

Abstract
Using light irradiation as a trigger, large-scale structural reconfiguration of DNA nanostructures is demonstrated. We incorporated photo-cleavable spacers at strategic locations within the short oligonucleotide strands connecting adjacent helices within a DNA origami sphere, and then used light to transform the sphere into two tethered hemispheres.
Self-assembly of DNA represents a powerful method of creating intricate two and three dimensional nanostructures.1,2 Recent advances in DNA nanotechnology have enabled the efficient fabrication of a large diversity of nanoscale objects in only one step.3–16 Utilization of these nanostructures for biomedical applications would be greatly advanced if they could undergo dynamic reconfigurations in response to specific stimuli.17–19 Several studies have demonstrated that chemicals such as oligonucleotides,7,20–23 bioactive small molecules,24,25 or acid26 can be used effectively to trigger such transformations. For example, chemicals have been used to induce DNA devices to travel on patterned surfaces,27,28 undergo topological reconfiguration,21 or expose encapsulated cargo.22,24,25 Light irradiation represents another attractive stimulus because it can be controlled with high spatial and temporal resolution without the limitations associated with chemical diffusion. Recent studies have demonstrated the feasibilty of attaching light sensitive oligonucleotides to the surfaces of DNA origami allowing the assembly and dissasembly between multiple 2-D DNA origami particles29 or the release of protein cargo tethered outside of a 3-D origami.30 The applications for dynamic DNA nanotechnology would be further expanded if light could be used to reconfigure the structures of DNA nanoparticles themselves, in addition to altering their connectivity to external factors.
Here we present a novel strategy to trigger large-scale structural reconfigurations within a DNA nanostructure via the strategic incorporation of photo-cleavable spacers as part of the structural component of a DNA origami nanoparticle. We designed an origami sphere containing photo-labile spacers within the crossovers (photo-crossovers) at desired locations without interfering with the assembly of the nanostructure. Upon light irridiation, these origami spheres were transformed to two tethered hemispheres. Because crossovers are commonly used as a structual component for connecting adjacent helices in DNA origami designs, the strategy presented here can be broadly applied to a variety of DNA nanostructures. In addition to the superb spatiotemporal precision of light irradiation, light is also capable of penetrating into environments inaccessible to chemical stimuli such as the interiors of nanoparticles. The ability to trigger large-scale structural transformation using light, in addition to chemical controls, will greatly facilitate the applicability of dynamic DNA nanotechnology in various biomedical applications.
To provide a demonstration of a light controllable structural transformation, we designed a hollow DNA origami sphere as described by Han et al10 (Fig. 1a). In this sphere, the circular, single stranded genomic DNA of the m13 bacteriophage (the scaffold) was folded into staggered rings held together by short oligonucleotide crossovers (Fig. 1a insert, red strands). We hypothesized that if photo-crossovers were positioned throughout the structure’s equator, light irradiation would sever the covalent connections between the two hemispheres leading to a large structural reconfiguration. For the photo-cleavable linkage, we used ortho-nitrobenzyl (o-NB) groups which are ideal for DNA nanotechnology because they are commercially available and can be readily incorporated at desired positions within a crossover (Fig. 1b).31 Although the size of o-NB groups in our photo-crossovers is bigger than the phosphate bonds in conventional crossovers, structural interference is expected to be minimal when they are introduced into all crossovers within one plane of the sphere.
Fig 1.

(a) Schematic diagram of the light reconfigurable DNA sphere. The cylinder represents the scaffold DNA strand, and the top and bottom hemispheres are shown as white and blue respectively. The excess scaffold is omitted. Insert: The red helices represent the crossover strands that connect the top (white) and bottom (blue) scaffold helices. (b) Depiction of the separation of adjacent helices that would result when photo-crossovers were cleaved with light.
We first investigated the structural transition occurring in the absence of any crossover that holds the two rings together at the sphere’s equator. Because our structure does not utilize the entirety of the m13 sequence, we utilized the unfolded excess scaffold DNA as a design criterion to maximize the structural reconfiguration and positioned two equal length portions of unfolded DNA between the two largest rings at the equator of the sphere (Fig. 2a–c). We created spheres with and without all equator crossovers, as illustrated in Fig. 2a. When all equator crossovers were included in the reaction mixture, uniform spheres were observed in transmission electron microscopy (TEM) images (Fig. 2d). The particle diameters and DNA density were in good agreement with the design. Next, we created structures in the absence of any equator crossovers to mimic the complete cleavage of all equator crossovers, and observed well separated hemispheres rather than closed spheres. The separated hemispheres were up to ~100 nm apart (Fig. 2e), a length consistent with the predicted 106 nm length of the 311 base pairs of DNA located between the two hemispheres.32,33 Interestingly, the DNA strands connecting the two hemispheres were clearly visible in the TEM images. Although these strands represents only a small percentage of the overall m13 DNA length (8.6%), this linearized portion can create a separation distance larger than the diameter of the hemispheres/spheres. While the unfolded excess scaffold DNA has not been broadly used as a design consideration in DNA origami (for an exception, see reference 34), our results highlight that the excess DNA can have important consequences in dynamic DNA nanotechnology.
Fig. 2.

DNA sphere reconfiguration upon modification of staple crossovers at strategic locations connecting the two equator scaffold strands. (a) Flattened, (b) unfolded, and (c) full depiction of the DNA sphere. Red double lines indicate the location of the crossovers at the hemisphere equator. Grey lines indicate the location of the unfolded scaffold DNA. TEM images of the nanostructure created in the presence (d) and absence (e) of the staple crossover pairs 1–9. Scale bars equal 50 nm. nt = nucleotides
We next minimized the number of equator crossovers required to produce fully closed spheres so that fewer o-NB groups would need to be photolyzed to initiate a structural reconfiguration upon light irradiation. We discovered that when only 3 of the 9 crossovers were used (2, 5, and 8 in Fig. 2a), closed spheres formed predominantly (Fig. S4). We then assembled light sensitized spheres containing photo-crossovers in the 3 identified locations, and found that closed spheres formed equally well with photo-crossovers compared to those formed with unmodified crossovers, suggesting that o-NB photo-spacer incorporation does not interfere with self-assembly despite the increase in distance they introduced between connected helices (Fig. S1, S5).
To test the efficiency of structural transitions, we illuminated the o-NB containing spheres with light. Upon 10 minutes of light irradiation at 302nm, most spheres underwent the predicted structural transformation from closed to open (Fig. 3a), as revealed by gel electrophoresis analysis (Fig. 3b) as well as TEM analysis (Fig. 3c, d). Upon close examination of the irradiated samples with TEM, we observed that the two hemispheres were well separated as predicted, and the excess scaffold DNA strands were clearly observable.
Figure 3.
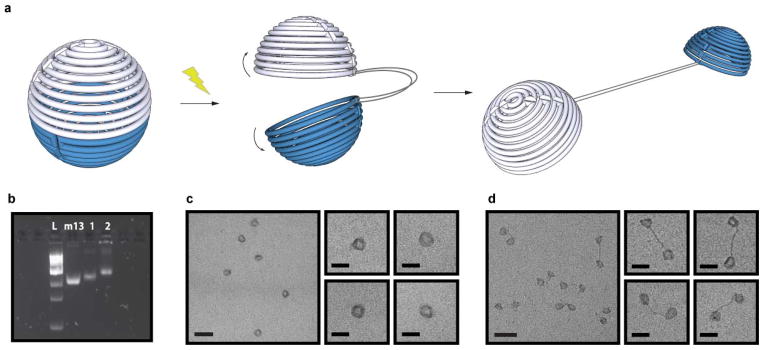
Light-triggered transformation of the DNA sphere into to two hemispheres. (a) Schematic depiction of the structural reconfiguration of the sphere upon exposure to light. (b) Fluorescent image of SYBR Safe stained 1.8% agarose gel showing successful photo-reconfiguration of the DNA sphere. L = 1kb ladder, m13 = m13 DNA scaffold, 1 = Closed sphere containing o-nb photo-crossovers, 2 = Sphere containing o-nb photo-crossovers after 10 min light irradiation. TEM images of the nanostructures before (c) and after light irradiation (d). Scale bars are 100 nm (zoom out) and 50 nm (zoom in).
To characterize the detailed kinetics of light actuated structural reconfiguration, light sensitized spheres were irradiated for varying lengths of time, and the sphere opening was quantified using TEM (Fig. 4). We identified closed, partially open, and fully open spheres in the TEM images and calculated percent opening as the number of open spheres compared to all identified nanostructures. Nanostructures were only considered open if the scaffold DNA was observed connecting the two separated hemispheres. Adjacent hemispheres without visible scaffold DNA were always classified as partially open, and therefore our calculation likely underestimated the percentage of open spheres (see SI for details). In general, we saw a consistent increase over time in the percentage of open structures, which plateaued after 12 minutes (Fig. 4, blue bars). The plateauing at about 95% most likely signified complete structural opening, as our quantification method was conservative. We then irradiated spheres that lacked photo-crossovers without o-NB groups to verify that the light induced reconfiguration was specific to photo-spacer cleavage rather than a general effect of light irradiation such as heat generation. We observed negligible opening of these spheres even after 14 minutes of constant irradiation (Fig. 4, red bars), with about 1–2% of open structures. Because the percent opening remained unchanged throughout the time course of the experiment, the small percentage of open structures observed were most likely due to incomplete structural formation rather than non-specific opening. Together these results confirmed that o-NB cleavage was responsible for the observed DNA nanostructure reconfiguration.
Fig. 4.

Kinetics of light induced structural reconfiguration. Fraction of total structures that were identified as in open configurations were quantified upon irradiation with varying period of light. Spheres containing either unmodified or photo-crossovers were irradiated with either 302 nm or 365 nm light. Only nanostructures containing photo-crossovers irradiated at 302 nm showed increased opening over time.
To further examine the wavelength specificity of the reconfiguration process, we irradiated control and sensitized spheres with 365nm light, a wavelength inefficient in cleaving our o-NB photo-spacers that are most sensitive to 300–350nm light.35 Control spheres showed no response to 365 nm light irradiation as expected (Fig. 4, red bars). However, we observed that 365nm light produced opening in approximately 9% of structures after 14 minutes of irradiation (Fig. 4 green bars). The slower kinetics observed were consistent with less efficient absorption of our o-NB groups with 365 nm light.35 Together, these results demonstrate that our photo-crossovers are more responsive to wavelengths of light near 300 nm and suggest that DNA nanostructures may be reconfigured using different wavelengths by using altered o-NB groups36 or different classes of photo-cleavable spacers.31 This would allow the use of longer wavelengths of light that are known to be less damaging to DNA or to have better tissue penetration. Indeed our structures began to show light induced damage after 10 minutes of illumination with 302 nm light, but not with 365 nm (Fig. S8).
In conclusion, we demonstrated a novel strategy using light to induce large structural reconfiguration in DNA origami nanostructures by strategically positioning photo-cleavable o-NB groups between adjacent DNA helixes. No reduction in folding efficiency was observed when these photo-cleavable spacers were used, despite the increased distance placed between the helices. Cleavage of the spacers with light irradiation led to the drastic structural transition from a sphere into two tethered hemispheres. This approach used commercially available DNA modifications and was accomplished through remote light irradiation. The use of photo-crossovers can be broadly applied to various DNA nanostructures that contain aligned crossovers between adjacent helices. It could also be applied to create light-initiated release of cargo within DNA structures with high spatial and temporal resolution. Because of the noninvasive nature of light, it is likely that this strategy can be used when structural targets are too sterically encumbered to be accessible to chemical reagents, when the addition of chemical reagents is impractical, or as an orthogonal stimulus in conjunction with chemical stimuli. Such criteria may be important in creating a new generation of dynamic DNA nanostructures that rely on moveable parts where precise control of a nanoparticle’s shape needs to be actuated in a predictable and controllable fashion.
Supplementary Material
Acknowledgments
This work was supported by the NIH Director’s new innovator award (1DP2NS082126), NINDS (1R01NS087950, 1R21NS078660, 1R01NS081716), NIMH (5R00MH085944), Pew Foundation, Alfred P. Sloan Foundation, Michael J. Fox Foundation, NARSAD, Boston University Biomedical Engineering Department, and Boston University Photonic Center. The authors would like to thank the W. M. Keck Microscope Facility at the Whitehead Institute for usage of the transmission electron microscope.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures, gel images, additional TEM images, DNA sequences, and caDNAno files & images. See DOI: 10.1039/c000000x/
Notes and references
- 1.Seeman NC. Nature. 2003;421:427–431. doi: 10.1038/nature01406. [DOI] [PubMed] [Google Scholar]
- 2.Seeman NC. Annual Review of Biochemistry. 2010;79:65–87. doi: 10.1146/annurev-biochem-060308-102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothemund PWK. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 4.Nangreave J, Han D, Liu Y, Yan H. Current Opinion in Chemical Biology. 2010;14:608–615. doi: 10.1016/j.cbpa.2010.06.182. [DOI] [PubMed] [Google Scholar]
- 5.Pinheiro AV, Han D, Shih WM, Yan H. Nat Nano. 2011;6:763–772. doi: 10.1038/nnano.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castro CE, Kilchherr F, Kim DN, Shiao EL, Wauer T, Wortmann P, Bathe M, Dietz H. Nat Meth. 2011;8:221–229. doi: 10.1038/nmeth.1570. [DOI] [PubMed] [Google Scholar]
- 7.Andersen ES, Dong M, Nielsen MM, Jahn K, Subramani R, Mamdouh W, Golas MM, Sander B, Stark H, Oliveira CLP, Pedersen JS, Birkedal V, Besenbacher F, Gothelf KV, Kjems J. Nature. 2009;459:73–76. doi: 10.1038/nature07971. [DOI] [PubMed] [Google Scholar]
- 8.Kuzuya A, Komiyama M. Chem Commun. 2009:4182–4184. doi: 10.1039/b907800b. [DOI] [PubMed] [Google Scholar]
- 9.Ke Y, Sharma J, Liu M, Jahn K, Liu Y, Yan H. Nano Lett. 2009;9:2445–2447. doi: 10.1021/nl901165f. [DOI] [PubMed] [Google Scholar]
- 10.Han D, Pal S, Nangreave J, Deng Z, Liu Y, Yan H. Science. 2011;332:342–346. doi: 10.1126/science.1202998. [DOI] [PubMed] [Google Scholar]
- 11.Douglas SM, Dietz H, Liedl T, Hogberg B, Graf F, Shih WM. Nature. 2009;459:414–418. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ke Y, Douglas SM, Liu M, Sharma J, Cheng A, Leung A, Liu Y, Shih WM, Yan H. J Am Chem Soc. 2009;131:15903–15908. doi: 10.1021/ja906381y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ke Y, Voigt NV, Gothelf KV, Shih WM. J Am Chem Soc. 2011;134:1770–1774. doi: 10.1021/ja209719k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dietz H, Douglas SM, Shih WM. Science. 2009;325:725–730. doi: 10.1126/science.1174251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iinuma R, Ke Y, Jungmann R, Schlichthaerle T, Woehrstein JB, Yin P. Science. 2014;344:65–69. doi: 10.1126/science.1250944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ke Y, Ong LL, Shih WM, Yin P. Science. 2012;338:1177–1183. doi: 10.1126/science.1227268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bath J, Turberfield AJ. Nat Nano. 2007;2:275–284. doi: 10.1038/nnano.2007.104. [DOI] [PubMed] [Google Scholar]
- 18.Liedl T, Sobey TL, Simmel FC. Nano Today. 2007;2:36–41. [Google Scholar]
- 19.Krishnan Y, Simmel FC. Angew Chem Int Ed. 2011;50:3124–3156. doi: 10.1002/anie.200907223. [DOI] [PubMed] [Google Scholar]
- 20.Yurke B, Turberfield AJ, Mills AP, Simmel FC, Neumann JL. Nature. 2000;406:605–608. doi: 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- 21.Han D, Pal S, Liu Y, Yan H. Nat Nano. 2010;5:712–717. doi: 10.1038/nnano.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lo PK, Karam P, Aldaye FA, McLaughlin CK, Hamblin GD, Cosa G, Sleiman HF. Nat Chem. 2010;2:319–328. doi: 10.1038/nchem.575. [DOI] [PubMed] [Google Scholar]
- 23.Zhang DY, Seelig G. Nat Chem. 2011;3:103–113. doi: 10.1038/nchem.957. [DOI] [PubMed] [Google Scholar]
- 24.Douglas SM, Bachelet I, Church GM. Science. 2012;335:831–834. doi: 10.1126/science.1214081. [DOI] [PubMed] [Google Scholar]
- 25.Banerjee A, Bhatia D, Saminathan A, Chakraborty S, Kar S, Krishnan Y. Angew Chem Int Ed. 2013;52:6854–6857. doi: 10.1002/anie.201302759. [DOI] [PubMed] [Google Scholar]
- 26.Modi S, Swetha MG, Goswami D, Gupta GD, Mayor S, Krishnan Y. Nat Nano. 2009;4:325–330. doi: 10.1038/nnano.2009.83. [DOI] [PubMed] [Google Scholar]
- 27.Lund K, Manzo AJ, Dabby N, Michelotti N, Johnson-Buck A, Nangreave J, Taylor S, Pei R, Stojanovic MN, Walter NG, Winfree E, Yan H. Nature. 2010;465:206–210. doi: 10.1038/nature09012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu H, Chao J, Xiao SJ, Seeman NC. Nature. 2010;465:202–205. doi: 10.1038/nature09026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Y, Endo M, Hidaka K, Sugiyama H. J Am Chem Soc. 2012;134:20645. doi: 10.1021/ja307785r. [DOI] [PubMed] [Google Scholar]
- 30.Derr ND, Goodman BS, Jungmann R, Leschziner AE, Shih WM, Reck-Peterson SL. Science. 2012;338:662–665. doi: 10.1126/science.1226734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brieke C, Rohrbach F, Gottschalk A, Mayer G, Heckel A. Angew Chem Int Ed. 2012;51:8446–8476. doi: 10.1002/anie.201202134. [DOI] [PubMed] [Google Scholar]
- 32.Berg JM, Tymoczko JL, Stryer L. Biochemistry. W. H. Freeman; Basingstoke: 2012. [Google Scholar]
- 33.To achieve maximal separation between the hemispheres using the Tethered Design, staple sequences complementary to the excess scaffold must be included.
- 34.Liedl T, Hogberg B, Tytell J, Ingber DE, Shih WM. Nat Nano. 2010;5:520–524. doi: 10.1038/nnano.2010.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.www.idtdna.com/Site/Catalog/Modifications/Product/1707
- 36.Holmes CP. J Org Chem. 1997;62:2370–2380. doi: 10.1021/jo961602x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.