although some patients may not be alive by now it is of note the estimated number of patients and actual number of genetically diagnosed patients are in a similar range.
Among all mutations identified, 95% are missense, as mentioned earlier. Three most frequent mutations are p.V603L, p.D207V, and p.C44S, respectively with allele frequency of 46.8%, 21.9% and 3.2% [2 26 27]. The p.V603L and p.C44S mutations were also identified in Korea and Northern part of China, probably being compatible with a hypothesis of historical migration of people from the continent to Japan through Korean peninsula [28 29].
In other parts of Asia, much fewer patients have been reported. Nevertheless, p.A662V and p.V727M seem to be common in South East Asia region: the former from Vietnam and Malaysia while the latter from Thailand and Malaysia, in addition to India [29–31]. The former has also been found also in the US and Australia. However, ethnically they appear to originate from Vietnam.
Israel and Middle East
The largest cluster of GNE myopathy is that of Jews originating from Iran and neighboring countries (Uzbekistan, Afghanistan, Iraq and Syria). They are all homozygous for the kinase mutation p.M743T, which is the commonest GNE mutation worldwide. About 150 such patients were identified in Israel over the years and the estimated carrier frequency is 1 in 20 in this ethnic group [1]. A survey in the large Persian (Iranian) Jewish community residing in Southern California suggested an even higher carrier rate of 1 in 11 [32].
Interestingly the p.M743T mutation has been identified not only in Middle Eastern Jews but also in Muslim Arabs in Israel (of both Beduin and Palestinian origins) who all (5 families) carry it in a homozygous genotype. Furthermore, this homozygous mutation has been reported in Muslim patients from North Africa (Egypt and Tunisia) [33]. Thus a regional founder mutation is strongly suggested and unpublished data suggest this mutation to be about 2500 years old. The origin of this high frequency p.M743T GNE mutation in Persian Jews coming from various regions of Iran is unclear, as no data on general population testing in Iran is available. However, a cluster of GNE myopathy patients due to p.M743T mutation was identified in a small town (Sangesar) in northern Iran. They all belonged to the Bahai religion (a relatively modern new religion originating in Persia during the 19th century) and a carrier rate of 1 in 25 was estimated [34]. It is unclear if this cluster is due to ‘spread’ of mutation from neighboring Jewish residents.
The importance of the knowledge about this common mutation is for easy diagnosis in patients originating from the Middle East residing outside this region. However, one should be cautious since although for more than a decade no patient with GNE myopathy having other mutations was identified in Israel, three families with different mutations were identified in 2013. One of those is a Jewish family from Mumbai, India. Both patients were homozygous to a mutation not reported in patients from other regions of India. This fact emphasizes the need for pattern recognition of the clinical features of GNE myopathy in order not to delay correct diagnosis.
North America
Many patients in North America have been identified as having GNE myopathy, mostly in the US and Canada. A significant portion of these patients are homozygous for the p.M743T mutation and are of Middle Eastern background. The remainder is comprised mostly by patients who are compound heterozygotes for private mutations of GNE, reflecting the mixed ethnic background in the USA. Mutations in these patients have been traced to various ethnic backgrounds such as German: p.V247A, p.D409Y, and p.F559C, British: p.G166V and p.R277W, Irish: p.A662V and p.D409Y, Indian p.V727M, and Cajun p.I618T [1 35–37]. Other mutations, such as p.R102W have only been described in America [37].
The only description of GNE myopathy in Hispanics is of a compound heterozygote patient (p.A555V/Y706H) whose ethnic background included Mexico [38].
Europe
Since the identification of the causative gene, patients with novel GNE mutations were identified in numerous European countries (e.g. Italy, Germany, Netherlands, France, Belgium). However because many European countries have large immigrant communities, including Asia, the recognition of the clinical pattern of GNE myopathy is critical for neuromuscular practice in this continent. Special attention should be given to mutations with possible founder effect. One such cluster was identified in Gypsies/Roma patients who are all homozygous for a kinase mutation p.I618T [39]. The mutation was not new when identified, however, at least 27 patients shared it. Two unusual features were mentioned: atrophy of thenar muscles and cardiac arrhythmias. Another region with relatively high GNE myopathy prevalence was recently identified in northern UK and Ireland. Point prevalence was estimated to be 0.19–0.44 in 100,000 for Scotland and Northern Ireland. Two mutations were the most frequent: p.A662V, which is a mutation described in other regions of the world and p.A409T which seemed to be of Northern British origin [40].
NATURAL HISTORY AND PATIENT REGISTRY
Patient monitoring program (Ultragenyx/TREAT-NMD)
The rate of progression of GNE myopathy is variable over few decades. There is a need for more accurate assessment of the clinical variability as well as identifying markers of progression that will optimize the design and interpretation of therapeutic trials. In addition there is a need for patients registry that will not only identify the patients worldwide but will also serve as a source for patients’ information. Such a program was developed by TREAT-NMD and Ultragenyx (HIBM patient monitoring program). There are two components to this program: the first is patients’ registry that will be open to all patients worldwide based on their willingness to add their data. This program will combine the physicians’ reported information with the patient’s personal report and will be conducted under the auspice of TREAD-NMD complying with Good Clinical Practice guidelines. This module has already been initiated (http://gnem-dmp.com/). The second part of this program looking at the natural history of GNE myopathy will be conducted in several sites with large cohorts of patients. These will be different from the sites running therapeutic trials and will have larger distribution in Europe and North America. This second module of the program is currently in progress as a sponsored clinical trial.
NIH study
In 2011, a longitudinal, prospective, single-center natural history study of patients with GNE myopathy was initiated at the National Institutes of Health (NIH study 11-HG-0218; ClinicalTrials.gov: NCT01417533). The objectives of the study are to delineate the natural history of GNE myopathy in a genetically diverse cohort, by characterizing the pattern and rate of progression of muscle weakness, its effect on patients’ function and quality of life of patients and its correlation with genotype and environmental factors; identify ideal outcome measures to be used in clinical trials and discover blood biomarkers that would allow for diagnosis and monitoring of patients. Patients are evaluated every 6–12 months during an inpatient visit that last 3–4 days at the NIH Clinical Center. Evaluations include confirmation of GNE mutations, blood and urine laboratory tests, electrocardiogram, echocardiogram, pulmonary function tests, muscle MRI and measures of strength, function and quality of life.
Remudy (Japanese registry)
Remudy (Registry of Muscular Dystrophy) is a national patient registry for muscle diseases in Japan that was originally established for dystrophinopathy [41] (http://remudy.jp). GNE myopathy patient registration began in June 2012. By the end of 2013,146 GNE myopathy patients have been registered. Registered items include personal information, family history, diagnostic information, and current clinical status. Registration form is filled and signed by both patients themselves and their physicians. This registry will be harmonized with international registry, which is run by TREAT-NMD and Ultragenyx as part of Patient monitoring program (see above).
MOUSE MODEL AND THERAPEUTIC DEVELOPMENT
As mentioned, the Gne knock-out mouse model is an embryonic lethal [16].
The NIH-USA group established a mouse model by knocking-in the p.M743T mutation. However, most mice die with 72 hours after birth due to renal disease and showed no myopathic phenotype; ManNAc administration rescued the neonatal lethal phenotype in these mice [42]. Similar results were obtained in other laboratories [43]. Interestingly the Gne M712T knock-in model developed by the Jerusalem group had a different phenotype. In some animals no renal disease was observed and animals survived more than 1 year without any therapy [43]. Those who die at later age did not show muscle abnormalities. The explanation for these variations in the model remain unclear but may be due to genetic background differences. A group in Kanazawa University in Japan developed Gne V603L knock-in model mouse. Their mice also showed renal phenotype with shorter life span but without myopathy, which was rescued by the administration of NeuAc [44].
The Tokyo group cross-mated heterozygote mice with a transgenic mouse model expressing human p.D207V mutant GNE, eventually obtaining mice overexpressing human mutant GNE protein and disrupting the production of their own Gne. This transgenic mouse model mouse recapitulated the phenotype GNE myopathy clinically, pathologically and biochemically. Mice developed muscle atrophy and weakness after 20 weeks of age, β-amyloid after 30 weeks, and rimmed vacuoles after 40 weeks while sialic acid level was persistently low [45]. NeuAc, ManNAc and sialylactose was administered presymptomatically to these mice and continued for 54–57 weeks, when all the clinicopathological features are supposed to have already developed. Treated mice showed improved survival, body weight, muscle pathology and muscle mass and strength comparable to that of their unaffected littermates [21]. Sialic acid content in muscle was increased but was still considerably lower than in littermates, indicating that even mild increase of muscle sialic acid level is efficacious at least in mice, and that we could expect even better efficacy if sialic acid level could be further increased. Overall, these results provided a proof-of-concept evidence supportive of initiating clinical trials in humans.
CLINICAL TRIALS
Metabolic supplementation with ManNAc, sialic acid and IVIG (as a source of sialic acid) has been evaluated (Table). It is not clear the extent to which metabolic supplementation can correct the defect or modify the course of the disease. Given the slow progression in GNE myopathy, significant changes in muscle strength may not be observed after a relatively short-term metabolic treatment. Because muscle is replaced by fibro-fatty tissue over time in GNE myopathy, stopping or slowing the progression of the disease is realistic, and can have a considerable impact in patients with this chronic debilitating myopathy.
Table.
Clinical trials for development of therapy in GNE myopathy
| Clinical trial ID | Sponsor | Drug | Phase | #Patient | Status | Outcomes |
|---|---|---|---|---|---|---|
| NCT00195637 | NHGRI | Immune Globulin | 1 | 4 | Completed | |
| NCT01236898 | Tohoku University | NeuAc | 1 | 6 | Completed | Safe, No ADE. |
| UMIN000011532 | Tohoku University | SA–ER tablet | 1 | 9 | Active | |
| NCT01359319 | Ultragenyx Pharmaceutical Inc | SA–ER tablet | 1 | 46 | Completed | |
| NCT01517880 | Ultragenyx Pharmaceutical Inc | SA–ER tablet | 2 | 46 | Completed | |
| NCT01830972 | Ultragenyx Pharmaceutical Inc | SA–ER/SA–IR capsule | 2 | 56 | Active, not recruiting | |
| NCT01634750 | TRND/NHGRI | ManNAc | 1 | 22 | Completed | Safe. |
IVIG trial (NIH)
In 2005, intravenous immunoglobulin (IVIG) was used to investigate the effects of sialic acid (Neu5Ac) in 4 patients with GNE myopathy at the NIH (ClinicalTrials.gov: NCT00195637), since IgG contains 8 μmol of Neu5Ac/g. IVIG was infused as a loading dose of 1 g/kg on 2 consecutive days followed by 3 doses of 400 mg/kg at weekly intervals, providing a total of 1.8 mmol (0.55 g) of Neu5Ac for an average subject weighing 70 kg, ie, roughly 6 days worth of normal Neu5Ac production (0.3 mmol/24h). IVIG administration improved objective measures of muscle strength (by 35% in the quadriceps and 46% in the shoulders), as well as function in GNE myopathy patients [46]. Patients lost the benefit of IVIG and its sialic acid contribution about 2 weeks after stopping its administration. The clinical improvements were not accompanied by demonstrable histological changes or increased sialylation of target glycoproteins (using available methods at that time), possibly because such changes require longer-term treatment or muscle regeneration. However, the finding of definitive improvements after IVIG treatment suggests that provision of sialic acid holds therapeutic promise.
NeuAc (Japan)
Phase 1 clinical trial was conducted at Tohoku University from November 2010 to June 2011 (ClinicalTrials.gov: NCT01236898). Three genetically confirmed patients were recruited and were given 800 mg of NeuAc 3 times a day up to 5 consecutive days. No significant adverse effects were observed.
SA-ER (Ultragenyx)
Since regular sialic acid is rapidly excreted after oral administration a slow release product (SA-ER), was developed by Ultragenyx, a company involved in developing metabolic treatments for rare diseases. A trial of 47 recruited patients for oral supplementation using this investigational new drug was started in 2012 (ClinicalTrials.gov: NCT01517880). Baseline serum sialic acid levels were reduced in patients and this highly correlated to their performance in several muscle functional measurements [47]. The trial design was 24 weeks of double blind administration of two doses of extended-release sialic acid (SA-ER) at a dose of 3gr/day or 6 gr/day and a placebo-control group. This was followed by continued administration of either the high or the low dose for an additional 24 weeks. Results of the first phase of the trial gave a modest positive sign in the upper limb functional measurements, compared to a decline in the placebo group (unpublished data presented at the GNE myopathy Consortium meeting, September 2013). Patients with greater walking ability at baseline had a better effect, suggesting that the degree of advancement of this myopathy may be a factor in the observed response. As expected the serum sialic acid levels rose significantly. There were no serious side effects and minimal adverse events were not dose related. Results of the Phase 2 are pending. All 46 of the continuing patients are now on an open label high dose SA-ER for additional 48 weeks.
ManNAc (NIH)
ManNAc is a naturally occurring uncharged monosaccharide and is the first committed precursor for the biosynthesis of Neu5Ac and a substrate of the GNE enzyme. Oral administration of ManNAc in 2 independent GNE myopathy mouse models improved muscle pathology and hyposialylation[21 42].
There is anecdotal evidence of GNE myopathy patients using ManNAc from a nonpharmaceutical source and without medical supervision in doses up to approximately 12 g/day and ranging from a period of 2 months to several years. The most common reported complaint is gastrointestinal symptoms such as abdominal cramps and diarrhea.
A first-in-human Phase 1a, randomized, placebo-controlled, double-blind, single-dose study (ClinicalTrials.gov NCT01634750; IND No.78,091) was conducted at the NIH in 2012–2013. The purpose of this study was to evaluate the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of ManNAc in subjects with GNE myopathy. A total of 22 subjects were enrolled in 3 cohorts. Cohort A included 6 subjects that were randomly assigned in a 2:1 ratio to receive ManNAc (n=4) or placebo (n=2) orally as a liquid solution. Cohorts B and C included 8 subjects randomly assigned in a 3:1 ratio to receive ManNAc (n=6) or placebo (n=2). The dose levels investigated were 3,000 mg, 6,000 mg, and 10,000 mg. ManNAc was safe and well-tolerated in all subjects that participated in this study.
A Phase Ib, escalating multiple-dose study and a Phase II efficacy study of ManNAc in subjects with GNE myopathy are being planned.
Liposomal systemic GNE delivery
A single patient with GNE myopathy due to two missense mutations (one in the kinase and one in the epimerase domains) was given 7 intravenous injections of incremental doses of wild type GNE over a period of 13 months [48]. The DNA vector was coupled to a CMV promoter and delivered systemically in a liposomal package (Lipoplex). The effect on muscle function was minimal but the patient was in an advanced phase of the disease and much strength recovery cannot be expected. However, 72 hours after the highest dose, expression of wild-type GNE and increased sialylation in muscle could be demonstrated. This single-patient trial for compassionate use showed proof-of-principle for this delivery method, although it is expected that infusions will have to be intermittently repeated, as the delivered gene is not expected to persist in the cell cytoplasm.
Future Therapeutic Development
While metabolic supplementation as therapy for GNE myopathy seems promising, there are still other strategies including developments of: 1) better GNE metabolites or sialic acid compounds [49] 2) drugs to block or modify degenerative process, and 3) gene or cell-based therapy. These may be combined with supplementation therapy in the future. Approaches should be explored as they may better correct all deleterious effects of decreased GNE function, although safety and feasibility will need to be established. The GNE research laboratory in Jerusalem (under S. Mitrani Rosenbaum) with collaboration of other laboratories is trying to develop an AAV-mediated gene vector for systemic administration of GNE. Initial results of this approach in animals are promising [50] but the final proof-of-principle of this approach will be only when human trials are started.
CONCLUSIVE REMARK
Much progress toward understanding and treating GNE myopathy has been achieved, but the final target of developing an efficacious therapy is still underway. However, this is one of the first human hereditary myopathies where a logical metabolic therapy is currently being evaluated and a gene therapy is actively developed.
As clinical trials for potential therapies for GNE myopathy are underway, it is necessary to provide a timely diagnosis for patients with GNE myopathy. An early diagnosis has the potential of maximizing the effect of such therapies and reducing anxiety and unnecessary testing in these patients.
Acknowledgments
Funding:
Studies reported in this review have been supported partly by Intramural Research Grant 23-5 for Neurological and Psychiatric Disorders of NCNP, Tokyo, Japan; Research on rare and intractable diseases from the Ministry of Health, Labour and Welfare, Japan; the Neuromuscular Disease Foundation (NDF) of Los Angeles; the Therapeutics for Rare and Neglected Diseases (TRND) Program of the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, Bethesda, Maryland, United States; Hadassah Southern Califonia groups (Malka and Haifa) and numerous patients’ support groups.
Footnotes
Competing Interests:
Dr. Argov is a co Pi and consultant for Ultragenyx. Dr. Nishino is a consultant for Ultragenyx.
Contributor Information
Ichizo Nishino, Email: nishino@ncnp.go.jp.
Nuria Carrillo-Carrasco, Email: carrilln@mail.nih.gov.
Zohar Argov, Email: zohara@ekmd.huji.ac.il.
References
- 1.Eisenberg I, Avidan N, Potikha T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29(1):83–7. doi: 10.1038/ng718. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 2.Nishino I, Noguchi S, Murayama K, et al. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002;59(11):1689–93. doi: 10.1212/01.wnl.0000041631.28557.c6. [DOI] [PubMed] [Google Scholar]
- 3.Huizing M, Carrillo-Carrasco N, Malicdan MC, et al. GNE myopathy: new name and new mutation nomenclature. Neuromuscul Disord. 2014;24(5):387–9. doi: 10.1016/j.nmd.2014.03.004. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Argov Z, Mitrani-Rosenbaum S. The hereditary inclusion body myopathy enigma and its future therapy. Neurotherapeutics. 2008;5(4):633–7. doi: 10.1016/j.nurt.2008.07.004. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Argov Z, Eisenberg I, Grabov-Nardini G, et al. Hereditary inclusion body myopathy: the Middle Eastern genetic cluster. Neurology. 2003;60(9):1519–23. doi: 10.1212/01.wnl.0000061617.71839.42. [DOI] [PubMed] [Google Scholar]
- 6.Park YE, Kim HS, Choi ES, et al. Limb-girdle phenotype is frequent in patients with myopathy associated with GNE mutations. J Neurol Sci. 2012;321(1–2):77–81. doi: 10.1016/j.jns.2012.07.061. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 7.Chai Y, Bertorini TE, McGrew FA. Hereditary inclusion-body myopathy associated with cardiomyopathy: report of two siblings. Muscle & nerve. 2011;43(1):133–6. doi: 10.1002/mus.21839. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 8.Mori-Yoshimura M, Oya Y, Hayashi YK, et al. Respiratory dysfunction in patients severely affected by GNE myopathy (distal myopathy with rimmed vacuoles) Neuromuscular disorders: NMD. 2013;23(1):84–8. doi: 10.1016/j.nmd.2012.09.007. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 9.Weihl CC, Miller SE, Zaidman CM, et al. Novel GNE mutations in two phenotypically distinct HIBM2 patients. Neuromuscular disorders: NMD. 2011;21(2):102–5. doi: 10.1016/j.nmd.2010.11.002. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mori-Yoshimura M, Monma K, Suzuki N, et al. Heterozygous UDP-GlcNAc 2-epimerase and N-acetylmannosamine kinase domain mutations in the GNE gene result in a less severe GNE myopathy phenotype compared to homozygous N-acetylmannosamine kinase domain mutations. J Neurol Sci. 2012;318(1–2):100–5. doi: 10.1016/j.jns.2012.03.016. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 11.Lamark T, Johansen T. Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol. 2012;2012:736905. doi: 10.1155/2012/736905. published Online First: Epub Date. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krause S, Schlotter-Weigel B, Walter MC, et al. A novel homozygous missense mutation in the GNE gene of a patient with quadriceps-sparing hereditary inclusion body myopathy associated with muscle inflammation. Neuromuscular disorders: NMD. 2003;13(10):830–4. doi: 10.1016/s0960-8966(03)00140-8. [DOI] [PubMed] [Google Scholar]
- 13.Kannan MA, Challa S, Urtizberea AJ, et al. Distal myopathy with rimmed vacuoles and inflammation: a genetically proven case. Neurol India. 2012;60(6):631–4. doi: 10.4103/0028-3886.105199. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 14.Fischer C, Kleinschnitz K, Wrede A, et al. Cell stress molecules in the skeletal muscle of GNE myopathy. BMC neurology. 2013;13:24. doi: 10.1186/1471-2377-13-24. published Online First: Epub Date. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitrani-Rosenbaum S, Yakovlev L, Becker Cohen M, et al. Sustained expression and safety of human GNE in normal mice after gene transfer based on AAV8 systemic delivery. Neuromuscular disorders: NMD. 2012;22(11):1015–24. doi: 10.1016/j.nmd.2012.03.013. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 16.Schwarzkopf M, Knobeloch KP, Rohde E, et al. Sialylation is essential for early development in mice. Proc Natl Acad Sci U S A. 2002;99(8):5267–70. doi: 10.1073/pnas.072066199. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yardeni T, Choekyi T, Jacobs K, et al. Identification, tissue distribution, and molecular modeling of novel human isoforms of the key enzyme in sialic acid synthesis, UDP-GlcNAc 2-epimerase/ManNAc kinase. Biochemistry. 2011;50(41):8914–25. doi: 10.1021/bi201050u. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schauer R. Sialic acids as regulators of molecular and cellular interactions. Curr Opin Struct Biol. 2009;19(5):507–14. doi: 10.1016/j.sbi.2009.06.003. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gagiannis D, Orthmann A, Danssmann I, et al. Reduced sialylation status in UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase (GNE)-deficient mice. Glycoconj J. 2007;24(2–3):125–30. doi: 10.1007/s10719-006-9019-7. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 20.Salama I, Hinderlich S, Shlomai Z, et al. No overall hyposialylation in hereditary inclusion body myopathy myoblasts carrying the homozygous M712T GNE mutation. Biochem Biophys Res Commun. 2005;328(1):221–6. doi: 10.1016/j.bbrc.2004.12.157. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 21.Malicdan MC, Noguchi S, Hayashi YK, et al. Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat Med. 2009;15(6):690–5. doi: 10.1038/nm.1956. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 22.Huizing M, Krasnewich DM. Hereditary inclusion body myopathy: a decade of progress. Biochim Biophys Acta. 2009;1792(9):881–7. doi: 10.1016/j.bbadis.2009.07.001. S0925-4439(09)00142-2 [pii] [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyden SE, Duncan AR, Estrella EA, et al. Molecular diagnosis of hereditary inclusion body myopathy by linkage analysis and identification of a novel splice site mutation in GNE. BMC Med Genet. 2011;12:87. doi: 10.1186/1471-2350-12-87. published Online First: Epub Date. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tasca G, Ricci E, Monforte M, et al. Muscle imaging findings in GNE myopathy. J Neurol. 2012;259(7):1358–65. doi: 10.1007/s00415-011-6357-6. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 25.Del Bo R, Baron P, Prelle A, et al. Novel missense mutation and large deletion of GNE gene in autosomal-recessive inclusion-body myopathy. Muscle & nerve. 2003;28(1):113–7. doi: 10.1002/mus.10391. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 26.Tomimitsu H, Shimizu J, Ishikawa K, et al. Distal myopathy with rimmed vacuoles (DMRV): new GNE mutations and splice variant. Neurology. 2004;62(9):1607–10. doi: 10.1212/01.wnl.0000123115.23652.6c. [DOI] [PubMed] [Google Scholar]
- 27.Cho A, Hayashi YK, Monma K, et al. Mutation profile of the GNE gene in Japanese patients with distal myopathy with rimmed vacuoles (GNE myopathy) J Neurol Neurosurg Psychiatry. 2013 doi: 10.1136/jnnp-2013-305587. published Online First: Epub Date. [DOI] [PubMed] [Google Scholar]
- 28.Kim BJ, Ki CS, Kim JW, et al. Mutation analysis of the GNE gene in Korean patients with distal myopathy with rimmed vacuoles. J Hum Genet. 2006;51(2):137–40. doi: 10.1007/s10038-005-0338-5. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 29.Lu X, Pu C, Huang X, et al. Distal myopathy with rimmed vacuoles: clinical and muscle morphological characteristics and spectrum of GNE gene mutations in 53 Chinese patients. Neurol Res. 2011;33(10):1025–31. doi: 10.1179/1743132811Y.0000000070. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 30.Liewluck T, Pho-Iam T, Limwongse C, et al. Mutation analysis of the GNE gene in distal myopathy with rimmed vacuoles (DMRV) patients in Thailand. Muscle & nerve. 2006;34(6):775–8. doi: 10.1002/mus.20583. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 31.Nalini A, Gayathri N, Nishino I, et al. GNE myopathy in India. Neurol India. 2013;61(4):371–4. doi: 10.4103/0028-3886.117609. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 32.Kaback M, Lopatequi J, Portuges AR, et al. Genetic screening in the Persian Jewish community: A pilot study. Genet Med. 2010;12(10):628–33. doi: 10.1097/GIM.0b013e3181edef5b. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 33.Amouri R, Driss A, Murayama K, et al. Allelic heterogeneity of GNE gene mutation in two Tunisian families with autosomal recessive inclusion body myopathy. Neuromuscular disorders: NMD. 2005;15(5):361–3. doi: 10.1016/j.nmd.2005.01.012. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 34.Khademian H, Mehravar E, Urtizberea J, et al. Prevalence of GNE p.M712T and hereditary inclusion body myopathy (HIBM) in Sangesar population of Northern Iran. Clin Genet. 2012;9999(9999) doi: 10.1111/cge.12086. published Online First: Epub Date. [DOI] [PubMed] [Google Scholar]
- 35.Eisenberg I, Grabov-Nardini G, Hochner H, et al. Mutations spectrum of GNE in hereditary inclusion body myopathy sparing the quadriceps. Human mutation. 2003;21(1):99. doi: 10.1002/humu.9100. published Online First: Epub Date. [DOI] [PubMed] [Google Scholar]
- 36.Vasconcelos OM, Raju R, Dalakas MC. GNE mutations in an American family with quadriceps-sparing IBM and lack of mutations in s-IBM. Neurology. 2002;59(11):1776–9. doi: 10.1212/01.wnl.0000039780.13681.ad. [DOI] [PubMed] [Google Scholar]
- 37.Saechao C, Valles-Ayoub Y, Esfandiarifard S, et al. Novel GNE mutations in hereditary inclusion body myopathy patients of non-Middle Eastern descent. Genet Test Mol Biomarkers. 2010;14(2):157–62. doi: 10.1089/gtmb.2009.0157. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 38.Darvish D, Vahedifar P, Huo Y. Four novel mutations associated with autosomal recessive inclusion body myopathy (MIM: 600737) Molecular genetics and metabolism. 2002;77(3):252–6. doi: 10.1016/s1096-7192(02)00141-5. [DOI] [PubMed] [Google Scholar]
- 39.Kalaydjieva L, Lochmuller H, Tournev I, et al. 125th ENMC International Workshop: Neuromuscular disorders in the Roma (Gypsy) population, 23–25 April 2004, Naarden, The Netherlands. Neuromuscular disorders: NMD. 2005;15(1):65–71. doi: 10.1016/j.nmd.2004.09.008. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 40.Chaouch A, Brennan KM, Hudson J, et al. Two recurrent mutations are associated with GNE myopathy in the North of Britain. J Neurol Neurosurg Psychiatry. doi: 10.1136/jnnp-2013-306314. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakamura H, Kimura E, Mori-Yoshimura M, et al. Characteristics of Japanese Duchenne and Becker muscular dystrophy patients in a novel Japanese national registry of muscular dystrophy (Remudy) Orphanet J Rare Dis. 2013;8(1):60. doi: 10.1186/1750-1172-8-60. published Online First: Epub Date. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galeano B, Klootwijk R, Manoli I, et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest. 2007;117(6):1585–94. doi: 10.1172/JCI30954. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sela I, Yakovlev L, Becker Cohen M, et al. Variable phenotypes of knockin mice carrying the M712T Gne mutation. Neuromolecular Med. 2013;15(1):180–91. doi: 10.1007/s12017-012-8209-7. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 44.Ito M, Sugihara K, Asaka T, et al. Glycoprotein hyposialylation gives rise to a nephrotic-like syndrome that is prevented by sialic acid administration in GNE V572L point-mutant mice. PLoS One. 2012;7(1):e29873. doi: 10.1371/journal.pone.0029873. published Online First: Epub Date. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malicdan MC, Noguchi S, Hayashi YK, et al. Muscle weakness correlates with muscle atrophy and precedes the development of inclusion body or rimmed vacuoles in the mouse model of DMRV/hIBM. Physiol Genomics. 2008;35(1):106–15. doi: 10.1152/physiolgenomics.90219.2008. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 46.Sparks S, Rakocevic G, Joe G, et al. Intravenous immune globulin in hereditary inclusion body myopathy: a pilot study. BMC Neurol. 2007;7:3. doi: 10.1186/1471-2377-7-3. 1471-2377-7-3 [pii] published Online First: Epub Date. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mayhew JE, Skrinar AM, Bronstein F, et al. Characterization of strength and function in adults with inclusion body myopathy (HIBM)/GNE myopathy. Neuromuscular Disorders; 18th International Congress of The World Muscle Society; Asilomar, California, USA. 2013. p. 755. [Google Scholar]
- 48.Nemunaitis G, Jay CM, Maples PB, et al. Hereditary inclusion body myopathy: single patient response to intravenous dosing of GNE gene lipoplex. Hum Gene Ther. 2011;22(11):1331–41. doi: 10.1089/hum.2010.192. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malicdan MC, Noguchi S, Tokutomi T, et al. Peracetylated N-acetylmannosamine, a synthetic sugar molecule, efficiently rescues muscle phenotype and biochemical defects in mouse model of sialic acid-deficient myopathy. J Biol Chem. 2012;287(4):2689–705. doi: 10.1074/jbc.M111.297051. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tal-Goldberg T, Lorain S, Mitrani-Rosenbaum S. Correction of the Middle Eastern M712T Mutation Causing GNE Myopathy by Trans-Splicing. Neuromolecular Med. 2013 doi: 10.1007/s12017-013-8278-2. published Online First: Epub Date. [DOI] [PubMed] [Google Scholar]