

Abstract
Since the discovery of Toll, in the fruit fly Drosophila melanogaster, as the first described pattern recognition receptor (PRR) in 1996, many families of these receptors have been discovered and characterized. PRRs play critically important roles in pathogen recognition to initiate innate immune responses that ultimately link to the generation of adaptive immunity. Activation of PRRs leads to the induction of immune and inflammatory genes, including proinflammatory cytokines and chemokines. It is increasingly clear that many PRRs are linked to a range of inflammatory, infectious, immune, and chronic degenerative diseases. Several drugs to modulate PRR activity are already in clinical trials and many more are likely to appear in the near future. Here, we review the different families of mammalian PRRs, the ligands they recognize, the mechanisms of activation, their role in disease, and the potential of targeting these proteins to develop the anti-inflammatory therapeutics of the future.
I. Introduction
In 1989, Charles Janeway, in his introduction to the Cold Spring Harbor Symposium on "Immune recognition," said that "a critical issue for future study is the analysis of microbial signals that induce second signaling capacity in antigen-presenting cells, and the receptors on antigen presenting cells that detect these microbial signals.… I term these receptors pattern recognition receptors (PRRs)" (Janeway, 1989). Many families of proteins have been confirmed or identified as PRRs, because this hypothesis was postulated to include C-type lectin-like receptors (CLRs), Toll-like receptors (TLRs), nucleotide-binding domain, leucine-rich repeat-containing receptors (NLRs), RIG-I (retinoic acid inducible gene I)-like receptors (RLRs), and AIM2 (absent in melanoma 2)-like receptors (ALRs). Activation of PRRs generates inflammatory responses that are critical to control pathogens (Fig. 1). Roles for individual PRRs in pathogen recognition are well recognized, with many pathogens being detected by combinations of different PRRs. PRRs on antigen-presentation cells are also critical for helping to drive adaptive immune responses against pathogens.
Fig. 1.

Schematic summary of innate signaling pathway components and filaments. In this schematic, example signaling domains are distinguished by shape. LRR domains are stacks of ellipses; TIR domains are circles; death domains and CARD domains are rectangles; PYRIN domains are triangles. Filaments and assemblies are elongated cylinders. (A) TLR signaling via MyD88. The example shows dimerization of TLR4 (and coreceptor MD-2) by bacterial LPS. Signaling is via TIR domain interactions and the MyD88 IRAK kinase death domain assembly: the MyDDosome (blue/green). (B) TLR signaling via TRIF. TRIF is a TIR-domain containing adaptor for TLR signaling from endosomes. The TRIF N-terminal domain "TriFFosome" assembly (gray) is speculative. (C) NLR signaling via the inflammasome and ASC. The example is assembly of NLRP3 into the inflammasome together with caspase 1 and the adaptor ASC. ASC pyrin domains also self-assemble as filaments (dark red). (D) Recognition of viral RNA by RLRs. RIG-I and MDA5 are CARD-domain containing ATP-helicases that bind double-stranded RNA. RIG-I specifically recognizing triphosphate (ppp) 5′ ends. CARD domains of helicases on longer RNA self-assemble into tetramers that interact with the MAVS protein. (E) MAVS signaling. MAVS CARD domains are displayed at the outer membrane of mitochondria. The MAVS CARD associates with the RLR helicase CARDs and also produces filaments (magenta). (F) CLR signaling. Dectin-1 is an example in which dimerization activates kinases and signals via the CARD9, Bcl10, and the death-domain containing paracaspase MALT1 to mediate NFκB activation. Bcl10 also forms filamentous assemblies (gray/black).
What has become increasingly clear is that PRRs also recognize endogenous ligands from damaged tissue to drive inflammatory responses. PRRs are now thought to be linked to a range of chronic inflammatory diseases, including autoimmunity, asthma, diabetes, obesity, atherosclerosis, hypertension, and cancer, as well as playing important roles in infectious disease. Ligand-bound structures of several PRRs have been solved, allowing rational therapeutic targeting of many of these proteins. Pharmacological manipulation of PRR activity and their associated signaling pathways provides multiple therapeutic opportunities not only for adjuvants and modulating infectious disease but also for targeting many chronic inflammatory diseases that are currently managed symptomatically with drugs, such as corticosteroids, with their accompanying side effects. In this review, we will systematically describe the different families of human PRRs and discuss their links to different diseases. We finish by speculating on the prospect of future targeting of PRRs and whether this therapeutic approach might be successful in a wide range of many diseases that currently have limited treatment options available.
Throughout the review we make nomenclature recommendations for the receptor families being discussed, and as such, the abbreviations used are a list of agreed-upon abbreviations. Further guidelines and nomenclature templates can be found in the Concise Guide to Pharmacology (Alexander et al., 2013) and online at http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=302.
II. Toll-Like Receptors
A. Toll-Like Receptors and Their Nomenclature
The first PRR to be identified was Toll in Drosophila, after which TLRs were rapidly identified in mammals. There are 10 TLRs in humans and they recognize different microbial- and damage-associated ligands (O’Neill and Bowie, 2007). The human TLRs, their ligands, and the signals they activate are shown in Fig. 1.
TLR4 was the first TLR identified (Medzhitov et al., 1997) and was characterized as a PRR through the study of the lipopolysaccharide (LPS)-resistant C3H/HeJ and C57BL/10ScCr mice strains. In C3H/HeJ mice the Tlr4 gene has a single adenine to cytosine point mutation, resulting in a P712H substitution in the TIR (Toll-IL1 receptor) domain of TLR4 (Poltorak et al., 1998; Qureshi et al., 1999), conferring dominant-negative activity on TLR4 in these mice (Vogel et al., 1999). The C57BL/10ScCr strain is homozygous for a null mutation of Tlr4 (Poltorak et al., 1998). The role of TLR4 in LPS signaling was confirmed in TLR4−/− mice, which were hyporesponsive to LPS (Hoshino et al., 1999). Mutations in the human Tlr4 gene, corresponding to D299G and T399I, were shown to associate with hyporesponsiveness to inhaled LPS (Arbour et al., 2000), and expression of these mutants in vitro show reduced activation in response to LPS (Rallabhandi et al., 2008).
To recognize LPS, TLR4 was found to require an additional protein, myeloid differentiation protein 2 (MD-2), with which it has to be associated (Shimazu et al., 1999) (Fig. 2A). Mice lacking MD-2 do not respond to LPS (Nagai et al., 2002). A number of MD-2 polymorphisms have been identified that alter LPS binding and/or activation (Hamann et al., 2004; Gu et al., 2007; Vasl et al., 2008). LPS interaction with MD-2/TLR4 involves at least two other proteins. LPS binds first to lipopolysaccharide binding protein in serum (Schumann et al., 1990) and is then transferred to CD14 (Wright et al., 1990). The major role for CD14 is to enhance the sensitivity of the MD-2/TLR4 signaling complex, dropping the binding affinity for LPS to picomolar concentrations (Gioannini et al., 2004). Mice without CD14 are resistant to endotoxic shock (Haziot et al., 1996). TLR4 can also signal in a heterodimer with TLR6 in response to oxidized low density lipoprotein and amyloid-β through a process regulated by CD36 (Stewart et al., 2010).
Fig. 2.
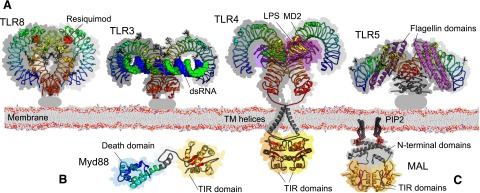
TLR and TIR-containing adaptor structures. Selected examples of dimeric structures in recognition and signaling by TLRs are shown as ribbon diagrams with space-filling shadows. (A) TLR extracellular LRR domains in rainbow scheme from blue (N terminus) to red (C terminus): TLR8 with the small molecule ligand Resiquimod (red atoms) is from PDB ID 3W3L; TLR3 with bound RNA is from PDB ID 2CIY; TLR4 with MD2 and LPS (magenta/violet ribbons; yellow/red atoms, respectively) from PDB ID 3VQ2 extended with transmembrane domains modeled from EGFR. TLR5 with Flagellin domains is from PDB ID 3V47. (B) The adaptor MyD88 is modeled from individual domains [PDB IDs 3MOP and 4DOM for its death (blue) and TIR (orange) domain, respectively], with the TIR domain directed toward the TLR TIR dimer (yellow/orange, based on the TLR10 example PDB ID 2J67). (C) The TIR domain containing adaptor MAL/TIRAP is shown as a putative TIR dimer (orange) anchored via modeled PIP2-binding motifs (gray).
TLR2 was originally identified as the LPS receptor (Kirschning et al., 1998; Yang et al., 1998), but the TLR2 activation by LPS was subsequently attributed to bacterial lipoprotein contamination of the LPS preparations (Lee et al., 2002; Hellman et al., 2003). TLR2 recognizes a wide range of ligands, many of which are from Gram-positive bacteria (Takeuchi et al., 1999), and it signals not as a homodimer but as a heterodimer with either TLR1, TLR6, or TLR10 (Ozinsky et al., 2000; Takeuchi et al., 2001; Hasan et al., 2005). Mice without TLR2 are hyporesponsive to Gram-positive bacterial cell wall components (Takeuchi et al., 1999). A number of single nucleotide polymorphisms (SNPs) in TLR2 have been reported in the extracellular domain (R753Q, Y715K, and Y715X) and the cytoplasmic domain (P631H) (Lorenz et al., 2000; Smirnova et al., 2003; Merx et al., 2007). Although the extracellular domain mutations result in decreased activity of TLR2 to Gram-positive bacterial ligands (Lorenz et al., 2000; Schroder et al., 2003), the genetic evidence linking these SNPs with a susceptibility to Gram-positive infections is unclear (Schroder and Schumann, 2005). Twelve SNPs in TLR1 (of these S248N, H305L, P315L, and I602S have defective signaling) and fourteen SNPs in TLR6 have also been identified (Johnson et al., 2007). The interaction between TLR10 and TLR2 is less well studied because tlr10 is expressed in humans but not mice (Roach et al., 2005). TLR10 heterodimers recognize Gram-positive bacterial ligands and some fungal products (Guan et al., 2010). TLR2 heterodimerization probably leads to the activation of differential signaling; in particular, this seems likely for TLR2/TLR10 heterodimers (Farhat et al., 2008; Guan et al., 2010). Genetic evidence links TLR10 to infectious and immune-related diseases (Kormann et al., 2008; Mailaparambil et al., 2008; Park et al., 2011). TLR2 also cooperates with other receptors for pathogen recognition, for example cooperation between TLR2 and Dectin-1 (dendritic cell–associated C-type lectin) allows recognition of the yeast particles, such as zymosan (Brown et al., 2003; Gantner et al., 2003). In a manner analogous to TLR4, other proteins have been implicated as coreceptors for TLR2, such as CD14 and CD36 (Hoebe et al., 2005; Jiang et al., 2005).
TLR5 recognizes monomeric flagellin, which is a constituent of the flagella filament from β- and γ-proteobacteria (Hayashi et al., 2001) (Fig. 2A). TLR11, a receptor most closely related to TLR5, is expressed in mice but not humans and has been suggested to detect bacterial and, along with another mouse TLR, TLR12, protozoal ligands (Andersen-Nissen et al., 2007a; Mathur et al., 2012; Gazzinelli et al., 2014). Genetic defects in TLR5 are increasingly linked to a number of diseases, and the lack of TLR11 in people is a possible explanation for the human susceptibility to urinary tract infections, toxoplasmosis and Salmonella typhi infections (Mathur et al., 2012; Gazzinelli et al., 2014). However, so little is currently known regarding TLR11 that these early observations should be treated with caution.
TLRs 1, 2, 4, 5, 10, 11, and 12 are all expressed in the plasma membrane, whereas TLRs 3, 7, 8, and 9 are expressed in the endosome. TLRs 3, 7, 8, and 9 are primarily viral sensors that sense nucleic acids (Blasius and Beutler, 2010). TLRs 4, 11, and 13 can translocate to the phagosomal-endosomal compartment (Blasius and Beutler, 2010; Kawai and Akira, 2011). Endosomal TLRs are trafficked to their intracellular location using different accessory proteins including Unc-93 homology B1, glycoprotein 96, and a protein associated with TLR4 A (Lee et al., 2012a).
B. Structural Biology of the Toll-Like Receptors
The molecular basis for agonist and antagonist binding at TLRs is increasingly well understood with the solving of a number of unbound and ligand-bound crystal structures of the extracellular leucine-rich repeat (LRRs) domains (Fig. 2A). The overall structures of LRR proteins resemble a horseshoe. The amino acid sequences of LRR family proteins have multiple copies of repeating LRR modules, with each module being 20- to 30-amino-acids long. The LRR module has an LxxLxLxxN sequence motif, with the central LxL part forming the core of a β-strand with the two leucines pointing toward the interior of the protein to make a hydrophobic core. The variable x residues are exposed to solvent, and some are involved in ligand interactions. The β-strands assemble into a large β-sheet to form the entire concave surface of the horseshoe. Most inactive or antagonist-bound TLRs usually exist in a monomeric state, taking up the characteristic horseshoe-like structure of the LRR (Song and Lee, 2012). The ligand recognition mechanisms of the TLRs vary, but agonist-bound TLRs exist as dimers with the two C termini of the extracellular domains converging in the center and the N termini facing outward to form an “m” shape. This is thought to bring the intracellular TIR signaling domains into close association to facilitate signaling (Song and Lee, 2012) (Fig. 2A).
There is wide diversity, however, in how agonists bind to TLRs. Triacylated lipoproteins, such as Pam3CSK4, bind the TLR1/TLR2 heterodimer (Jin et al., 2007). The lipid chains of Pam3CSK4 bridge the two TLRs, therefore playing a crucial role in the formation of the heterodimer. Two of the three lipid chains of Pam3CSK4 interact with a hydrophobic pocket in TLR2, and the amide-bound lipid chain lies in a hydrophobic channel within TLR1. The ligand-bound complex of TLR1 and TLR2 is stabilized by protein-protein interactions at the interface near the ligand binding pocket (Jin et al., 2007). TLR2/TLR6 binds to diacylated lipoproteins, such as Pam2CSK4, with the two ester-bound lipid chains inserting into the same TLR2 pocket as seen in the TLR2-TLR1 triacyl lipopeptide complexes. In the TLR2/6 heterodimer the hydrophilic glycerols and peptide backbones of the lipoproteins form hydrogen bonds with amino acid residues of both TLR2 and TLR6 to stabilize the complex. The TLR2-TLR6 complex cannot bind to triacylated lipoproteins because the hydrophobic channel responsible for interaction with the amide-bound lipid chain in TLR1 is blocked by two bulky phenylalanines in TLR6 (Song and Lee, 2012). In crystallographic studies, a weak agonist form of lipoteichoic acid from Streptococcus pneumoniae binds to TLR2 but cannot crosslink to either TLR1 or TLR6 to induce heterodimerization (Kang et al., 2009).
Crystallization of LPS bound to MD-2/TLR4 showed that 5 of the LPS acyl chains are fully accommodated in MD-2 and only the sixth acyl chain is exposed to interact with TLR4. The partial exposure of the sixth acyl chain is critical for forming the dimerization interface. The LPS phosphate groups interact with the positively charged residues in TLR4, and the main dimerization face of TLR4 is between LRRs 15-17 (Park et al., 2009). A zebrafish fragment of TLR5 (up to LRR14) shows that the flagellin D1 domain drives ligand binding and dimerization of this receptor (Yoon et al., 2012). One face of D1 interacts with a large surface area of the LRRs (from LRRNT to LRR9) and, at the same time, it forms a bond with the second TLR5 molecule in the dimer bridging the two TLRs. The use of a short fragment of TLR5 in this structure means it is unclear whether the C-terminal part of the receptor contributes to dimerization or to ligand interaction, although mutagenesis experiments suggest it probably plays only a minor role (Yoon et al., 2012).
Of the endosomal TLRs, agonist-bound structures are currently available for TLR3 and TLR8 (Fig. 2A), although ligand bound TLR9 structures are present in the unreleased section of the Protein Data Bank. TLR3 exists as a monomer that, upon binding to dsRNA, forms a dimer with two RNA binding sites located in the N-terminal and C-terminal regions. The distance between the two binding sites is about two helical turns of RNA, with the backbone phosphates and sugars of the ligand playing an important role in TLR3 binding at both sites. The extracellular domains of TLR7, TLR8, and TLR9 are much larger than the other TLRs consisting of 26 LRRs and they contain an insertion loop (Z-loop) of approximately 30–40 amino acids between LRR14 and LRR15. When the full-length ectodomain of human TLR8 was expressed in Drosophila melanogaster Schneider 2 cells, it was found to be cleaved at the Z-loop by an unidentified protease, but the resulting N-terminal (residues 27–455) and C-terminal (residues 456–827) fragments remained associated during purification, suggesting a strong interaction between them (Ohto et al., 2014). The extracellular domain structure of TLR8 forms more of a ring-shaped structure than the other TLRs, in which each half of the ring is produced by the N- and C-terminal fragments. This is caused by a combination of factors including several hydrogen bonds formed between the N-terminal and C-terminal domains of the protein, the latter half of the Z-loop (residues 458–481) forming an ordered structure in the concave surface of the N-terminal fragment through extensive hydrophobic interactions from LRR3 to LRR18 and the formation of a continuous β-sheet in the concave face of the ring-like structure from LRR14 in the N-terminal fragment to LRR15 in the C-terminal fragment. When synthetic TLR8 agonists, such as imidazoquinoline or thiazoquinoline, bind the receptor they do so between the two TLR8 protomers and interact with the N-terminal LRR11–14 of one protomer and the C-terminal LRR16–18 of the other (Fig. 2A). One of the most interesting features of the TLR8 structure is that this receptor exists as a dimer in the presence or absence of a ligand. When the TLR8 dimer binds an agonist it undergoes structural rearrangement such that the ligand penetrates the dimerization interface of the unliganded dimer, which causes disruption of protomer-protomer interactions leading to complete reorganization of the dimer to bring the C termini into close proximity (Ohto et al., 2014). There is evidence to suggest that TLR9 also exists as preformed dimers (Latz et al., 2007), and it is likely, therefore, that both TLR7 and TLR9 will behave similarly to TLR8.
The ligand specificity of TLR7/8/9 raises some interesting questions about the molecular and pharmacological activation of the receptors. TLR7 and TLR8 are both stimulated by the small molecule imidazoquinolines. However, Imiquimod (3M Pharmaceuticals, St. Paul, MN) only activates TLR7, whereas the derivatives resiquimod and CL097 stimulate both TLR7 and TLR8 and the thiazoloquinoline derivative CL075 only turns on TLR8 (Ohto et al., 2014). On the basis of amino acid conservation, it is believed that TLR7 and TLR8 are likely to use the same ligand binding pocket. So how does specificity happen? Structural studies of TLR8 reveal that the entrance to the pocket contains an acidic aspartic acid residue that forms stacking interactions with the aromatic ring of the ligands (Tanji et al., 2013). In TLR7, the equivalent residue is replaced by a leucine, a hydrophobic amino acid, that may well alter either, or both, the accessibility to the binding site or the stability and affinity of the receptor–ligand interaction (Ohto et al., 2014). Whether the nucleic acid ligands for TLR8 or TLR9, which differ extensively in their physical and chemical properties, use the same binding surface remains to be determined.
Ligand binding and activation of TLR3, 7, 8, and 9 are dependent on an acidic pH. TLR3 dimerization requires an acidic environment presumably because several histidines make critical bonds with the phosphate backbones of the RNA so the stability of this interaction depends on protonation of these amino acids (Liu et al., 2008). The endosomal pH is also critical for TLR 7, 8, and 9 ligand binding and receptor activity (Gibbard et al., 2006) possibly due to ssRNA and activating CpG-containing oligonucleotides forming secondary short G–C duplex structures. These regions could change from a Watson–Crick base pairing to a Hoogsteen conformation at pH 5, allowing the protonated cytosine N3 group to make a strong electrostatic bond with the critical Asp543 in TLR8 (which is conserved as Asp534 in TLR9) (Gay et al., 2014). Several studies have suggested that cleavage of the Z-loop in TLR 7–9, through proteases including asparagine endopeptidase and cathepsin, is required for nucleic acid sensing and the proper functioning of these proteins. Some reports suggest only the C terminus of the TLRs is required for full activity, whereas others suggest that both C and N termini are required (reviewed in Ohto et al., 2014).
Currently it is unclear how coreceptors such as CD14 or CD36 fit in with the structural data. CD36 contributes to diacylglyceride recognition only at TLR2/6 (Hoebe et al., 2005). CD14 has long been known to be important for TLR4 signaling, yet how it structurally interacts with the MD-2/TLR4 complex is unclear. It may be some time before it is clear how coreceptors interact with TLR signaling complexes.
C. Activation and Signal Transduction at Toll-Like Receptors
1. Ligand Recognition at Toll-Like Receptors.
The best characterized ligand for the MD-2/TLR4 complex is lipid A (the biologically active component of LPS). The lipid A domain of LPS consists of a disaccharide to which various substituents, including acyl chains of variable length and number, are attached (Raetz and Whitfield, 2002). Escherichia coli lipid A is usually hexa-acylated, whereas a tetra-acylated lipid A, lipid IVa, is also produced by E. coli as an intermediate in the lipid A biosynthetic pathway (Raetz and Whitfield, 2002). Different lipid A structures may be agonists or antagonists at the MD-2/TLR4 (Walsh et al., 2008). Subtle alterations in Lipid A structure profoundly alter its biologic activity such that a synthetic compound CRX-527 [(2S)-2-[[(3R)-3-decanoyloxytetradecanoyl]amino]-3-[(2R,3R,4R,5S,6R)-3-[[(3R)-3-decanoyloxytetradecanoyl]amino]-4-[(3R)-3-decanoyloxytetradecanoyl]oxy-6-(hydroxymethyl)-5-phosphonooxyoxan-2-yl]oxypropanoic acid] is an agonist, but decreasing the secondary acyl chain length below 6 or increasing it above 14 results in a loss of agonist activity (Stover et al., 2004).
A number of other putative TLR4 ligands have been identified. Examples include endogenous ligands (such as high mobility group box 1 protein, heat shock protein (HSP) 60, HSP70, type III repeat extra domain of fibronectin, hyaluronic acid oligosaccharides, heparin sulfate polysaccharide fragments, fibrinogen), other pathogen-derived ligands [such as S. pneumoniae pneumolysin, Chlamydia pneumoniae HSP60, mouse mammary tumor virus envelope proteins, respiratory syncytial virus (RSV) fusion protein], allergen proteins, and plant ligands such as taxol (Gay and Gangloff, 2007). The molecular nature of how these ligands interact with TLR4 and whether MD-2 is required is not well understood. The type III repeat extra domain of fibronectin (Okamura et al., 2001), C. pneumoniae HSP60, and RSV fusion protein (Rallabhandi et al., 2012) do appear to require MD-2 for activation of TLR4, although the molecular basis for this is unclear. Until crystallographic or biophysical evidence shows that protein ligands bind to the receptor to induce a conformational change and activate signaling it remains controversial as to whether these proteins are true ligands for TLR4.
A wide range of structurally diverse ligands are recognized by TLR2 and its associated proteins (Zahringer et al., 2008). Many of these ligands are glycolipids, lipopeptides, or glycosylphosphatidylinositol-anchored structures, for example lipoteichoic acid from Gram-positive bacteria, lipoarabinomannan from mycobacteria, and glycosylphosphatidylinositol anchored lipids from Trypanosoma cruzi, which, like LPS, contain a significant hydrophobic component (Zahringer et al., 2008). Well characterized ligands of TLR2 include Mycoplasma fermentan 2-kDa macrophage-activating lipopeptide, which interacts with TLR2/TLR6 (Takeuchi et al., 2001), triacylated lipopeptides (such as Pam3CSK4 at TLR2/TLR1), and diacylated lipids/lipopeptides (lipoteichoic acid, Pam2CSK4) (Ozinsky et al., 2000; Takeuchi et al., 2001). Like the recognition of LPS by TLR4, the number of acyl chains linked to a lipoprotein plays an important role in ligand recognition by TLR2. Other properties of lipoproteins such as the ester bonds present in the acyl chains and the nature of the amino acids are also discriminated by TLR2 heterodimers (Buwitt-Beckmann et al., 2005a,b), and some lipopeptides may be recognized by TLR2 independently of TLR1 or TLR6 (Buwitt-Beckmann et al., 2006). Lipoteichoic acid, like LPS, is a glycolipid with repeating carbohydrate units from Gram-positive bacteria, and it is also recognized by TLR2 (Morath et al., 2002).
Ligands other than lipoproteins are also recognized by TLR2. TLR2 is one of several receptors believed to be important for peptidoglycan recognition, but the structural motif in peptidoglycan recognized by TLR2 is unknown. It is possible that the peptidoglycan recognition attributed to TLR2 may be due to the peptidoglycan preparation being contaminated with lipoproteins (Zahringer et al., 2008). A range of LPS structures has also been suggested to be TLR2 ligands such as lipid A from Porphormonas gingivalis (Hirschfeld et al., 2001), Leptospira interrogans (Werts et al., 2001), and Legionella pneumophila (Girard et al., 2003). This remains a controversial topic. Contaminating lipoproteins in the LPS from P. gingivalis has been suggested to be the reason why the LPS from this bacterium can activate TLR2, but the literature on this subject remains confusing (Hashimoto et al., 2004). The molecular mechanisms underlying how ligands such as whole bacteria [for example, Francicella tularensis (Cole et al., 2007)] and fungal ligands [such as zymosan (Gantner et al., 2003)] are recognized by TLR2 or TLR2/Dectin 1, respectively, are unclear.
TLR5 is the only TLR that exclusively binds protein (flagellin) and is conserved from fish to mammals (Hayashi et al., 2001; Iqbal et al., 2005; Stockhammer et al., 2009). TLR5 has been shown to be important in recognizing bacteria such as Salmonella enterica serovar Typhimurium in the gut epithelial cells and E. coli in the urinary tract (Uematsu et al., 2006; Andersen-Nissen et al., 2007a,b). TLR11 detects uropathogenic E. coli, enteric salmonellae (both serovars Typhimurium and Typhi), and the parasite Toxoplasma gondii (Mathur et al., 2012; Gazzinelli et al., 2014).
TLR9 detects microbial DNA (Hemmi et al., 2000) that contains 2′-deoxyribose phosphate backbone, the efficiency of which can be enhanced by phosphorothioate linkages or specific sequences (e.g., CpG motifs, which are four times less abundant in mammalian DNA) (Haas et al., 2008). TLR9 recognizes dsDNA viruses, such as mouse cytomegalovirus and herpes simplex viruses (HSV) 1 and 2 (Blasius and Beutler, 2010). TLR7 recognizes guanosine- and uridine-rich ssRNA (Hemmi et al., 2002), synthetic polyuridines, imidazoquinoline derivatives, such as resiquimod (R848) and Imiquimod, and guanine analogs, such as loxoribine (Blasius and Beutler, 2010). A number of pathogens are recognized by TLR7, including ssRNA viruses [human immunodeficiency virus (HIV), influenza, and vesicular stomatitis virus], Borrelia burgdorferi, some small interfering RNAs (siRNAs), and RNA released into endolysosomes by phagosomal bacteria (Blasius and Beutler, 2010). TLR8 in humans, but not mice, recognizes a similar range of ligands to TLR7 (Jurk et al., 2002). TLR3 detects dsRNA (Alexopoulou et al., 2001) and the synthetic analog polyinosinic-polycytidylic acid (poly I:C) but recognizes a wide range of viral pathogens including dsRNA viruses (reovirus), some ssRNA viruses, including West Nile virus, RSV, and encephalomyocarditis virus and some dsDNA viruses such as HSV and mouse cytomegalovirus. This is probably due to viral replication during an infection cycle resulting in the production of dsRNA intermediates that can be sensed by TLR3, suggesting a level of redundancy in viral sensing by endosomal TLRs (Blasius and Beutler, 2010).
2. Signal Propagation at Toll-Like Receptors.
TLR dimerization brings the juxtamembrane sequences at the C terminus of the two ectodomains and the TIR signaling domains into close proximity. The association of the receptor TIR domains provides a new scaffold that allows the recruitment of specific adapter proteins to form a postreceptor signaling complex. Inflammatory signaling through TLRs uses five different adapter proteins recruited in different combinations to different TLRs, allowing for tailored responses to each stimulus (O’Neill and Bowie, 2007). There are several protein kinases downstream of these adapters, notably the interleukin (IL)-1 receptor-associated kinase (IRAK) family and TBK1 (TANK-binding kinase 1). These activate pathways leading to the activation of the respective transcription factors nuclear factor κB (NFκB) and interferon response factor (IRF)3, which, in turn, induce various immune and inflammatory genes.
Activated TLR4 recruits two distinct adapter protein pairs, Mal/MyD88 (myeloid differentiation primary response gene 88) and TRAM (TRIF-related adapter molecule)/TRIF (TIR-domain protein inducing interferon) (Figs. 1, A and B, and 2, B and C). Mal (also known as TIRAP) and TRAM (also known as TICAM2) are thought to engage directly with the receptor and to act as "bridging adapters" for the recruitment of MyD88 and TRIF, respectively (Gay et al., 2014). Mal was thought to be required only for signaling through TLR2 and TLR4, but recent evidence suggests that it is required for endosomal as well as cell membrane TLR signaling (Bonham et al., 2014). TLR5 appears not to have a requirement for Mal and signals directly through MyD88. Endosomal TLRs 7–9 signal through MyD88, whereas TLR3 signals through TRIF (Gay et al., 2014). Signaling through MyD88 is required for rapid activation of the NFκB transcription factor and also to IRF5 and IRF7 from the endosome, resulting in the production of proinflammatory cytokines such as tumor necrosis factor (TNF)α. TRIF stimulates sustained NFκB activation and activation of IRF3 (Gay et al., 2014). IRF3 induces expression of a distinct set of genes to NFκB, such as interferon (IFN)β and chemokine (C-C motif) ligand 5 (O’Neill and Bowie, 2007). The molecular interactions between the TLR TIR complex and the TIRs of the adaptor proteins appear to be weak, and the precise details about how these molecular interactions occur are currently unknown (Gay et al., 2014) (Figs. 2 and 3).
Fig. 3.

Structures and interactions of death, pyrin, and CARD domains. Death domains and related domains share a common arrangement of six alpha helices with conserved types of packing interaction. (A) Comparison of death domain, PYD, and CARD structures colored from blue (N terminus) to red (C terminus) presented in equivalent orientations. Also shown is the TRIF N-terminal domain that is modified and has additional helices (gray). (B) Types of domain interaction. Contacts between domains in assemblies are classified type I (blue), II (orange), and III (green). Helical lattices have repeats in three directions shown schematically by the colored contacts between the gray hexagons (central subunit a, type I contact to b, type II to c, type III to d). The larger schematic shows details from death domains in the MyDDosome (PDB ID 3MOP). Helices contacting subunit a are colored [scheme as in (A)] or gray otherwise. Examples of helical assemblies: full death domain assembly of MyDDosome (C); ASC pyrin domain filament (D); MAVS CARD domain filament (PDB IDs 3MOP, 3J63, 3J6J, respectively) (E). MyDDosome layers are MyD88, IRAK4, and IRAK2.
The signaling complexes formed by MyD88 and TRIF are, however, better understood. Biophysical and crystallographic work have revealed that 6–8 MyD88 molecules form an oligomeric macromolecular platform with 4 IRAK4 and 4 IRAK 2 molecules through death domain (DD) interactions to form a MyDDosome complex (Lin et al., 2010; Gay et al., 2014) (Fig. 3). This process of MyDDosome assembly is thought to be highly allosteric. The engagement of the MyD88 DD by IRAK-4 and IRAK-1 permits their association with each other, an interaction thought to result in IRAK-4 mediated phosphorylation of IRAK-1. This, in turn, stimulates a cascade of IRAK-1 autophosphorylation (Jain et al., 2014). The importance of phosphorylation in the assembly of the signaling complex raises interesting possibilities for the pharmacological manipulation of MyDDosome assembly. This may also be affected by the presence of tissue-specific splice variants of IRAK-1 and IRAK-4, which could also influence TLR signaling. IRAK-1 is ultimately released from the active receptor complex and, in conjunction with TRAF-6, binds to TAK (transforming growth factor-β–activated kinase)-1 binding protein 1 followed by binding of TAK-1 itself and then TAK-1 binding protein 2. This leads to the ubiquitination and degradation of IRAK-1 and ultimately activation of NFκB-mediated proinflammatory genes.
Less information is available about the TRIF signaling platform, but it is likely a TRIFFosome forms involving an oligomeric complex of TRIF with some or all of its downstream signaling partner proteins, although the stoichiometry that these complexes may form is unknown (Lin et al., 2010; Gay et al., 2014) (Fig. 1B). Pharmacologically the formation of oligomeric signaling platforms raises some interesting questions about how partial agonism may be achieved. For example does the stoichiometry of an oligomeric signaling complex alter or do the number of signaling complexes per cell change? Of course it remains possible that partial agonism observed in cell populations reflects binary "on/off" responses at the level of the individual cells. Improvements in single-cell analysis techniques will allow this to be resolved.
D. Toll-Like Receptor Mutations and Disease
Research on TLR-deficient mice implicated them in multiple pathologies that have subsequently been confirmed by many studies of human diseases (Table 1) (O’Neill et al., 2009; Lin et al., 2012; Netea et al., 2012). Primary immunodeficiencies are documented in patients with complete deficiencies in TLR signaling. Patients lacking MyD88-IRAK4 are very susceptible to pyogenic bacteria, whereas those with TLR3, Unc-93 homology B1, or TRIF deficiencies are hypersusceptible to herpes viruses (Sancho-Shimizu et al., 2011; Netea et al., 2012).
TABLE 1.
TLR targets in different diseases
| TLR | Tissue | Therapeutic Potential/Use | Action |
|---|---|---|---|
| TLR2 | Heart /vasculature | Atherosclerosis | Antagonistic |
| Lung | Asthma | Antagonistic | |
| Kidney | Ischemia/reperfusion | Antagonistic | |
| Pancreas | Diabetes | Antagonistic | |
| TLR3 | Systemic | Vaccine adjuvant | Agonistic |
| Breast | Anticancer | Agonistic | |
| Renal system | Anticancer | Agonistic | |
| Eye | CNV inhibition in AMD | Agonistic | |
| Brain | Antiviral (WNV) | Antagonistic | |
| TLR4 | Heart /vasculature | Atherosclerosis | Antagonistic |
| Lung | Asthma | Antagonistic | |
| Kidney | Ischemia/reperfusion | Antagonistic | |
| Pancreas | Diabetes | Antagonistic | |
| Joints | Rheumatoid arthritis | Antagonistic | |
| Brain | Alzhiemer’s disease, Parkinson's disease | Antagonistic | |
| Systemic | Sepsis | Antagonistic | |
| Vaccine adjuvant | Agonistic | ||
| TLR5 | Hematopoietic | Radioprotective | Agonistic |
| system and GI-tract | Vaccine adjuvant (influenza virus) | Agonistic | |
| Systemic | Treatment of Crohn’s Disease | Antagonistic | |
| GI-tract | |||
| TLR7/8 | Skin | Treatment of viral induced lesions | Agonistic |
| Blood | (caused by papilloma virus and herpes simplex virus) | Agonistic | |
| Systemic | Primary tumors | Agonistic | |
| Cutaneous metastases | Agonistic | ||
| Antiviral (HCV) | Agonistic | ||
| Chronic lymphocyte leukemia | Antagonistic | ||
| SLE | |||
| TLR9 | Prostate | Anticancer | Agonistic |
| Systemic | Vaccine adjuvant | Agonistic | |
| Systemic | Tumor vaccine adjuvant | Agonistic | |
| Lung/systemic | Asthma/allergy | Agonistic |
More subtle phenotypes are seen in patients with mutations that lead to alterations in TLR functionality. TLR4 is linked to a range of diseases, including infectious disease, atherosclerosis, asthma, allergic disease, cardiac disease, liver disease, renal disease, cancer, inflammatory bowel disease, sepsis, obesity, diabetes (type I and type II), rheumatoid arthritis, Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, ischemia/reperfusion injury, and many diseases of inflammatory origin (O’Neill et al., 2009; Lin et al., 2012; Netea et al., 2012). The important roles of TLR4, and increasingly TLR2, in recognizing endogenous damage-associated ligands explain why these receptors are so important in noninfectious chronic inflammatory diseases, such cardiac dysfunction, ischemia/reperfusion injury, obesity, and central nervous system degenerative disorders. Polymorphisms in TLR1 have been linked to leprosy and candidaemia susceptibility, whereas TLR2 mutations are associated with leprosy, tuberculosis, Borerelia, and Lyme disease susceptibility. TLR3 mutations may be linked to cardiac disease, colorectal cancer, macular degeneration, HIV-1 infection, and HSV encephalitis. TLR5 polymorphisms have been linked to susceptibility to L. pneumophila infection, systemic lupus erythematosis (SLE), and Crohn’s disease. Polymorphisms in TLR6 may be linked to responsiveness to bacillus Calmette-Guérin vaccination, asthma, aspergillosis, and cardiac disease. TLR7 mutations are linked to hepatitis C virus (HCV), HIV, and SLE. Polymorphisms in TLR8 are linked with Crimean-Congo hemorrhagic fever and tuberculosis, whereas TLR9 is linked to many inflammatory disorders including non-Hodgkin’s lymphoma, cervical cancer, lupus nephritis, and cerebral malaria. TLR10 polymorphisms may be associated with asthma and Crohn’s disease (O’Neill et al., 2009; Lin et al., 2012; Netea et al., 2012).
Interpretation of the genetic data is complicated, however. Selection pressure exerted by infectious agents should result in removal of mutations that suppress the immune response against a pathogen unless another protective advantage is conferred upon that individual (Netea et al., 2012). For example two receptor polymorphisms were originally identified in TLR4 (D299G and T399I) as decreasing responsiveness to inhaled LPS (Arbour et al., 2000). The D299G phenotype is associated with protection against mortality due to cerebral malaria, but an increased susceptibility to Gram-negative infection such that while the prevalence of this allele is high in Africa it has nearly been lost in Europe (Netea et al., 2012). Interpretation of the genetic data on TLRs is also complicated by the accuracy of the patient phenotype reported in the studies. Much of the early data on the two TLR4 polymorphisms generated a number of studies with conflicting data (Schroder and Schumann, 2005; Ferwerda et al., 2008) because most of the studies considered either the D299G or the T399I polymorphism, but neglected the fact that these polymorphisms also exist in a cosegregated (D299G/T399I) way which leads to 4 potential haplotypes, namely wild type/wild type, D299G/wild type, T399I/wild type, and D299G/T399I (Ferwerda et al., 2008). Large sample sizes are required when considering these types of issues. None the less TLRs are clearly associated with many diseases so therapeutic targeting of either the TLRs themselves or the signals they generate is likely to be very useful.
III. The Nucleotide-Binding Domain and Leucine-Rich Repeat-Containing Receptors
A. Nucleotide-Binding Domain and Leucine-Rich Repeat-Containing Receptors and Their Nomenclature
The NLR family is a diverse set of cytoplasmic receptors. There are 22 human and 34 murine NLRs. The receptors possess a conserved domain architecture consisting of an N-terminal effector domain, a central nucleotide-binding and oligomerization domain, and a variable number of C-terminal LRRs. A standardized nomenclature for the NLR family has recently been adopted (Ting et al., 2008) in which the nature of the effector domain results in the classification of each receptor into one of five subfamilies (Table 2). For historical reasons and in recognition of the scope of the literature prior to 2008, four of the NLRs—NOD1 (nucleotide oligomerization domain) (NLRC1), NOD2 (NLRC2), class II, major histocompatibility complex, transactivator (NLRA), and NAIP (neuronal apoptosis inhibitory protein) (NLRB)—are still generally and acceptably known by their common names. As a result of this accepted nomenclature, we refer throughout to NLRP and not NALP. Even within subfamilies the functionality of the NLRs is diverse, covering biologic processes as varied as pathogen recognition, homeostatic regulation, transcriptional control, inflammatory suppression, microbiota regulation, and embryonic development. In relation to proinflammatory signaling the NLRs can be broadly split between those (NOD1 and NOD2) that activate NFκB after engagement of receptor interacting protein 2 (RIP2) (Philpott et al., 2014) and those (NLRP1, NLRP3, NLRC4, NAIP) that form an inflammasome to activate caspase-1 and process the cytokine precursors pro–IL-1β and pro–IL-18 (Davis et al., 2011) (Fig. 1C).
TABLE 2.
Gene composition of NLR subfamilies
Murine genes are shown in parentheses.
| NLR Subfamily | Type of Effector Domain | Family Gene Members |
|---|---|---|
| NLRA | Acidic activation domain | CIITA; (CIIta) |
| NLRB | Baculovirus inhibitor of apoptosis repeats | NAIP; (Naip1-7) |
| NLRC | Caspase activation and recruitment domain | NOD1, NOD2, NLRC3, NLRC4, NLRC5;(Nod1, Nod2, Nlrc3, 4, 5) |
| NLRP | Pyrin domain | NLRP1-14; (Nlrp1a-c, 2, 3, 4a-4g, 5, 6, 9a-c, 10, 12, 14 |
| NLRX | Unknown domain | NLRX1; (Nlrx1) |
CIITA, class II, major histocompatibility complex, transactivator.
B. Structural Biology of the Nucleotide-Binding Domain and Leucine-Rich Repeat-Containing Receptor
Structural studies of the NLR proteins are notoriously difficult because of their tendency for protein aggregation and precipitation when overexpressed or purified as either full-length receptors or isolated domains. This has hindered attempts to solve receptor structures and although much progress has been made recently, particularly with the effector domains, the levels of full-length receptor that can be purified still make structural determination at the atomic level exceptionally difficult. Consequently, there is limited structural information available for the NLR family, and this represents an area of much needed progression, particularly in relation to multidomain and full-length constructs.
Electron microscopy has been successfully used to visualize the formation of wheel-like structures, analogous to the apoptosome (Qin et al., 1999), for purified full-length NLRP1, NLRC4, and NAIP5 (Faustin et al., 2007; Halff et al., 2012). The stoichiometry of these structures varies between either 5 and 7 (NLRP1) or 11 or 12 (NLRC4/NAIP5) protomers per complex. In the past, researchers often assumed that this organizational arrangement reflects that within the inflammasome itself and have ignored the difference in size between the wheel-like structures (roughly 30 nm) and the inflammasome (about 1 μm). The recent observations using electron microscopy of helical filaments formed by inflammasome components (Baroja-Mazo et al., 2014; Franklin et al., 2014; Lu et al., 2014) (Fig. 3D) provide a potential framework for the formation of larger molecular structures. However, they need reconciling with the super-resolution microscopy of the endogenous inflammasome, which reveals a concentric ring-like structure with an outer layer of ASC (apoptosis-associated speck-like protein containing a caspase activation and recruitment domain [CARD]), an inner NLR ring, and a central caspase ring (Man et al., 2014).
At the atomic level, structural data exist for the nucleotide-binding domain (NBD)-LRR of NLRC4 (Hu et al., 2013), the LRRs of NLRX1 (Hong et al., 2012) and NLRP1, and the effector domains of NOD1 (CARD), NLRC5 (atypical CARD), NLRP1 (CARD and pyrin domain [PYD]) (Hiller et al., 2003), and NLRP3, 4, 7, 10, 12, and 14 (all PYD) (Coussens et al., 2007; Manon et al., 2007; Srimathi et al., 2008; Pinheiro et al., 2010, 2011; Bae and Park, 2011; Eibl et al., 2012, 2014; Su et al., 2013; Jin et al., 2013; Gutte et al., 2014). Although we still await the structure of an NLR in complex with either its ligand or a downstream signaling adaptor, these structures have provided important insight into the molecular functionality of NLR signaling regulation and transduction.
The similarity of the NLR family to other NBD-LRR proteins led to the proposal that the receptors are maintained in an autoinhibited state before activation similar to that seen with apoptosis protease activating factor-1 or cell death protein-4 (Danot et al., 2009). The recent crystal structure of murine NLRC4, lacking both the CARD and residues 622–644, in the inactive conformation has confirmed, at least in the case of NLRC4, this theory (Hu et al., 2013) (Fig. 4). Consistent with the proposed models of autoinhibition, the inactive conformation is maintained through two discrete mechanisms: first, by the presence of ADP in the Walker A site of the NBD, particularly via interactions between histidine 443 and the β-phosphate of the ADP; and second, contact between helical domain (HD) 2 and the NBD facilitate the folding over of the LRRs and their subsequent interaction with the NBD (Fig. 4). Disruption of ADP binding or mutation of HD2 released NLRC4 inhibition, resulting in an autoactive receptor (Hu et al., 2013). Although it is tempting to transfer this mechanism to all NLRs, further experimentation is needed to confidently claim a unified mechanism of autoinhibition. Meanwhile the structure of the LRRs of NLRX1 led to the identification of positively charged surfaces centered around Arg669 (Fig. 4C), mutation of which disrupted binding of the RNA ligands (Hong et al., 2012). Whether the LRRs are a universal location for ligand binding by NLR proteins remains controversial. A comparison of the structure of the NLRP1 LRRs (Fig. 4D) with those of NLRX1, NLRC4, and a model of the NOD2 LRRs led the authors to propose that muramyl dipeptide (MDP), a putative NLRP1 ligand, would not bind the LRRs (Reubold et al., 2014).
Fig. 4.

Functional insights from NLR LRR structures. (A) Structure of the NBD (HD1 in yellow; HD2 in blue) and LRR domain (red) from NLRC4 (adapted from PDB ID 4KXF). Bound ADP is shown in red. (B) The inhibitory interaction between His443 (orange) and ADP (red). Protein domains are colored as in (A). (C) Cartoon (left) and electrostatic map (right) of an NLRX1 LRR promoter (adapted from PDB ID 3UN9). The LRRs are shown in gray, the C-terminal capping structure in yellow and the N-terminal capping structure in magenta. Mutation of Arg699 (highlighted in orange) disrupts the binding of RNA to the LRR. A basic patch surrounding Arg699 is predicted to be responsible for RNA binding. (D) Crystal structure of the LRR domain from NLRP1 (adapted from PDB ID 4IM6).
The NLR effector domains play a vital role in the assembly of the multiprotein complexes required for signal transduction. As members of the DD superfamily they are involved in homotypic interactions (Fig. 3), such as NOD1 CARD:RIP2 CARD and NLRP3 PYD:ASC PYD, involving multiple interfaces in a manner likely to be analogous to that seen with the PIDDosome and MyDDosome complexes involved in caspase-2 activation and TLR signaling, respectively (Fig. 3). The structures of a range of NLR effector domains and the inflammasome forming adaptor ASC (de Alba, 2009) have been solved, enabling insight into the interactions involved in signal transduction.
The solution structure of the NOD1 CARD is a monomer (Manon et al., 2007), but the crystal structure is a dimer, stabilized by an interprotomer disulfide between Cys39 residues, in which helix 6 of one protomer swaps with helix 1 of the other (Coussens et al., 2007; Srimathi et al., 2008). Interestingly the crystal structure of the NLRP14 PYD also shows a similar process of helix swapping (Eibl et al., 2014) (Fig. 5). The physiologic relevance of NOD1 CARD dimerization has yet to be proven, but a recent study suggests an increasing propensity to form dimers results in a reduction in NOD1 signaling despite retaining the interaction with RIP2, presumably as a result of changes in the downstream modification of RIP2 or through conformational interference in the assembly of the signaling complex (Mayle et al., 2014). NOD2 also interacts with RIP2 but possesses a tandem set of CARDs that have been shown to interact with one another by isothermal titration calorimetry (Fridh and Rittinger, 2012). The structure of the NOD2 CARDs has yet to be solved, and it will be interesting to see how their tandem nature influences the structural basis of NOD2 signaling. For example, do they adopt a similar tetrameric assembly to that recently reported for the tandem CARDs of RIG-I (Peisley et al., 2014)?
Fig. 5.

CARDs and PYDs. (A) Monomeric structures of the NOD1 CARD (left, PDB ID 2DBD) and NLRP14 PYD (right, PDB ID 4NIL). First helix is shown in blue and sixth helix in green. (B) Dimeric structures of the NOD1 CARD (left, PDB ID 2NZ7) and NLRP14 PYD (right, PDB ID 4NIJ). Helix 6 (green) is extended compared with folds in (A). (C) PYDs from NLRP1 (left, PDB ID 1PN5) and AIM2 (right, PDB ID 4O7Q). Helix 1 is colored in blue and helix 6 in red. Helix 3 adopts a loop conformation in NLRP1. (D) NLRP10 PYDs from mouse (left, PDB ID 2DO9) and human (right, PDB ID 2M5V). Domains are colored as in (C).
The categorization of NLRC5 as a CARD-containing receptor was controversial as structural predictions of its effector region were unable to clearly characterize a discrete domain fold within the death domain superfamily. The recent solution structure of the NLRC5 effector domain does, however, support the presence of a CARD, albeit an atypical one (Gutte et al., 2014). Specifically the NLRC5 CARD lacks the usual kink observed in α-helix 1, and α-helix 3 is replaced with an ordered loop. The functional rationale for these structural characteristics remains unknown, but it is entirely plausible that interaction with a downstream binding partner, such as a transcription factor involved in MHC I transcriptional regulation, might result in stabilization of the loop into an ordered helix. The NLRP1 PYD, which is missing in mice and may therefore not be crucial to receptor function, also lacks an ordered third helix (Hiller et al., 2003), indicating that this is not a characteristic unique to NLRC5 (Fig. 5). In fact, helix 3 in the PYDs of other NLR proteins are structurally the most variable part of the protein fold, with both the length and orientation of the helix differing between NLR PYDs (Bae and Park, 2011; Su et al., 2013). This raises the possibility that this region of the PYD is critical for mediating interaction with downstream binding partners and may help to explain the different dynamics and protein interactions present in NLR signaling. For example, studies of the PYD of murine and human NLRP10 have highlighted cross-species differences in the conformation of the helix 3 region that are proposed to account for the different behavior of murine and human NLRP10 in mediating aggregation of ASC (Su et al., 2013) (Fig. 5). The generation of helix 3 PYD chimeras would be an interesting step in the functional characterization of NLRP signaling pathways.
C. Activation and Signal Transduction in the Nucleotide-Binding Domain and Leucine-Rich Repeat-Containing Receptor Family
In a reflection of the varied functionality of the NLR family, the methods of receptor activation and the signaling pathway used are both diverse and extensive. In this review we will focus on the mechanisms that enable activation and initiation of the major proinflammatory pathways controlled by NOD1, NOD2, and the inflammasome. For further details on these and other NLR signaling pathways, we refer the reader to the following reviews (Schroder and Tschopp, 2010; Davis et al., 2011; Franchi et al., 2012; Lupfer and Kanneganti, 2013; Wen et al., 2013; Lamkanfi and Dixit, 2014; Philpott et al., 2014).
1. Activation and Signaling at Nucleotide Oligomerization Domain 1 and 2.
NOD1 and NOD2 are proinflammatory NLRs activated by fragments of bacterial peptidoglycan. Specifically, NOD1 responds to γ-d-glutamyl-meso-diaminopimelic acid (ie-DAP) produced by Gram-negative and a few Gram-positive bacteria (Chamaillard et al., 2003a; Girardin et al., 2003a), whereas NOD2 responds to MDP that is found in all bacteria (Girardin et al., 2003b). NOD2 is activated by the common N-acetyl form of MDP and the N-glycolyl form found in mycobacteria and related Actinomycetes. Disruption of namH, the gene responsible for conversion of N-acetyl MDP to the N-glycolyl form, in Mycobacterium smegmatis resulted in a loss of NOD2-dependent TNFα secretion, which was restored upon genetic complementation with namH (Coulombe et al., 2009). This study also demonstrated that N-glycolyl MDP was both a more potent activator of NOD2 and, consistent with the immunologic efficacy of mycobacteria, produced a stronger T-cell mediated immune response as an adjuvant than N-acetyl MDP. The ability to specifically modulate NOD2 activation is an attractive proposition for enhancing vaccine adjuvant efficacy. Natural and synthetic peptidoglycan analogs and derivatives show a spectrum of efficacy dependent upon factors such as the length and cleavability of the associated glycan sequence, the precise nature of the amino acid in the first and second positions of the dipeptide, the presence and nature of chemical modifications on individual amino acids and at the N terminus, and the precise length of the tripeptide stem (Girardin et al., 2003c; Rubino et al., 2013; Wang et al., 2013).
The majority of bacteria remain extracellular, whereas NOD1 and NOD2 are cytoplasmic. Multiple complementary mechanisms have been described to explain how the ligands can come into contact with the receptors. Active import of ie-DAP and MDP through solute carrier protein 15A1 (SLC15A1; also known as peptide transporter 1) occurs in intestinal epithelial cells and macrophages (Vavricka et al., 2004; Dalmasso et al., 2010). This may have clinical relevance as upregulation of SLC15A1 expression in inflammatory bowel disease (IBD) patients has been reported (Wojtal et al., 2009). SLC15A2 (also known as peptide transporter 2), a close homolog of SLC15A1, shows more fastidious ligand selectivity. Swaan et al. (2008) used computational modeling to calculate the SLC15A2 pharmacophore and predicted that only ie-DAP and not MDP would be imported. Using primary epithelial airway cells and transfected Chinese hamster ovary cells they confirmed these predictions and demonstrated that the affinity constant for ie-DAP uptake was approximately 200 μM. The importance of the solute carrier protein transporter family in mediating NOD1 and NOD2 ligand import has been supported by the observation that release into the cytosol of MDP internalized into endosomes is dependent on the family members SLC15A3 and SLC15A4 (Lee et al., 2009; Nakamura et al., 2014). Before cytosolic release, endosomal internalization of ie-DAP and MDP, at least in a human embryonic kidney 293 cell line reconstitution system, occurs in a dynamin-dependent and caveosome-independent manner. Internalization is optimal at a pH between 5.5 and 6.0 and does not require the activity of the vacuolar ATPase complex (Lee et al., 2009). SLC15A3 and SLC15A4 also serve to provide a focal point for the formation of active signaling complexes at the endosomal membrane (Irving et al., 2014; Nakamura et al., 2014).
Entry of ie-DAP and MDP into the cytoplasm can also be mediated directly by bacteria. Shigella flexneri invasion stimulates NOD1 after the shedding of peptidoglycan during replication (Nigro et al., 2008), whereas invasion by Listeria monocytogenes is proposed to result in cytosolic peptidoglycan fragments after cleavage by the autolysins p60 and NamA (Lenz et al., 2003). Listeria minimizes the immunostimulatory impact of its peptidoglycan by deacytlation of N-acetylglucosamine residues. Deletion of the pgdA gene, which encodes a peptidoglycan N-deacetylase, served to increase NOD1 activation and impaired bacterial survival. The importance of peptidoglycan deacetylation as an immune evasion strategy was confirmed by the loss of NOD1 and NOD2 stimulatory activity after artificial deacetylation of peptidoglycan from Helicobacter pylori (Boneca et al., 2007). Peptidoglycan ligands can also be directly delivered to the cytosol as a result of the action of bacterial secretion systems or through fusion between the cell membrane and spherical fragments of bacterial membranes known as outer membrane vesicles released from Gram-negative bacteria (Kaparakis et al., 2010; Bielig et al., 2011). The H. pylori type IV secretion system, encoded on the cagPAI pathogenicity island, facilitates intracellular ligand delivery and subsequent activation of NOD1 (Viala et al., 2004).
The importance of the LRRs of NOD1 and NOD2 in ligand recognition has been demonstrated through chimeric receptors in which swapping the LRR regions of the two receptors swapped their ligand specificity (Girardin et al., 2005) and also through point mutations in the LRRs that impair receptor activation (Tanabe et al., 2004). However, whether NOD1 and NOD2 bound their activating ligands directly remained uncertain. Recent evidence suggests, albeit with certain caveats, that NOD1 and NOD2 do indeed directly sense their ligands (Monie, 2013). The first evidence of direct interaction was provided using the biophysical techniques atomic force microscopy and surface plasmon resonance to show that ie-DAP bound to immobilized commercially prepared NOD1 but that no interaction was detected between NOD1 and MDP or after removal of the NOD1 LRRs (Laroui et al., 2011). Although this provided some indication of interaction specificity and supported a critical role in ligand recognition for the LRRs of NOD1, a failure of NOD1 to interact with a wider range of small molecules would strengthen the interpretation of the data. Soon afterward two groups reported a direct interaction between NOD2 and MDP. Mo et al., (2012) used biotinylated-MDP to pull down NOD2 from cellular lysates, however, this interaction was dependent on the presence of the nucleotide-binding domain of NOD2, and an LRR-only construct was not pulled down. Rather than refuting the importance of the LRRs, this observation may reflect a need for the nucleotide-binding domain to facilitate proper folding of the NOD2 LRRs. Simultaneously, surface plasmon resonance studies reported direct binding between NOD2 and MDP (Grimes et al., 2012). Confirming the need for further studies to characterize the precise nature of the interaction of NOD2 with MDP, this work reported competitive binding, at nanomolar affinities, for both the agonist and antagonist stereoisomers of MDP with NOD2. This may reflect a subtle pharmacokinetic relationship between the receptor and the ligands or may be indicative of nonspecific interactions under the experimental conditions. Similar experiments will be needed testing a wider range of small molecules and MDP analogs before a firm conclusion can be made.
NOD2 was recently suggested to play an important role in the response to viral infections such as those caused by the ssRNA viruses RSV and influenza A virus and the DNA virus human cytomegalovirus (Sabbah et al., 2009; Lupfer et al., 2013; Kapoor et al., 2014). RSV or human cytomegalovirus infection upregulated NOD2 expression and resulted in IRF3 activation and IFN-β production (Sabbah et al., 2009; Kapoor et al., 2014), whereas lack of Nod2 led to an increased susceptibility to influenza A virus possibly through alterations in the regulation of inflammasome function (Lupfer et al., 2013). However, given the dramatic differences between MDP and potential viral ligands, further characterization of the antiviral role of NOD2 is needed to determine whether the changes in NOD2 expression are a direct, or a bystander, physiologic response.
Before ligand recognition, NOD1 and NOD2 adopt an inactive conformation in the cytosol in which interactions between the LRRs and the NBD domain are believed to lock the receptor in a closed conformation likely to be analogous to that observed in the recent crystal structure of NLRC4 (Hu et al., 2013). Both NOD1 and NOD2 are known to associate with cellular chaperones such as SGT1 (suppressor of G2 allele of Skp1), HSP70 and HSP90, which are hypothesized to play an important role in maintaining the receptors in an inactive form and to contribute to protein stability (da Silva Correia et al., 2007; Mayor et al., 2007; Lee et al., 2012c; Mohanan and Grimes, 2014). In a process dependent upon ATP binding and hydrolysis, NOD1 and NOD2 adopt an open conformation, most likely displacing the bound chaperones, undergo NBD-mediated oligomerization, and make their CARD available for interaction with downstream signaling adaptors. Disruption of either nucleotide hydrolysis or the formation of an inactive conformation, as observed by NOD2 polymorphisms connected to the inflammatory disorder Blau syndrome, results in receptor hyperactivation (Mo et al., 2012; Zurek et al., 2012; Parkhouse et al., 2014b). Recognition of ligand results in an increase in the membrane-associated fraction of NOD1 and NOD2 and precipitates the formation of a signaling competent complex (Barnich et al., 2005; McDonald et al., 2005; Lécine et al., 2007; Kufer et al., 2008; Irving et al., 2014; Mayle et al., 2014; Nakamura et al., 2014). Early studies identified the plasma membrane as the site of NOD1 and NOD2 relocalization, whereas more recent work highlights the relocalization of the receptors to the endosomal membrane. Given the multiple entry routes for the activatory ligands and the role of NOD1 and NOD2 in the induction of autophagy as a defense against bacterial infection (Cooney et al., 2010; Homer et al., 2010; Travassos et al., 2010), these locations may well dictate the downstream functionality of the receptors. It does however raise interesting, and as yet unanswered questions, about the spatial nature and timings of receptor activation and relocalization.
The primary adaptor protein recruited to both NOD1 and NOD2 is RIP2. RIP2 consists of a kinase domain and a C-terminal CARD. Originally described as a serine/threonine kinase, RIP2 has also been shown to function as a tyrosine kinase with the capability of autophosphorylating Tyr474 within its own CARD (Tigno-Aranjuez et al., 2010). The interaction between RIP2 and NOD1 and NOD2 is mediated by homotypic CARD–CARD interactions. CARDs are members of the death domain protein superfamily, which also includes the DD and the PYD. Studies of the PIDDosome, a caspase-2 activating complex formed between the DDs of receptor interacting protein-associated ICH-1/cell death protein-3 homologous protein with a death domain and PIDD, identified three different interfaces (Types I, II, and III) involved in DD interaction (Park et al., 2007). Further studies have shown that these interfaces are used across the DD superfamily (Kersse et al., 2011). In the case of NOD1 and RIP2, early studies used site-directed mutagenesis and coimmunoprecipitations to identify residues (Glu53, Asp54, Glu56) forming an acidic patch in the third helix of the NOD1 CARD and basic residues (Arg444, Arg483, Arg488) in RIP2 critical for the interaction (Manon et al., 2007). More recently, further mutagenic studies and comparative computational modeling based on the apoptosis protease activating factor 1:procaspase-9 complex (Qin et al., 1999) and the tetrameric RIG-I CARD complex (Peisley et al., 2014) have indicated that the interactions between NOD1 and RIP2 are likely to involve multiple interfaces and involve Tyr474 in the RIP2 CARD (Mayle et al., 2014).
Given the broad similarities between NOD1 and NOD2, it might be expected that they would both interact with RIP2 in the same way. However, NOD2 has two CARDs that have been shown to interact directly with a 1:1 stoichiometry and an affinity of approximately 1 μM as determined by isothermal titration calorimetry (Fridh and Rittinger, 2012). The association of the tandem NOD2 CARDs alters the interfaces available for interaction with RIP2, and indeed studies so far suggest that it is basic residues on NOD2 (Arg38, Arg86) and acidic residues on RIP2 (Asp461, Glu472, Glu475, Asp492) that are most important for this interaction (Fridh and Rittinger, 2012). How these differences will ultimately influence the macromolecular signaling complexes that form after receptor activation remains to be seen.
NOD1 and NOD2 also exhibit differences in the repertoire of adaptors used and the role of nucleotide hydrolysis in the cycling of the receptor between active and inactive forms. In addition to signaling via RIP2, NOD2 can also interact with the adaptor protein CARD9, leading to upregulation of the stress kinase signaling pathways (Hsu et al., 2007), a pathway that has yet to be connected with NOD1 activation. Somewhat surprisingly the interaction between NOD2 and CARD9 is independent of the CARD domains of the two proteins; instead it involves the NOD2 NBD and the linker region between the NOD2 CARDs and NBD domain (Parkhouse et al., 2014a).
2. Activation and Signaling at the Inflammasome.
Inflammasome formation can be initiated by the NLR family members NLRP1, NLRP3, and a cooperative alliance of NLRC4 and NAIP. The inflammasome itself is a large, roughly 1-μm in diameter, multiprotein complex in which the basic composition consists of an NLR, the adaptor protein ASC, and an effector caspase such as caspase-1. The physiologic function of the inflammasome is to activate caspase-1, thereby facilitating processing and secretion of the inflammatory cytokines IL-1β and IL-18 (Fig. 1C). The activation of caspase-1 can also result in cell death via pyroptosis. Inflammasome formation can also be initiated by AIM2, and it has been reported that NLRP6, NLRP7, and NLRP12 can form inflammasomes (Wen et al., 2013). However, these latter NLRP-related observations require further verification because their physiologic function or relevance is currently unclear. This part of the review will focus on the cellular events required for activation of the NLRP1, NLRP3 and NLRC4/NAIP inflammasomes.
a. Nucleotide-binding domain and leucine-rich repeat-containing receptor containing a pyrin 1.
NLRP1 was the first functional inflammasome forming protein described (Martinon et al., 2002) and has a unique domain organization that in humans is sequentially composed of an N-terminal PYD, an NBD, a LRR, a function to find domain, and a C-terminal CARD. Mice have three polymorphic NLRP1 genes, Nlrp1a, Nlrp1b, and Nlrp1c, all of which lack the PYD. The functional implications of the different domain organization between humans and mice have yet to be fully interrogated.
It has been suggested that activation of human NLRP1 is achieved via direct interaction with the NOD2 ligand MDP (Faustin et al., 2007). However, clear physiologic confirmation of this specificity remains absent, and a recent structure of the NLRP1 LRRs suggested that MDP binding would not occur within the LRRs (Reubold et al., 2014). Work using mice lacking Nlrp1b has, however, provided definitive confirmation that murine NLRP1b is activated by the lethal toxin from Bacillus anthracis (Kovarova et al., 2012). How lethal toxin activates NLRP1b still requires full elucidation, but a number of recent works have suggested a direct role for lethal toxin-mediated cleavage of murine NLRP1b (and rat NLRP1a) near the N terminus (Hellmich et al., 2012; Levinsohn et al., 2012; Chavarría-Smith and Vance, 2013). Polymorphic and engineered variation to the identified cleavage site led to alterations in cellular sensitivity to lethal toxin and subsequent changes in NLRP1 inflammasome activation. It has also been suggested that NLRP1 undergoes spontaneous self-cleavage in the C-terminal function to find domain and that this process is required for full activation of the inflammasome (D’Osualdo et al., 2011; Finger et al., 2012). To date similar requirements for receptor cleavage have not been described in other inflammasome-forming proteins, highlighting the diverse cellular processes that have evolved to activate caspase-1.
b. Nucleotide-binding domain and leucine-rich repeat-containing receptor containing a caspase activation and recruitment domain 4/neuronal apoptosis inhibitory protein.
NLRC4 and NAIP interact with one another to form a functional inflammasome complex. Flagellin was the first ligand shown to activate the NLRC4 inflammasome complex, and this was originally believed to be the result of direct ligand recognition by NLRC4. However, it was recently shown that the NAIP members of the NLR family provide specific ligand recognition and work in conjunction with NLRC4 to form a functional inflammasome. NAIP is present as four functional isoforms in mice (NAIP1, 2, 5, and 6) but only one in humans. Murine NAIP5 and 6 recognize bacterial flagellin (e.g., FlaA from Listeria pneumophila or FliC from Salmonella typhimurium), murine NAIP2 responds to bacterial type 3 secretion system rod proteins (e.g., PrgJ from S. typhimurium), whereas murine NAIP1 and human NAIP are activated in response to the needle component of type 3 secretion systems (Lightfield et al., 2008; Kofoed and Vance, 2011).
The molecular basis underlying the specifics of NAIP-mediated ligand recognition has yet to be fully elucidated. The C-terminal 35 amino acids of flagellin were initially identified as crucial for NLRC4/NAIP inflammasome activation (Lightfield et al., 2008), with subsequent work narrowing the key region to the last seven residues (Miao et al., 2010). Although the C terminus of flagellin is undisputedly crucial for NLRC4/NAIP activation, inflammasome reconstitution assays have recently indicated that the N-terminal region of flagellin, which interacts with the C-terminal portion to form the D0 motif of full-length flagella, can also induce NAIP5-NLRC4 complex formation and caspase-1–dependent cell killing (Halff et al., 2012). Consequently there may be a contextual element to flagellin recognition by NAIP5/NLRC4. This study also provided the first clear evidence that NAIP5 interacted directly with flagellin and that this interaction precipitated the formation of wheel-like complexes between NAIP5 and NLRC4 (Halff et al., 2012).The specificity of the molecular events involved in flagellin recognition by NLRC4/NAIP is aptly demonstrated by Listeria. L. monocytogenes is a poor activator of the NLRC4/NAIP inflammasome, and death occurs in only about 10% of infected cells. However, forcing L. monocytogenes to ectopically express the flagellin protein FlaA from L. pneumophila initiates robust activation of NLRC4/NAIP and death in around 70% of infected cells (Sauer et al., 2011a).
Sequence similarity between the C terminus of flagellin, FliC, and the PrgJ in S. typhimurium led to the discovery that the NAIP/NLRC4 inflammasome also recognized both rod protein and needle protein components of bacterial T3SSs (Malhas et al., 2002; Miao et al., 2010; Kofoed and Vance, 2011; Zhao et al., 2011; Yang et al., 2013a; Rayamajhi et al., 2013). To date the region of the NAIP protein needed for ligand recognition has not been pinpointed. Vance and colleagues (Tenthorey et al., 2014), however, used chimeras of NAIP2 and NAIP5 to implicate regions of the murine NAIP proteins encompassing both HD1 and HD2 in the specificity of ligand recognition. This would move away from the accepted role of the LRR domain in direct ligand detection as highlighted by the TLRs and NOD1/2. It may, however, suggest the involvement of more subtle molecular processes involving ligand-mediated release of the NAIP proteins from an inactive conformation permitting subsequent interaction with NLRC4 and inflammasome formation.
c. Nucleotide-binding domain and leucine-rich repeat-containing receptor containing a pyrin 3.
In general activation of the NLRP3 inflammasome requires two distinct signals. Signal 1, or priming, invokes the upregulation and production of inflammasome components, such as NLRP3, and the substrate pro–IL-1β. Expression of pro-IL-18 is also upregulated by signal 1 at the mRNA, but not the protein, level (Schroder et al., 2012; Lin et al., 2014). Signal 1 is induced by NFκB signaling from receptors such as the TLRs or NOD1/2. Experimentally cells are often primed using LPS. Infectious agents do not usually require an independent priming step, because they provide ligands for both priming and the subsequent signal 2. Signal 2 results from the exposure of NLRP3 to the activating ligand, leading to cleavage of caspase-1 and the processing of pro–IL-1β and pro–IL-18 followed by secretion of the active cytokines. Currently identified activators of the NLRP3 inflammasome are diverse in both origin and chemical structure. They include endogenous and exogenous molecules, such as: ATP, uric acid, silica, cholesterol crystals, nigericin and other pore-forming proteins, viral nucleic acid, and mitochondrial DNA. In addition, cellular processes such as potassium efflux and lysosomal rupture also induce NLRP3-mediated inflammasome activation (Davis et al., 2011; Wen et al., 2013; Lamkanfi and Dixit, 2014). NLRP3 cannot plausibly bind all these activators directly and should be viewed as a general sensor of homeostatic disruption. Here we will briefly summarize some of the main theories currently underpinning NLRP3 activation, including the roles of lysosomal rupture, mitochondria, reactive oxygen species (ROS), and ionic balance.
Phagocytosis of particulate and crystalline material can result in activation of the NLRP3 inflammasome after rupture of lysosomes and the release of their contents into the cytoplasm (Cassel et al., 2008; Eisenbarth et al., 2008; Halle et al., 2008; Hornung et al., 2008; Dostert et al., 2009; Duewell et al., 2010). The lysosomal protease cathepsin B was initially proposed to mediate inflammasome activation because addition of the cathepsin B inhibitor CA-074-Me (methyl 1-[3-methyl-2-[[3-(propylcarbamoyl)oxirane-2-carbonyl]amino]pentanoyl]pyrrolidine-2-carboxylate) causes a reduction in caspase-1 activation after lysosomal disruption (Hornung et al., 2008). However, mice lacking cathepsin B still show caspase-1 activation after exposure to the particulate and crystalline ligands uric acid, hemozoin, and alum (Dostert et al., 2009). This suggests that as with other small-molecule inhibitors, CA-074-Me may have other targets resulting in a nonspecific impact on NLRP3 activity. Although lysosomal disruption clearly has the capacity to activate NLRP3, many of the activatory ligands, including ATP and the ionophore nigericin, do not involve phagocytosis or lysosomes. Hence, this is not an overarching mechanism of NLRP3 inflammasome activation, although it may be particularly relevant for some forms of inflammation, such as those connected with neurodegeneration.
The production of ROS is a common response to cell danger and pathogen attack. Generated by many cellular components including phagosomes, mitochondria, and nicotinamide adenine dinucleotide phosphate oxidases, ROS have been a popular choice in attempts to provide a common theme to the mechanism of NLRP3 activation (Allen et al., 2009; Schroder et al., 2010; Zhou et al., 2010, 2011). It has been suggested that ROS activity upregulates the redox related protein thioredoxin-interacting protein, which subsequently interacts with and activates the NLRP3 inflammasome (Zhou et al., 2010). However this theory remains highly controversial because other researchers detect no role for thioredoxin-interacting protein in the activation of IL-1β (Masters et al., 2010). In fact, the role of ROS in inflammasome activation may need revisiting in more general terms because of the observation that ROS inhibitors serve to block the priming and not the activation step of the NLRP3 inflammasome (Bauernfeind et al., 2011).
As well as acting as a potential source for ROS, dysfunction and perturbation of the mitochondria have been associated with NLRP3 activation. In particular, one study postulated that mitochondrial DNA functions as a direct ligand for NLRP3 (Shimada et al., 2012). However, Nakahira et al. (2011), while also identifying an important role for mitochondrial DNA in NLRP3 activation, suggested that the release of mitochondrial DNA occurs downstream of inflammasome activation. Release of the mitochondrial phospholipid cardiolipin also results in NLRP3 activation, and this has also been suggested to act as a direct ligand for NLRP3 (Iyer et al., 2013). The precise role of the mitochondria in NLRP3 activation remains controversial (Lawlor and Vince, 2014), and it is unlikely that both, or maybe even either, mitochondrial DNA and cardiolipin act as direct NLRP3 activators. However, mitochondrial dysfunction and damage may well prove to be an important component of inflammasome activation.
A further recurring theme in NLRP3 activation is the role of ion concentration. High levels of extracellular calcium have been suggested to drive NLRP3 activation, potentially through calcium sensing G protein–coupled receptors, leading to an increase in intracellular calcium concentration (Lee et al., 2012b; Rossol et al., 2012). Similarly increased intracellular calcium concentrations, after the release from intracellular stores such as the endoplasmic reticulum, may stimulate mitochondrial damage and subsequent inflammasome activation (Murakami et al., 2012). Potassium efflux, not influx, also activates the NLRP3 inflammasome. Potassium efflux is a mediated through the formation of pores in the plasma membrane or via the activation of the P2X7 purigenic receptors by ATP (Kanneganti et al., 2007; Muñoz-Planillo et al., 2009, 2013). Indeed it was recently proposed that potassium efflux is the unifying process involved in all steps leading to NLRP3 inflammasome activation (Muñoz-Planillo et al., 2013). In this work, Muñoz-Planillo et al. (2013) demonstrate that neither mitochondrial perturbation nor the production of ROS is needed for NLRP3 activation and that both high levels of extracellular calcium and the phagocytosis of crystalline material result in potassium efflux. They therefore propose that a reduction in intracellular potassium concentration is all that is required to activate the NLRP3 inflammasome. Although this fits with early models for NLRP3 inflammasome activation, extensive validation is needed to clarify whether potassium efflux is indeed a stimuli-independent inflammasome activator.
3. Post-Translational Regulation of Nucleotide-Binding Domain and Leucine-Rich Repeat-Containing Receptor Signaling.
Post-translational modifications such as phosphorylation or the addition of ubiquitin are important regulators of inflammatory signaling. Although this still represents a relatively unexplored area of NLR research, it is becoming increasingly clear that post-transcriptional modifications play an important role in the control of NLR signaling. Dixit and coworkers reported phosphorylation of serine 533 in murine NLRC4, probably mediated by protein kinase C (PKC) δ, as an essential requirement for activation of the NLRC4 inflammasome after infection with S. typhimurium (Qu et al., 2012). However, a more recent study from the Núñez laboratory suggests that PKCδ is dispensable for NLRC4 activation by both shigella and salmonella (Suzuki et al., 2014), raising questions about the precise requirements of, or kinases involved in, NLRC4 Ser533 phosphorylation. These differences may result from the different experimental systems or might represent alternative activation pathways for NLRC4. The structure of NLRC4 suggests that phosphorylated Ser533 would stabilize HD2-LRR interactions and favor a monomeric form of the receptor (Hu et al., 2013), making the mechanistic functionality of Ser533 phosphorylation unclear.
The adaptor proteins ASC and RIP2 also undergo phosphorylation during NLR signaling. ASC is phosphorylated in a spleen tyrosine kinase (SYK)- and c-Jun N-terminal kinase (JNK)–dependent manner and although both kinases appear to operate upstream of ASC, it is currently unclear whether ASC is a direct substrate for them. Phosphorylation of Tyr144 (Tyr146 in humans) in the ASC CARD was crucial for the formation of ASC specks, implying a critical role in inflammasome activation (Hara et al., 2013). Similarly phosphorylation of Tyr474 in the RIP2 CARD is required for maximal signaling after NOD2 stimulation. In this instance, phosphorylation is mediated by the kinase domain of RIP2 itself (Tigno-Aranjuez et al., 2010). Tyr474 is also involved in the binding of RIP2 to NOD1 (Mayle et al., 2014), but the impact of phosphorylation on this interaction has yet to be determined.
Post-translational modification via both ubiquitination and deubiquitination can directly modulate NLR signaling. For example, activation of the NLRP3 inflammasome requires the action of various deubiquitinases to remove ubiquitin molecules that would inhibit formation of the inflammasome (Juliana et al., 2012; Lopez-Castejon et al., 2013; Py et al., 2013). Inflammasome activation also requires ubiquitination of ASC by the linear ubiquitin assembly complex (Rodgers et al., 2014). Whether NLR-mediated inflammasomes can be deactivated via ubiquitin-mediated pathways has yet to be resolved, but at least in the case of the AIM2 inflammasome there is evidence that ubiquitin modifications of inflammasome components can result in recruitment of p62 and inflammasome degradation in autophagosomes (Shi et al., 2012). Ubiquitination also plays a crucial role in NOD1 and NOD2 signaling. Studies of the adaptor RIP2 have revealed extensive ubiquitination by a range of E3 ligases including cellular inhibitor of apoptosis protein 1, cellular inhibitor of apoptosis protein 2, X-linked inhibitor of apoptosis protein, Pellino3, and itchy E3 ubiquitin protein ligase (Bertrand et al., 2009; Krieg et al., 2009; Tao et al., 2009; Yang et al., 2013b; Tigno-Aranjuez et al., 2013). RIP2 ubiquitination enhances NFκB and JNK-mediated signaling, except when mediated by itchy E3 ubiquitin protein ligase, in which case the JNK and mitogen-activated protein kinase (MAPK) pathways are favored. Ubiquitin modification of NOD1 and NOD2 may competitively inhibit RIP2 binding (Ver Heul et al., 2013) but also acts as a direct signal for proteosomal degradation of the receptor thereby regulating the inflammatory response (Lee et al., 2012c; Zhang et al., 2014a).
D. Nucleotide-Binding Domain and Leucine-Rich Repeat-Containing Receptor Mutations and Disease
1. Inflammatory Disorders and Nucleotide-Binding Domain and Leucine-Rich Repeat-Containing Receptor Hyperactivation.
Excessive production of IL-1 is commonly associated with autoinflammatory and chronic inflammatory disorders (Zhong et al., 2013). SNPs in NLR family members have been associated with ligand-independent hyperactivation of the receptors and subsequent dysregulated secretion of IL-1β. The best characterized are the Cryopyrin-associated periodic syndromes, which can be caused by point mutations in the NBD of NLRP3 leading to receptor autoactivation. Mutations in the NBD of NOD2 also cause receptor hyperactivation and lead to development of the inflammatory disease Blau syndrome. Although drugs such as anakinra, an IL-1R antagonist, have been successfully used to treat many of these inflammatory conditions, the long-term nonspecific suppression of IL-1 has negative effects, particularly in relation to protection against disease (Dinarello and van der Meer, 2013). The development of new therapeutics targeted at specific disease-associated receptors or their signaling pathways is therefore an attractive proposition.
2. Nucleotide Oligomerization Domain 2 and Susceptibility to Crohn’s Disease.
Crohn’s disease is a form of IBD involving dysregulation of the intestinal microbiota that can affect anywhere in the gastrointestinal tract, although in adults it is usually restricted to the ileum of the small intestine (Van Limbergen et al., 2009). Crohn’s disease is a complex condition associated with contributory environmental and genetic factors. The first gene to be associated with Crohn’s disease was NOD2 (Hugot et al., 1996), but since then 70 additional loci have been discovered spread over 20 chromosomes (Franke et al., 2010). Within NOD2 at least 66 SNPs spread throughout the receptor have been isolated in patients with Crohn’s disease. Most are extremely rare, but three, R702W, G908R, and fs1007insC located in and around the LRR domain, are found in 81% of all Crohn’s disease cases for which NOD2 is accountable (Lesage et al., 2002). These SNPs do not confer disease phenotypes but increase the risk of Crohn’s disease developing.
Somewhat counterintuitively for a proinflammatory disorder NOD2 Crohn’s disease-associated SNPs are correlated with a loss of receptor function (Chamaillard et al., 2003b). This may result in disease via dysregulation of the intestinal microbiota because NOD2-deficient mice have increased commensal bacterial loads and are characterized by an inability to remove newly colonizing bacteria (Petnicki-Ocwieja et al., 2009; Ramanan et al., 2014). Linked with, and possibly causing the change in microbiotal balance, Paneth cells harboring NOD2 loss-of-function SNPs fail to produce normal levels of defensins (Kobayashi et al., 2005; Petnicki-Ocwieja et al., 2009; Biswas et al., 2010, 2012). NOD2 dysfunction may also alter self-tolerization and TLR2/4 crosstolerization that normally results from chronic MDP exposure (Hedl et al., 2007). Wild-type macrophages, but not those carrying the NOD2 SNP fs1007insC pretreated with MDP, exhibited reduced proinflammatory cytokine production in response to subsequent NOD2 or TLR2/TLR4 stimulation (Hedl et al., 2007). Crohn’s disease may therefore result from a failure to induce inflammatory tolerance in the intestinal tract.
Traditionally Crohn’s disease has been treated with immunomodulators in the form of nonspecific anti-inflammatory or immunosuppressive drugs with the intention of sufficiently reducing inflammation without causing prolonged or excessive immunosuppression. However, the efficacy of these drugs can be variable, long-term administration has undesirable side effects, and some patients fail to respond. As the importance of the intestinal immune response in Crohn’s disease becomes more apparent, an increasing number of approaches have been taken to modulate intestinal homeostasis using biologics and drugs targeted at altering the activity and efficacy of intestinal immunity. These have included: dampening proinflammatory cytokine responses with for example anti–IL-12, anti–IFN-γ, and anti–TNF-α antibodies; the use of antibiotics such as metranidazole, ciprofloxacin, and rifaxim; probiotic delivery; treatment with thiopurines and corticosteroids such as prednisone; administration of folic acid antagonists such as methotrexate; and the inhibition of cell movement (Bamias and Cominelli, 2006; Taylor and Irving, 2011). Looking forward there is interest in the possible application of stem cell based therapies, though much work remains to be done in this respect.
IV. The C-Type Lectin-Like Receptors
A. C-Type Lectin-Like Receptors and Their Nomenclature
The C-type lectin-like family is a large group of proteins (∼1000 members) with a diverse range of functions. Various terms, including "C-type lectin,", "carbohydrate recognition domain" (CRD), and "C-type lectin domain”/”C-type lectin-like domain" (CTLD) are used to describe members of this superfamily. The term C-type lectin initially referred to Ca2+-dependent (C-type) carbohydrate-binding (lectin) animal proteins (Zelensky and Gready, 2005), and a compact CRD (<200 amino acids) was found to be responsible for the carbohydrate-binding activity (Weis and Drickamer, 1996). Various C-type lectin sequences were compared leading to the identification of conserved residues within the domain, thereby facilitating the further identification of CRDs in additional proteins. Some of these newly identified proteins did not bind carbohydrates or Ca2+; therefore, the more inclusive term C-type lectin-like domain was introduced to refer to this protein fold. The term C-type lectin-like is now used to describe any protein that contains a CTLD regardless of whether it binds carbohydrate or Ca2+ (Zelensky and Gready, 2005).
The CLRs have been have classified into subgroups based on structural and functional characteristics (Drickamer and Fadden, 2002; Zelensky and Gready, 2005; https://www.imperial.ac.uk/animallectins/ctld/mammals/humanvmousedata.html). Many of these proteins have a "C-type lectin-like domain-containing" (referred to as CLEC) name; however, they have several other "aliases" based on their expression and function. Several of these proteins recognize ligands on pathogens including fungi, viruses, bacteria, and helminths. Some of these CLRs contain a transmembrane region, whereas others are soluble (Osorio and Reis e Sousa, 2011). Because of space restrictions, we will not be able to discuss each protein in detail in this review; therefore, we will focus on select CLRs that are involved in the recognition of pathogen-associated molecular patterns and are potential drug/vaccine targets. Various CLRs have endocytic activity and they deliver their ligands to intracellular vesicles for antigen processing and presentation. This excellent "delivery system" makes these CLRs potential candidates to deliver antigen to dendritic cells (DCs) for immunotherapy. Additionally, ligands for some CLRs display potential for use as adjuvants in the development of vaccine strategies (Caminschi et al., 2009).
B. Structural Biology of the C-Type Lectin-Like Receptors
Structural data exist for the major CLRs Dectin-1, Mincle (macrophage-inducible C-type lectin), MCL (macrophage C-type lectin), DC-SIGN (dendritic cell specific intracellular adhesion molecule-3 grabbing nonintegrin), and DC-SIGNR (Feinberg et al., 2001; Brown et al., 2007; Furukawa et al., 2013). The CRDs of the closely related DC-SIGN and DC-SIGNR were solved in complex with bound pentasaccharide, which in the case of DC-SIGN resulted in the carbohydrate-mediated crosslinking of two monomers. Both receptors have an extended second alpha helix compared with other CRDs and show specificity for high-mannose N-linked oligosaccharides. The interaction between the ligand and receptor involves one of the Ca2+-binding sites and unusually an internal sugar residue rather than a terminal one (Feinberg et al., 2001).
Mincle and MCL both bind the mycobacterial cord factor trehalose dimycolate (TDM) and this is reflected in the similarities of their CRDs (root mean square deviation of 1.5 Å of 124 Cα atoms). Unlike the DC-SIGNR structure the CRD of Mincle has only two Ca2+-binding sites, whereas MCL only possesses one. However, ligand binding remains Ca2+ dependent and is likely to be assisted by the presence of a shallow hydrophobic groove next to the likely ligand-binding site. This groove is smaller in MCL, which may account for the lower affinity of MCL for TDM binding (Furukawa et al., 2013).
The structure of the CTLD of Dectin-1 (Fig. 6) is similar to that of many other carbohydrate binding proteins but possesses a more variable long-loop region (residues 184–218 in the murine sequence) that lacks the presence of stabilizing metal ions. As with the structures of Mincle and MCL, a shallow hydrophobic groove between the side-chains of Trp221 and His223 is believed to contribute to ligand binding. Interestingly the surface of the Dectin-1 CTLD also contains an acidic groove with an as yet undetermined function (Brown et al., 2007)
Fig. 6.

CLR receptor structure and kinases. The structure of the extracellular domains of the CLR Dectin-1 (PDB ID 2CL8) are shown as ribbons colored blue (N terminus) to red (C terminus). The bound glycan is shown in space-filling atoms. The structure is extended by modeling the dimer of transmembrane helices (colored in rainbow colors and based on PDB ID 2HAC). In the lower panel representing the cytoplasm, the tyrosine kinase Syk recognizes hemITAM phospho-tyrosine motifs (phospho-Tyr atoms in gold) with its tandem SH2 domains (based on combining PDB IDs 4FL3 and 1A81 for the kinase (green) and SH2 domains (blue) respectively).
C. Activation and Signal Transduction at the C-Type Lectin-Like Receptors
CLRs mediate their responses through various signaling motifs. Some CLRs associate with an immunoreceptor tyrosine-based activation motif-containing [ITAM: Yxx(L/I)x6-12Yxx(L/I)] adaptor protein, such as Fc receptor γ chain (FcRγ) or DNAX-activating protein of 12 kDa, to mediate their signals, although other CLRs signal through a partial ITAM (YxxL; commonly known as hemITAM) in their cytoplasmic tail. Engagement of these CLRs results in tyrosine phosphorylation of the ITAMs/hemITAMs, and Syk is recruited and activated. The two tyrosines in a canonical ITAM provide docking sites for the two Src homology 2 domains of Syk, although it is thought that two hemITAM receptors dimerize to bring the hemITAMs in close enough proximity to facilitate binding of the two Src homology 2 domains of Syk. This leads to the initiation of a signaling cascade that can result in the activation of MAPK, nuclear factor of activated T cells, NFκB, and CARD9 (Osorio and Reis e Sousa, 2011; Sancho and Reis e Sousa, 2012). On the other hand, this family also contains members with inhibitory potential. Several CLRs contain an immunoreceptor tyrosine-based inhibitory motif (ITIM: S/I/V/LxYxxI/V/L), and the phosphatases SHP-1 (Src homology region 2 domain-containing phosphatase 1), SHP-2, and/or src homology 2-containing inositol phosphatase are recruited to the phosphorylated tyrosine residue within their ITIM. SHP-1, SHP-2, and src homology 2–containing inositol phosphatase then dephosphorylate downstream substrates and dampen activation. In addition, several CLR family members lack canonical signaling motifs, and they mediate their effects independently of ITAM, hemITAM, or ITIM signaling motifs (Osorio and Reis e Sousa, 2011).
1. Immunoreceptor Tyrosine-Based Activation Motif-Coupled Receptors.
a. Dendritic cell–associated C-type lectin-2.
Dectin-2 is expressed on various myeloid cell types. It recognizes high mannose structures on fungal cell walls (e.g., Candida albicans, Aspergillus fumigatus) and allergens from house dust mites. Dectin-2 has a short cytoplasmic tail, which associates with the ITAM-containing FcRγ adaptor (Robinson et al., 2009). For detailed reviews on Dectin-2, please see Graham and Brown (2009) and Kerscher et al. (2013). Dectin-2 initiates various signaling pathways through this canonical ITAM, including activation of a Syk, PKCδ, and CARD9-Bcl10-MALT1 pathway that mediates cytokine production (Robinson et al., 2009; Saijo et al., 2010; Strasser et al., 2012), NLRP3 activation, and ROS production (Ritter et al., 2010; Gorjestani et al., 2011). We showed recently that the adaptor protein Linker for activation of B cells is phosphorylated downstream of Dectin-2, but not Dectin-1, and that Linker for activation of B cells can modulate pathogen-associated molecular pattern–induced β-catenin activation and subsequent cytokine and Th1 responses. β-catenin activation is induced by fungal ligands; however, whether Dectin-2 or other CLRs mediate this response is currently unknown (Orr et al., 2013). Dectin-2 signaling also involves phospholipase C γ 2, leading to MAPK and NFκB activation and cytokine production (Gorjestani et al., 2011).
Interestingly, although another CLR, Dectin-1, was shown to activate all NFκB subunits, Dectin-2 specifically activated c-Rel downstream of MALT1 (mucosa-associated lymphoid tissue lymphoma translocation protein 1), which is essential for the production of Th17 polarizing cytokines (Gringhuis et al., 2011). In agreement with this, Dectin-2 promotes Th17 responses to C. albicans, and Dectin-2−/− and IL-17RA−/− mice are highly susceptible to infection with C. albicans (Saijo et al., 2010). Recently, Dectin-2 has been shown to form heterodimers with another CLR, MCL, and these heterodimers bind C. albicans α-mannans more effectively than homodimers, resulting in potent antifungal responses. In support of this, one study found that MCL−/− mice were susceptible to C. albicans infection and they displayed reduced IL-17 production (Zhu et al., 2013), although another study did not observe any defect in MCL−/− mice in response to C. albicans infection (Graham et al., 2012), suggesting that this needs further clarification. Dectin-2 is also involved in the generation of both Th1 and Th17 responses to Candida glabrata (Ifrim et al., 2014) and it contributes to C. albicans–induced Th1 responses in collaboration with Dectin-1 (Robinson et al., 2009; Saijo et al., 2010). In addition to its role in antifungal responses, Dectin-2 also induces allergic inflammation in response to house dust mites through the production of cysteinyl leukotrienes and subsequent development of Th2 and Th17 responses (Barrett et al., 2011; Clarke et al., 2014; Norimoto et al., 2014; Parsons et al., 2014). Interestingly, Dectin-2 is also involved in regulating an autocrine IL-17 loop that controls the activation of a subset of neutrophils (Taylor et al., 2014). These data indicate that promotion/regulation of T cell responses, in particular IL-17 responses, is an important aspect of Dectin-2 signaling that could be important for vaccination strategies involving Dectin-2. Indeed, vaccine-induced immunity against various endemic mycosis in the United States requires Th17 cells, and in line with the data described here, Dectin-2 is involved in these responses. Dectin-1 and Dectin-2, but not Mincle, were required for vaccine-induced acquired immunity to Histoplasma capsulatum and Coccidioides posadasii, whereas Blastomyces dermatitidis required Dectin-2 (Wang et al., 2014).
b. Mincle.
Mincle is predominantly a myeloid cell receptor with expression on some B cell subsets (Kawata et al., 2012). Mincle recognizes endogenous and exogenous ligands including Sin3A-associated protein, 130 kDa in necrotic cells, α-mannan on fungal cell walls (e.g., Malassezia, Fonsecaea, Candida), and mycobacterial cord factor, TDM. Similar to Dectin-2, Mincle associates with the ITAM-containing FcRγ adaptor and it signals through the Syk, PKCδ, and CARD9-Bcl10-MALT1 pathway, culminating in cytokine production in response to fungal pathogens (Wells et al., 2008; Yamasaki et al., 2009; Strasser et al., 2012). Mincle has been shown to relocate to the phagocytic cup, although it was not essential for phagocytosis on macrophages (Wells et al., 2008); however, a recent study on human cells provided some evidence that Mincle may be involved in phagocytosis. The authors demonstrated a reciprocal pattern of Mincle expression on monocytes and neutrophils, an inverse correlation of Mincle expression on monocytes with phagocytosis and candidacidal activity, and a correlation of Mincle expression on neutrophils with phagocytosis and candidacidal activity (Vijayan et al., 2012). In addition, a recent study showed that Mincle−/− mice were highly susceptible to infection with Klebsiella pneumonia. They had an increased bacterial burden, exaggerated hyperinflammation, and severe lung pathology associated with a massive influx of neutrophils. Interestingly, the Mincle−/− neutrophils showed defective phagocytosis of nonopsonized bacteria and they were unable to form extracellular traps (Sharma et al., 2014). Together, these data suggest a role for Mincle in phagocytosis, although it may be redundant in circumstances when other PRRs can compensate.
After a long search, Mincle was found to be the receptor for mycobacterial cord factor, TDM, and its synthetic analog trehalose-6,6′-dibehenate (TDB). TDM/TDB induce the Syk-CARD9 pathway discussed above and NLRP3 activation (Schweneker et al., 2013). Mincle signaling in response to TDM promotes inflammatory cytokine and nitric oxide production and granuloma formation (Ishikawa et al., 2009a). Similar to Dectin-2, Mincle was recently shown to form heterodimers with MCL on the cell surface (Lobato-Pascual et al., 2013) and MCL recognizes TDM and subsequently drives Mincle expression via the CARD9/Bcl10/MALT1-NFκB pathway (Zhao et al., 2014). Mincle expression was not induced in MCL−/− mice, and TDM-induced immune responses such as granuloma formation were severely diminished in Mincle−/− and MCL−/− mice (Miyake et al., 2013). Mincle−/− mice showed decreased proinflammatory responses to mycobacteria bacillus Calmette-Guérin vaccination and increased mycobacterial loads (Behler et al., 2012, 2015; Lee et al., 2012d). In contrast, Mincle−/− mice developed a normal Th1/Th17 response to M. tuberculosis H37Rv and controlled mycobacterial growth similar to wild-type mice (Heitmann et al., 2013).These divergent results may be due to the use of different strains of M. tuberculosis, different amounts of TDM in the mycobacterial cells, or different culture conditions and further study is required to determine the role of Mincle during mycobacterial infections (Lang, 2013). In terms of vaccination strategies, TDM and TDB display potent adjuvant activity for Th1/Th17 vaccination, and this is mediated by Mincle/CARD9 signaling (Schoenen et al., 2010; Shenderov et al., 2013).
An important role for Mincle in the chronic skin infection chromoblastomycosis was recently discovered. Fonsecaea pedrosoi, a causative agent, signals through Mincle to produce TNF; however, this is not sufficient to clear the infection. Interestingly, a potential immunotherapy was highlighted in this study, as exogenous addition of a TLR ligand resulted in synergistic TNF production and led to resolution of the infection in mice (Sousa et al., 2011). This has recently translated to a marked improvement in the lesions of four patients treated with topical application of Imiquimod (3M Pharmaceuticals ) (de Sousa et al., 2014). In agreement with this, coactivation of Mincle and TLR2 with TDM and Pam3CSK4 was shown to result in the synergistic induction of ROS and TNF (Lee et al., 2012d). However, a recent study showed that Fonsecacaea monophora, another causative agent of chromoblastomycosis, engages Dectin-1, resulting in the activation of IRF1 and IL12A transcription. Mincle is simultaneously engaged, which induces the E3 ubiquitin ligase mouse double minute 2-dependent proteasomal-mediated degradation of IRF1, via Syk-CARD9-mediated protein kinase B signaling, thereby blocking IL12A transcription. This leads to impaired Th1 responses and increased Th2 responses. In contrast to the findings of Sousa et al. (2011), the authors also observed that Mincle could inhibit TLR-induced IRF-1 activation and IL12A transcription in a similar manner. The differences between these studies could be due to different Fonsecaea strains or differences in Mincle expression and function between human and mice cells; however, these discrepancies need to be investigated further to determine whether it would be beneficial to target these pathways for immunotherapy and whether using Mincle ligands (TDM or TDB) as vaccine adjuvants would be beneficial or detrimental (Wevers et al., 2014).
2. hem-Immunoreceptor Tyrosine-Based Activation Motif Receptors: Dendritic Cell–Associated C-Type Lectin-1.
Dectin-1 is expressed by various myeloid cells (DCs, monocytes, macrophages) and B cells. Dectin-1 recognizes mycobacteria, an unknown endogenous ligand on T cells, and β-glucans on fungal cell walls. Several signaling pathways have been identified downstream of Dectin-1 that result in the induction of phagocytosis, cytokine and ROS production, and induction of Th1 and Th17 responses. Syk is recruited to the hemITAM of Dectin-1 (Fig. 6) and a scaffold complex of CARD9, B-cell lymphoma/leukemia 10, and MALT-1 is formed. This complex activates p65 and c-Rel, whereas NIK activation by Syk results in the subsequent activation of transcription factor RelB. Independent of Syk, Dectin-1 activates Raf-1, resulting in the phosphorylation and acetylation of p65. This complex web of activation of the different subunits of NFκB determines which cytokines will be produced. Engagement of Dectin-1 also involves phospholipase C γ 2–induced activation of MAPK, activator protein-1, and NFκB, resulting in cytokine production and Th1 and Th17 differentiation. A Dectin-1 ligand, curdlan (a β−1,3-glucan), acts as an adjuvant, resulting in the promotion of Th17 responses in vitro and in the production of Th1 and Th17 responses in vivo (LeibundGut-Landmann et al., 2007). Interestingly, Dectin-1 signaling decreases Th1 responses, thereby facilitating Th17 differentiation in response to a pulmonary infection with A. fumigatus (Rivera et al., 2011). These data demonstrate that Dectin-1 is important for cytokine production and T cell responses, and the nature of the ligand can determine which type of T cell response will occur. For more detailed reviews on these pathways, please see Geijtenbeek and Gringhuis (2009) and van den Berg et al. (2012).
Several interesting “new” signaling pathways were recently identified downstream of Dectin-1. Dectin-1 induces IRF5 activation, resulting in IFN-β production in response to C. albicans (del Fresno et al., 2013) and it induces IRF1 activation and IL12A transcription in response to F. monophora (Wevers et al., 2014). Additionally, Dectin-1 activates the noncanonical caspase-8 inflammasome, independently of pathogen internalization, resulting in IL-1β transcription and processing. Dectin-1–mediated activation of the canonical NLRP3/caspase-1 inflammasome and subsequent production and processing of IL-1β requires pathogen internalization (Gringhuis et al., 2012). Dectin-1 is important for phagocytosis, phagolysosomal maturation, and cytotoxic responses (Li et al., 2011; Mansour et al., 2013; Strijbis et al., 2013; Ma et al., 2014). Dectin-1 activates macrophage-1 antigen in a pathway that involves the guanine nucleotide exchange factors Vav1 and Vav3, resulting in cytotoxic responses of neutrophils (Li et al., 2011). In macrophages, Dectin-1 interacts with Bruton’s tyrosine kinase and Vav1, and macrophages deficient in either Bruton’s tyrosine kinase, Vav1, or Dectin-1 display defective phagocytosis (Strijbis et al., 2013). These "new" pathways require further study to fully identify the signaling involved and to determine whether they are activated downstream of other CLRs.
Dectin-1 interacts with and collaborates with several other receptors. Synergistic induction of TNF occurs in response to collaboration between Dectin-1 and TLR2 (Dennehy et al., 2008). Interestingly, a recent study showed that priming cells with C. albicans, acting through the Dectin-1/Raf-1 pathway, resulted in enhanced proinflammatory cytokine production in response to TLR ligands/bacteria (Ifrim et al., 2013). A Dectin-1 and Galactin-3 complex and a TLR2 and Galactin-3 complex have been identified in response to stimulation with fungal ligands, suggesting that Galactin-3 may be involved in these collaborative responses (Esteban et al., 2011). Additionally, the tetraspanins CD37 and CD63 interact with Dectin-1. CD37 can stabilize Dectin-1 expression and inhibit cytokine production; however, whether these complexes are involved in Dectin-1/TLR collaboration is currently unknown (Mantegazza et al., 2004; Meyer-Wentrup et al., 2007). Dectin-1 also induces SOCS1 production via a Src, Syk, Pyk2, MAPK pathway. SOCS1 does not affect Dectin-1 signaling but it can attenuate TLR-induced cytokine responses (Eberle and Dalpke, 2012). Dectin-1 is thus involved in a complex cross-talk with TLRs and other receptors and in determining its role in immune responses to pathogens, Dectin-1 signaling needs to be considered in the context of its interactions with other receptors.
Dectin-1 null mice were more susceptible to infections with C. albicans, Pneumocystis carinii, and A. fumigatus, and they displayed defects in ROS production and cytokine production (Saijo et al., 2007; Taylor et al., 2007; Gessner et al., 2012). Additionally, Dectin-1 is involved in cytokine and ROS production in vitro in response to mycobacteria (Rothfuchs et al., 2007; Shin et al., 2008); however, in vivo Dectin-1 appears to play a minor role because Dectin-1−/− mice showed a slightly decreased pulmonary bacilli burden without affecting pulmonary pathology or cytokine production (Marakalala et al., 2011). Interestingly, Dectin-1−/− mice were recently shown to display increased susceptibility to dextran sodium sulfate-induced colitis. This has been attributed to altered responses to the intestinal fungal microflora (Iliev et al., 2012). Therefore, Dectin-1 is an important PRR and Dectin-1 ligands could be used as adjuvants or Dectin-1 could be a potential target for immunotherapy. In line with this, a Dectin-1 ligand, laminarin, was recently added to a β-mannan tetanus toxoid conjugate vaccine to induce a protective response against C. albicans, and immunization of mice with this vaccine resulted in increased titers of C. albicans β-mannan antigen recognizing antibody (Lipinski et al., 2013). Another study found that delivery of influenza hemagglutinin 1 to DCs via Dectin-1 was not sufficient to induce hemagglutinin 1–specific Th17 responses, and future vaccine strategies should involve multiple signals from other PRRs to promote robust Th17 responses against pathogens (Duluc et al., 2014).
3. Immunoreceptor Tyrosine-Based Inhibitory Motif Receptors: Dendritic Cell Immune Receptor.
Humans encode one dendritic cell immune receptor (DCIR) protein, whereas mice have four DCIR-like proteins. DCIR is expressed on various myeloid cells (monocytes, macrophages, DCs) and on B cells and HIV-1 infected CD4+ T cells (Meyer-Wentrup et al., 2009). DCIR binds to HIV-1, Schistosoma mansoni and to endogenous ligands (Bloem et al., 2014). DCIR possesses an ITIM in its cytoplasmic tail that can recruit the tyrosine phosphatases SHP-1 and SHP-2. With a chimeric receptor approach, DCIR was shown to inhibit tyrosine phosphorylation and Ca2+ mobilization upon coligation of the B-cell receptor (Kanazawa et al., 2002). In addition, DCIR engagement specifically inhibited TLR8-induced cytokine production (IL-12 and TNF) and TLR9-induced IFN-α production but not TLR2-, TLR3-, or TLR4-induced cytokine production. DCIR is internalized in a clathrin-dependent manner after engagement, and DCIR promotes robust antigen presentation (Meyer-Wentrup et al., 2008, 2009). DCIR surface expression is downregulated by various inflammatory stimuli, suggesting that this may be a mechanism to facilitate activation (Richard et al., 2003; Meyer-Wentrup et al., 2008).
DCIR promotes cerebral malaria infection but plays a protective role during infection with Chikungunya virus, because it limits the viral-induced inflammatory response and subsequent tissue/joint damage (Long et al., 2013; Maglinao et al., 2013). DCIR has also been implicated in autoimmune responses. Dcir−/− mice spontaneously developed late onset sialadenitis and enthesitis, and in a model of collagen-induced arthritis, Dcir−/− mice displayed increased cytokine production (IL-4, IL-10, and IL-17) and antibody production. This was associated with an excessively expanded DC population, and Dcir−/− bone marrow–derived DCs displayed increased proliferation and STAT5 phosphorylation, suggesting that DCIR is important for homeostasis (Fujikado et al., 2008). In human studies, abundant DCIR expression on a variety of immune cells has been associated with rheumatoid arthritis (Eklöw et al., 2008).
One of the best studied roles for DCIR is in the capture of HIV-1. DCIR can promote trans and cis infection of CD4+ T cells and it is involved in viral replication (Lambert et al., 2008). After ligation of DCIR by HIV-1, signaling pathways involving SHP-1, SHP-2, Syk, Src kinases, PKC-α, and MAPK are activated. These signaling intermediaries were required to enhance HIV-1 binding and infection (Lambert et al., 2011). DCIR therefore represents an attractive target for HIV-1 and, based on three-dimensional modeling of the extracellular domain of DCIR, chemicals were screened to determine whether they affected HIV-1 binding and infection. From this study, two inhibitors were recently identified that reduce HIV-1 binding and propagation (Lambert et al., 2013).
4. Immunoreceptor Tyrosine-Based Activation Motif/Immunoreceptor Tyrosine-Based Inhibitory Motif-Independent Receptors.
a. Dendritic cell–specific intracellular cell adhesion molecule-3 grabbing nonintegrin.
DC-SIGN is expressed on DC and macrophage populations. It recognizes high mannose and fucosylated proteins and it binds fungal pathogens, mycobacteria, helminthes, and viruses (Garcia-Vallejo and van Kooyk, 2013). Although the human DC-SIGN family has two members (DC-SIGN and L-SIGN), mice have eight homologs of DC-SIGN (Signr1–8), one of which is a psuedogene. DC-SIGN consists of an extracellular region and a cytoplasmic tail containing internalization motifs. Upon ligand binding, DC-SIGN is rapidly internalized to late endosomes/lysosomes, and internalized ligands are efficiently processed and presented to T cells (Engering et al., 2002). In addition to endocytosis, DC-SIGN induces intracellular signals that can modulate cytokine production. Interestingly, different signaling pathways are induced by fucose- and mannose-containing ligands. DC-SIGN associates with a scaffold complex containing lymphocyte-specific protein 1, kinase suppressor of Ras (KSR) and connector enhancer of KSR, which facilitates constitutive recruitment of Raf-1 to DC-SIGN. After ligand binding by mannose-expressing pathogens, leukemia-associated Rho guanine nucleotide exchange factor and RhoA are recruited to the DC-SIGN complex and Raf-1 is activated, and the p65 subunit of NFκB is subsequently phosphorylated and acetylated. This leads to increased transcription of cytokines (IL-10, IL-12, and IL-6) after TLR4 engagement. Conversely, upon binding of fucose-containing pathogens, DC-SIGN dissociates from the KSR1/connector enhancer of KSR/Raf-1 complex and enhances IL-10 production while inhibiting IL-12 and IL-6 production in a Raf-1-independent manner (Gringhuis et al., 2007, 2009). Although DC-SIGN does not induce cytokine production, it modulates cytokine production from other receptors including the TLRs through the regulation of p65 activation (Gringhuis et al., 2007, 2010). For more detailed reviews on DC-SIGN, please see Geijtenbeek and Gringhuis (2009) and Garcia-Vallejo and van Kooyk (2013).
Interestingly, DC-SIGN interaction with HIV-1 (gp120) leads to transmission of HIV-1 from DCs to T cells. HIV-1 engages TLR8, resulting in the activation of NFκB and transcription of the provirus by RNA polymerase II. Engagement of DC-SIGN results in activation of Raf-1 and phosphorylation of p65, leading to the recruitment of the transcription elongation factor pTEF-b to the HIV-1 transcription complex and subsequent generation of full-length HIV-1 transcripts (Gringhuis et al., 2010). Interestingly, DC-SIGN targeting by glycodendrimers bearing carbohydrates or glycomimetic DC-SIGN ligands led to the identification of a product that inhibits DC-SIGN-mediated infection of T cells by HIV-1 and uptake of Dengue virus by Raji cells (Varga et al., 2014).
b. C-Type lectin-like receptor mutations and disease.
In recent years, SNPs in these CLRs and their signaling proteins have been associated with various diseases, and we will discuss some of these in this next section. Several SNPs have been associated with an increased risk of developing fungal infections. One of the most studied SNPs has been the Y238X early stop mutation in Dectin-1, which results in poor expression of Dectin-1 and defective IL-17, TNF, and IL-6 production. This SNP was first described in a family who were affected by recurrent vulvovaginal candidiasis or onychomycosis (Ferwerda et al., 2009). This SNP has since been associated with susceptibility to invasive aspergillosis in stem cell transplant patients (Cunha et al., 2010) and increased oral and gastrointestinal colonization with Candida species in stem cell transplant patients (Plantinga et al., 2009); however, it does not affect the incidence of oropharyngeal candidiasis in HIV-infected patients (Rosentul et al., 2011). An association between other SNPs in Dectin-1 (rs3901533 T/T; rs7309123 G/G) and DC-SIGN (rs4804800 G; rs11465384 T; 7248637 A; 7252229 C) and an increased risk of invasive pulmonary aspergillosis infection was recently described. The authors examined 27 SNPs in a range of CLRs and associated proteins and although most of these SNPs individually did not affect risk of invasive pulmonary aspergillosis infection, interaction analysis revealed synergistic genetic effects of SNPs in Dectin-1, Dectin-2, CCL2, and CCR2 genes (Sainz et al., 2012). CARD9 is found downstream of Dectin-1 and other CLRs. A SNP resulting in an early stop codon (Q295X) in CARD9 has been identified in humans, and homozygous individuals suffer from chronic mucocutaneous candidiasis, other dermatophyte infections, and some of these patients suffered from fatal invasive candidiasis. These patients display a much lower Th17 response than healthy controls (Glocker et al., 2009). In addition, patients with SNPs in IL-17RA and IL-17F were susceptible to chronic mucocutaneous candidiasis (Puel et al., 2011). Other SNPs that disrupt the Th17 pathway have also been associated with increased fungal infections, indicating the key importance of the CLR-CARD9-Th17 pathways during antifungal immunity. For further details on these, please see Lilic (2012).
In addition to fungal infections, SNPs in DC-SIGN have been associated with a variety of infectious diseases including HIV-1, Dengue virus, and tuberculosis. A SNP in the promoter of DC-SIGN-336T increased the risk of parenterally acquired HIV-infection, although it did not affect the risk for mucosal infection (Martin et al., 2004). The -336G allele was associated with protection against tuberculosis in a sub-Saharan African population. This variant caused downregulation of mRNA expression of DC-SIGN (Vannberg et al., 2008). Similarly the -336G allele was associated with downregulation of the symptoms of a Dengue virus infection (de Oliveira et al., 2014). In addition, a SNP at -939 demonstrated an association with tuberculosis in Indonesian populations but not in Vietnamese populations (Kobayashi et al., 2011), whereas another SNP (rs4804803) increases liver disease severity during chronic HCV infection (Ryan et al., 2010). These data demonstrate the importance of DC-SIGN during various infectious diseases.
IBD is another area of interest with regard to CLR disease association. A SNP in Dectin-1 does not affect the risk of developing colitis; however, in agreement with murine studies, it is associated with the severity of ulcerative colitis (Iliev et al., 2012). Initially, a SNP in DC-SIGN was not found to associate with ulcerative colitis or Crohn’s disease; however, upon stratification of ulcerative colitis patients by a protective HLA-DR3 allele, the SNP increased susceptibility in the HLA-DR3–positive population (Núñez et al., 2007). In addition, a CARD9 (rs10870077) SNP has also been associated with ulcerative colitis and Crohn’s disease (Zhernakova et al., 2008). These data indicate gut interactions with fungi in the gut microflora may be important for the development and/or severity of IBD.
Finally, rheumatoid arthritis (RA) is another area of interest with regard to the CLRs. Recently, Dectin-2, monocyte chemoattractant protein 1, and DC-SIGN SNPs have been linked with sex-associated differences in susceptibility to rheumatoid arthritis (Cáliz et al., 2013). In addition, a SNP in DCIR (rs2377422) has been associated with RA in a Han-Chinese population (Guo et al., 2012), and DCIR expression is increased in RA, particularly in the rheumatic joint (Eklöw et al., 2008). In contrast, SNPs in Dectin-1 were not found to be associated with susceptibility or severity of RA; however, high Dectin-1 expression was observed in synovial tissue of RA patients (Plantinga et al., 2010). These data indicate that the CLRs are important during a range of diseases, and these genetic studies validate several of the in vitro or murine findings.
V. Cytoplasmic Nucleic Acid Sensors
PRRs for nucleic acids (NA) are not restricted to endosomes, where NA sensing TLRs predominate, but also exist in the cytoplasm. The intracellular NA sensors are a more recent discovery than other PRRs but have been the subject of intensive research. Foreign and self DNA, RNA, and DNA/RNA hybrids are all ligands for PRRs in the cytoplasm, and recent functional and structural details of these receptors, as well as their signaling mechanisms, has begun to unravel their importance in a range of immunologic contexts (Fujita et al., 2007; Goubau et al., 2013; Paludan and Bowie, 2013; Bhat and Fitzgerald, 2014). It is now well established that sensing of pathogen genomes and NA replication intermediates contributes to the innate immune response against viruses, bacteria, and parasites (Goubau et al., 2013; Bhat and Fitzgerald, 2014). Indeed, current dogma indicates that sensing viral genomes is essential for a robust type I IFN response to infection with either DNA or RNA viruses, indicating the importance of these PRRs in the context of pathogen sensing (Ablasser et al., 2013b; Paludan and Bowie, 2013). In addition to sensing pathogen-derived NAs, cytoplasmic PRRs can also detect and respond to mislocalized or excessive levels of self-NAs, initiating a sterile-inflammatory response (Ablasser et al., 2013b)
A. Cytoplasmic Nucleic Acid Sensors and Their Nomenclature
1. RNA Sensors.
The cytoplasmic PRRs that sense RNA predominantly belong to the RLR family, which consists of three proteins: RIG-I, melanoma differentiation associated antigen 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2). The RLRs are a subset of the superfamily 2 family of RNA helicases and are related to the DExD/H box helicases (Goubau et al., 2013). The superfamily 2 family has a broad range of biologic functions and includes regulators of transcription and translation, microRNA processing, and innate immunity (Fairman-Williams et al., 2010). RLR proteins consist of a core helicase domain appended by signaling modules that link RNA binding to activation of the inflammatory transcription factors IRFs, AP-1, and NFκB and hence IFN-I and cytokine production (Goubau et al., 2013). The RLRs are essential for host defense against many pathogens and can sense dsRNA from various sources to carry out their function (Fujita et al., 2007).
RIG-I has also been suggested to function as a component of an inflammasome that can initiate caspase-dependent cleavage of IL-1β in response to cytoplasmic RNA (Poeck et al., 2010), although this role has subsequently also been ascribed to both NLRP3 and a member of the DExD/H box helicase family, DHX (DEAH (Asp-Glu-Ala-His) box polypeptide) 33 (Mitoma et al., 2013). Extending the function of DExD/H box helicases in RNA sensing is a further report describing a complex of three such proteins, DDX (DEAD (Asp-Glu-Ala-Asp) box helicase) 1, DDX21, and DHX36, that may function to activate IFN-I in response to cytoplasmic RNA in a cell specific manner (Zhang et al., 2011b).
2. DNA Sensors.
The nomenclature of DNA sensors is complicated by the existence of many different candidate PRRs. Two of these proteins belong to the ALR family and are expressed from a specific gene locus encoding the pyrin and HIN (hemopoietic expression, interferon-inducibility, nuclear localization) domain-containing family of genes, so named as they contain a DNA-binding HIN domain and a PYD to mediate homotypic protein–protein interactions. AIM2, the eponymous member of the ALRs, binds intracellular DNA and forms an inflammasome to drive IL-1β cleavage and secretion (Bürckstümmer et al., 2009; Fernandes-Alnemri et al., 2009; Hornung et al., 2009; Roberts et al., 2009). A second ALR, γ-interferon–inducible protein 16 (IFI16), also binds intracellular DNA but then activates IFN-I production via IRF3 and NFκB (Unterholzner et al., 2010).
DNA-dependent activator of interferons (DAI) was among the first candidate DNA sensing PRRs (Takaoka et al., 2007). DAI is not an ALR but is alternatively named Z-DNA binding protein 1 as it contains a Z-DNA binding domain, although its exact function remains controversial (Ishii et al., 2008; Wang et al., 2008; Upton et al., 2012). There are several proteins associated with cytoplasmic DNA sensing that are also associated with DNA repair in the nucleus. DNA-dependent protein kinase (DNA-PK), Ku70, Ku80, meiotic recombination 11, and Rad50 all function in the cellular response to double-strand DNA breaks but have also been described as essential for innate immune DNA sensing (Zhang et al., 2011a; Ferguson et al., 2012; Kondo et al., 2013; Roth et al., 2014). Another enzyme with a well established nuclear function is RNA-polymerase III (RNA-polIII). RNA-polIII is responsible for the transcription of small RNAs but can also function as a DNA sensor in the cytoplasm by transcribing AT-rich DNA into RNA that then acts as a RIG-I ligand (Ablasser et al., 2009; Chiu et al., 2009). More recently came the discovery of an enzyme that catalyzes the production of a cyclic-dinucleotide second messenger, cGAMP-synthase (cGAS) that drives innate immune responses to cytoplasmic DNA and certain viruses (Sun et al., 2013). The second messenger cGAMP binds directly to an adaptor protein stimulator of interferon genes (STING) to stimulate IFN-I production (Ishikawa and Barber, 2008; Ablasser et al., 2013a). A second molecule, and also a member of the DExD/H box helicase family, DDX41, has been found to act in concert with STING as a protein that can bind DNA and cyclic dinucleotides to activate IFN-I production in DCs (Zhang et al., 2011c; Parvatiyar et al., 2012).
B. Structural Biology of Cytoplasmic Nucleic Acid Sensors
Members of both the RNA and DNA sensors have proven receptive to structural studies, and these have been informative in developing a mechanistic understanding of the mode of ligand detection and receptor activation. RIG-I contains a helicase domain, two CARDs, and a C-terminal domain (CTD) that are all required for RNA binding and signaling complex-formation. Structures of RIG-I from several groups in ligand-free and -bound states have provided insights into the mechanism of receptor activation and the mode of RNA ligand binding (Lu et al., 2010; Wang et al., 2010; Civril et al., 2011; Jiang et al., 2011; Kowalinski et al., 2011; Luo et al., 2011) (Fig. 7). In the absence of ligand RIG-I adopts a relatively open structure in which the tandem CARDs interact with the α-helical helicase domain forming the basis of receptor autoinhibition (Kowalinski et al., 2011; Luo et al., 2011). Mutating Phe540 within this interface results in constitutive activation of RIG-I and presumably works as a mechanistic mirror of the conformational change that occurs upon ligand binding to the CTD and the subsequent release of the tandem CARDs (Kowalinski et al., 2011). The E3 ubiquitin ligase TRIM25 mediates K63-linked ubiquitination of the RIG-I CARDs that facilitates their tetramerization and the formation of a so-called "lock-washer" structure that serves as a scaffold for interaction with the CARDs of MAVS (mitochondrial antiviral-signaling protein) to initiate downstream signaling (Peisley et al., 2014) (Fig. 7). Although the minimum length of RNA bound by a single RIG-I molecule is only 10–15 RNA bases, RIG-I and MDA5 molecules can cooperatively interact to bind sequentially along longer RNA strands (Binder et al., 2011; Peisley et al., 2011). Both the RIG-I and MAVS CARDs have been shown to be capable of forming filamentous structures, and a recent structural analysis proposes that the RIG-I CARDs serve as a nucleator for filament formation by the MAVS CARD (Peisley et al., 2011, 2013; Wu et al., 2014) (Figs. 3 and 7). This process is analogous to that seen with the AIM2 or NLRP3 PYD and ASC (Lu et al., 2014).
Fig. 7.
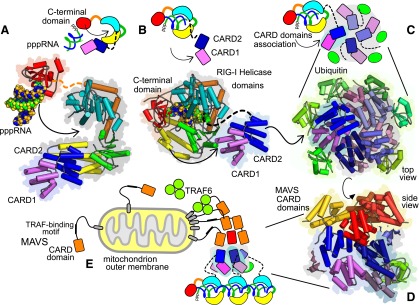
RLR domains and signaling. Recent structures of domains in RLR signaling are combined here to present the mechanisms involved. Cartoon schematics adjacent to the structures show domain associations and movements in equivalent colors. (A) Structure of RIG-I before RNA binding. RIG-I C-terminal domain (red, PDB ID 4A2X) recognizes 5′-triphosphate RNA (blue and green atoms) to engage with open form (PDB ID 4A2W) of the helicase (yellow, green, cyan) and associated N-terminal CARD domains (violet and blue). (B) RIG-I with pppRNA engaged. RNA association with helicase domains (PDB ID 3TMI) leads to displacement of CARD domains (modeled from PDB ID 4A2W, linker shown as dotted line). (C) Association of CARD domains and ubiquitin binding. Top view of the CARD domain tetramer (PDB ID 4NQK, violet and blue with other 3 copies gray shades) with associated ubiquitin molecules (green). The tetramer has a right-handed helical twist, down going clockwise. (D) Interaction with MAVS CARD domains (PDB ID 4P4H). Side view of the RIG-I CARD domain tetramer (violet, blue) with associated ubiquitin chains (green). Four MAVS CARD domains (yellow/orange) associate with the top surface. (E) Schematic view of signaling. Full-length MAVS is associated with the outer mitochondrial membrane (transmembrane helix in gray) and association with RIG-I cards propagates assembly of MAVS and TRAF signaling.
Activation of AIM2 and IFI16 by dsDNA involves direct interaction between the ligand and the HIN domain of the receptors (Fig. 8). Interestingly in structures of these complexes short DNA sequences are lined up along the binding surfaces to potentially mimic a longer sequence and recreate the presence of major and minor grooves (Jin et al., 2012; Ru et al., 2013). This suggests a mechanism for the oligomerization of the receptors during signaling. The linear arrangement of molecules is also important for inhibition of AIM2 signaling in the mouse via the action of the protein p202. Although p202 competes with AIM2 to bind DNA, it arranges its HIN domains in a different orientation to AIM2 and could feasibly intercalate into the minor groove of AIM2-bound DNA to inhibit subsequent AIM2 oligomerization and inflammasome formation (Ru et al., 2013).
Fig. 8.

Cytoplasmic DNA sensing by IFI16. Crystal structure of four HIN200 domains from IFI16 interacting with DNA (adapted from PDB ID 3RNU). IFI16 HIN200 domains interact with the backbone of the DNA molecule in a sequence-independent manner, in a similar way to AIM2 HIN200 domains.
Cytosolic DNA is also recognized by cGAS. The precise stoichiometry of the interaction is under debate; however, structural studies have shown that DNA binding occurs on the protein surface and results in a significant conformational change to the adjacent cGAS activation loop. The conformational change occurs only with B-form DNA and results in a rearrangement of the active site of cGAS and leads to the production of the second messenger 2′-3′ cGAMP, which then acts as a ligand for downstream signaling (Civril et al., 2013; Gao et al., 2013b; Kato et al., 2013; Kranzusch et al., 2013; Zhang et al., 2014b).
C. Activation and Signal Transduction of the Cytoplasmic Nucleic Acid Sensors
1. RNA Sensors.
a. Retinoic acid inducible gene I.
RIG-I was first described as a PRR in 2004 when its ability to sense RNA and the RNA viruses Newcastle disease virus and vesicular stomatis virus was discovered (Yoneyama et al., 2004). RIG-I’s role in host defense against infection has been unequivocally confirmed using both knockout mouse models and siRNA experiments in human cells. RIG-I–deficient mice are highly susceptible to numerous, although not all, RNA virus infections as well as some intracellular bacteria (summarized in Goubau et al., 2013) and are defective in cytokine and IFN-I production in response to RNA and synthetic RNA analogs (Kato et al., 2006).
Because RIG-I is such a critical PRR for antiviral host defense and one of the earliest intracellular NAs to be discovered there has been significant work in understanding the structural, biochemical, and cellular basis for RIG-I activation and signaling. Analysis of RIG-I ligand binding has revealed a unique characteristic of this PRR that allows it to discriminate between foreign and self-RNAs. RIG-I binds small double-stranded RNAs (dsRNAs) that contain either a 5′-triphosphate motif (Hornung et al., 2006; Pichlmair et al., 2006; Goubau et al., 2013) (Fig. 7) or, as has been recently postulated, a 5′-diphosphate (Goubau et al., 2014), but such RNA species are not normally present in mammalian cells thanks to RNA end capping. RIG-I contains a deep binding pocket in its CTD that allows the 5′−-triphospate dsRNA ligand to bind and make extensive contacts with its receptor. This binding, in combination with ATP hydrolysis by the helicase domain (Gee et al., 2008), allows gross structural changes in RIG-I, freeing the CARDs and initiating the next steps of the pathway (Jiang et al., 2011; Kowalinski et al., 2011; Luo et al., 2011; Peisley et al., 2014).
After dsRNA binding, interaction of RIG-I CARDs with the CARD of mitochondrion-resident adaptor protein MAVS allows further recruitment of signaling molecules. Formation of multiprotein complexes that include the adaptors TRAF2, -3, and -6 results in activation of a series of kinases that, in turn, switch on inflammatory transcription factors (Kowalinski et al., 2011; Goubau et al., 2013; Peisley et al., 2014). Both RIG-I and MAVS have been shown to form long filaments, at least in vitro, that are required for signaling activity (Patel et al., 2013; Peisley et al., 2013, 2014). The formation of these filaments is CARD dependent and full activation of signaling also involves binding to both K63-ubiquitin chains as well as covalent ubiquitin modifications by tripartite motif containing 25 and possibly other E3 ligases (Hou et al., 2011; Jiang et al., 2012; Peisley et al., 2013, 2014).
b. Melanoma differentiation associated antigen 5.
MDA5 is also cytoplasmic, functions to sense foreign RNA, consists of similar protein domains to RIG-I, and activates almost identical downstream signaling pathways. The major difference between RIG-I and MDA5 is that different RNA ligands activate these functionally similar proteins. RIG-I binds directly to small 3′-triphosphate dsRNA ends, whereas MDA5 shows a preference for long dsRNA species, allowing the functions of MDA5 and RIG-I to be nonredundant for pathogen sensing (Kato et al., 2006; Pichlmair et al., 2009; Loo and Gale, 2011). As with RIG-I, MDA5 is critical for the initiation of antiviral immune responses and is especially critical for the recognition of picornaviruses but also other RNA viruses (Kato et al., 2006; Feng et al., 2012; Goubau et al., 2013). There are both overlapping and independent sets of pathogens that can be sensed by MDA5 and RIG-I, consistent with the RNA ligands that activate these two receptors being distinct species. The consensus for viruses is that negative-stranded RNA viruses signal through RIG-I and positive-stranded viruses signal through MDA5 (Kato et al., 2006; Loo and Gale, 2011; Goubau et al., 2013). Each pathogen may produce RNA species that activates one or the other or both receptors to induce interferon production. Indeed certain large DNA viruses, such as vaccinia virus, are also detected by RNA sensors because they produce dsRNA during infection (Delaloye et al., 2009; Goubau et al., 2013). Unlike RIG-I, however, the precise RNA ligands that activate MDA5 during virus infection are less well characterized. Evidence suggests that the activation of MDA5 requires the formation of filament structures that can form along the length of a long dsRNA and that long dsRNA/MDA5 complexes are then stable enough to activate downstream signaling (Peisley et al., 2011; Wu et al., 2013a). The precise mode of self/non–self-discrimination by MDA5 is unclear, however, because there is additional evidence showing that RNAs with 2′-O-methylated 5′ caps can activate MDA5, suggesting specific modifications may be required and not only RNA length (Züst et al., 2011). After activation, the CARDs of MDA5 interact with the CARD of MAVS, and hence, activate the same range of transcription factors as RIG-I.
c. Laboratory of genetics and physiology 2.
The function of LGP2 is more enigmatic than RIG-I or MDA5, and whether it is a bona fide PRR is unclear. LGP2 also contains a helicase domain and a CTD but lacks the CARDs present in the other RLR family members (Rothenfusser et al., 2005; Yoneyama et al., 2005). Initial data indicated that LGP2 is a negative regulator of RLR signaling and may act by sequestration of RNA away from the other RLRs, consistent with its ability to bind RNA but its lack of CARDs (Rothenfusser et al., 2005; Yoneyama et al., 2005). Later information from LGP2 knockout mice and cells showed that the situation is more complex than initially thought. The physiologic function of LGP2 appears to be cell-type dependent and also may vary depending on the level of expression of the protein (Kato et al., 2006; Komuro and Horvath, 2006; Satoh et al., 2010; Childs et al., 2013; Rodriguez et al., 2014). Further analysis is required for the complete understanding of the LGP2 as a modulator of MDA5, possibly RIG-I, and how its ability to bind RNA activates its physiologic functions.
d. DEAD (Asp-Glu-Ala-Asp) box helicase 1, 21, and 36.
Although not RLRs, these three proteins are members of the DExD/H-box helicase family and have been reported to act as a complex in dendritic cells capable of activating IRF and NFκB signaling in response to foreign RNA (Zhang et al., 2011b). This proposed RNA-sensing pathway was shown to act via the signaling adaptor TRIF acting in a complex with MAVS to stimulate IFN-I production (Zhang et al., 2011b). How this protein complex affects the innate immune response to pathogens in vivo will be interesting to understand.
e. RNA inflammasomes.
Beyond the function of RNA PRRs that drive the transcription of interferons and inflammatory cytokines, there are several proteins that have been shown to act as inflammasomes activating RNA-dependent caspase-1/11 cleavage and secretion of IL-1β and IL-18. Although it is clear that RNA can activate inflammasomes and that this response can be crucial for host defense against RNA pathogens, the PRRs involved remain somewhat unclear. NLRP3 responds to multiple stimuli and can be activated by foreign RNA, although not via direct binding (Allen et al., 2009; Thomas et al., 2009; Ichinohe et al., 2010; Rajan et al., 2011). The identification of DHX33, a DExD/H-box helicase, that can bind viral and bacterial dsRNA and activate the canonical NLRP3 inflammasome pathway goes someway to explaining these original observations (Mitoma et al., 2013). However, a further pathway has also been described whereby RIG-I can itself act as an inflammasome scaffold for the detection of RNA and secretion of IL-1β (Poeck et al., 2010). Further characterization is required to dissect the physiologic functions of these RNA inflammasomes in response to pathogens.
2. DNA Sensors.
Despite historic links between DNA and inflammation, the initial mechanistic information to link the two came from studies of TLR9 and its ability to sense hypomethylated CpG DNA, a predominantly bacterial ligand (Hemmi et al., 2000). However, later studies, particularly from DNase II knockout mice and from the use of CpG-free DNA ligands, uncovered a second, TLR9-independent pathway of DNA sensing that is present in the cytoplasm rather than endosomes or plasma membranes (Kawane et al., 2014). DNase II–deficient mice die in utero of massive hepatocyte apoptosis driven by excessive levels of IFNβ (Yoshida et al., 2005; Kawane et al., 2006). This phenotype was shown to be dependent on a build-up of DNA and the presence of the interferon receptor (Yoshida et al., 2005). Later work showed that the production of IFNβ by DNA that lacks CpG motifs is driven by a cytoplasmic pathway that activates IRF3 and NFκB and is dependent on a endoplasmic reticulum–resident protein, STING (Stetson and Medzhitov, 2006; Ishikawa and Barber, 2008; Zhong et al., 2008). Cells lacking STING, the kinase TBK1, or IRF3 are defective in IFNβ production in response to cytoplasmic DNA (Ishii et al., 2006; Stetson and Medzhitov, 2006; Ishikawa and Barber, 2008), and crossing the STING knockout mouse with the DNaseII knockout resulted in rescue of the embryonic lethality of these mice (Ahn et al., 2012). STING null cells are also defective in responding to intracellular DNA-containing pathogens, such as DNA viruses (Ishikawa et al., 2009b), intracellular bacteria like L. monocytogenes (Sauer et al., 2011b), and certain parasites, such as Plasmodium sp. (Sharma et al., 2011). As with RNA sensing, the ability to detect and respond to intracellular DNA is especially critical for the production of interferons, cytokines, and chemokines in response to DNA virus infection. Indeed, STING knockout mice are highly susceptible to infection with, for example, herpes viruses (Ishikawa et al., 2009b). Signaling downstream of STING involves engagement of TBK1 and subsequent recruitment and activation of IRF3 to the C-terminal region of STING (Tanaka and Chen, 2012). TBK1 can also activate NFκB in this context, allowing the full range of IFN-I and cytokines to be produced by intracellular DNA detection (Abe and Barber, 2014). After the establishment of a TLR-independent intracellular DNA sensing pathway there followed a gold rush for the elusive PRRs that could activate the STING-dependent signaling pathway. A number of candidates have since been put forward as cytoplasmic DNA sensors.
a. DNA-dependent activator of interferons.
DAI/Z-DNA binding protein 1 was discovered as an interferon-stimulated gene (ISG) capable of sensing DNA and initiating an IRF3-dependent response in murine fibroblasts (Takaoka et al., 2007). These initial observations were carried out in specific cell lines where DAI had been knocked down by siRNA. Later work, however, using a knockout mouse and including a range of primary cell types, indicated that the production of IFN-I was not affected by loss of DAI and suggested redundancy in the DNA sensing systems (Ishii et al., 2008; Lippmann et al., 2008; Wang et al., 2008). Further observations showed that DAI is only present at a very low level, if at all, in most cell types in vivo and that to function it is required to be upregulated by an initial wave of IFN-I. These observations gave rise to the theory of primary or sentinel sensors, whose function may be enhanced by the presence of secondary sensors. DAI may therefore exist as a secondary sensor, a backup system that is part of the antiviral state initiated by IFN-I production. Another possibility is that DAI recognizes specific DNA ligands that have yet to be fully identified that may include specific sequences of Z-DNA, although the physiologic context of this remains unknown. Alternatively, DAI may not function in vivo as a DNA PRR that activates IRF-3 but may be involved in the initiation of receptor-interacting kinase-dependent NFκB activation and necroptosis (Kaiser et al., 2008; Rebsamen et al., 2009; Upton et al., 2012).
b. RNA-polymerase III.
RNA-pol III was identified in two independent studies as a DNA PRR that functions by providing RNA ligands for RIG-I. Transcription of AT rich DNA by RNA-pol III into dsRNA in the cytoplasm leads to RIG-I–dependent IFNβ production (Ablasser et al., 2009; Chiu et al., 2009). However, the DNA ligand used in this discovery was poly(dA:dT), a very specific form of synthetic DNA, and the only cell type in which this pathway has been shown to function is HEK293, which are probably neuroepithelial in origin (Shaw et al., 2002). As such a physiologic function of RNA-polII–dependent DNA sensing has yet to be defined (Unterholzner, 2013).
c. γ-Interferon–inducible protein 16.
IFI16’s function in DNA sensing was initially described in macrophages where it contributes to IFN-I production in response to cytosolic DNA and HSV-1 infection (Unterholzner et al., 2010). Further analysis of this response indicated its importance in the detection of HSV-1 and its ability to sense the cDNA intermediates of retrovirus replication (Paludan et al., 2011; Jakobsen et al., 2013; Monroe et al., 2014). IFI16 can activate IRF3 and NFκB-dependent signaling downstream of synthetic and viral dsDNA (Unterholzner et al., 2010) and functions via the HIN200 domain binding to DNA (Jin et al., 2012) (Fig. 8) and forming filaments along the length of the DNA molecule in a manner dependent on the PYRIN domain (Jin et al., 2012; Morrone et al., 2014). The signaling mechanism initiated by IFI16 binding DNA is unclear and may involve transcriptional regulation at multiple levels (Thompson et al., 2014). IFI16 is present in the nucleus where it also functions in DNA repair and p53-dependent stress responses, suggesting it may function as a nuclear DNA PRR (Johnstone et al., 2000; Aglipay et al., 2003). Indeed some evidence indicates IFI16 can detect HSV-1 genomes in the nucleus before shuttling to the cytoplasm in a manner dependent on acetylation to activate STING-dependent signaling (Li et al., 2012a; Orzalli et al., 2012). Dissecting the precise physiologic functions of IFI16 in vivo is complicated by large differences in the PHYIN family gene locus between humans and other mammals, particularly mice where this locus has undergone a large expansion to include at least twice as many genes with overlapping functions (Brunette et al., 2012; Cridland et al., 2012).
d. Absent in melanoma-2.
The ALR AIM-2 is a second member of the PHYIN family of proteins that functions as a DNA PRR. Unlike IFI16, AIM-2 is not implicated in IFN-I production but rather activates inflammasome-dependent IL-1β production (Bürckstümmer et al., 2009; Fernandes-Alnemri et al., 2009; Hornung et al., 2009; Roberts et al., 2009). AIM2 knockout mice are highly susceptible to infection with certain intracellular bacteria and lack induction of IL-1β in response to DNA, cementing the place of this ALR as an essential DNA PRR for host defense against infection (Fernandes-Alnemri et al., 2010; Jones et al., 2010; Rathinam et al., 2010). The mechanism of AIM2 signaling is well defined and characteristic of classic inflammasome activation. Upon DNA binding to the HIN200 domain of AIM2 (Jin et al., 2012), its PYD interacts with the ASC PYD, possibly serving as a nucleation point for ASC oligomerization (Lu et al., 2014), resulting in speck formation and caspase-1 recruitment and activation (Schroder et al., 2009; Latz et al., 2013).
e. DNA-dependent protein kinase.
DNA-PK is a heterotrimeric protein complex consisting of a catalytic subunit DNA-PKcs and two Ku proteins, Ku70 and Ku80. Together these three proteins form a critical component of the nonhomologous end joining pathway of double-stranded DNA break repair in the nucleus (Lieber et al., 2003). More recently reports of DNA-PK functioning as a cytoplasmic PRR for DNA sensing have re-enforced an emerging link between DNA damage responses and innate immunity (Zhang et al., 2011a; Ferguson et al., 2012). DNA-PK was found to activate DNA-dependent IFN-I production in murine fibroblasts and a type-III interferon response in human embryonic kidney 293 cells (X Zhang et al., 2011a; Ferguson et al., 2012). Furthermore, DNA-PK was found to regulate chemokine and cytokine production in response to DNA in vivo and to vaccinia virus and HSV-1 in fibroblasts and in mice (Ferguson et al., 2012).
f. cGAMP-synthase.
cGAS was identified more recently as a cytoplasmic enzyme that can detect and respond to foreign DNA and activate the IRF-dependent production of IFNβ (Sun et al., 2013). DNA binding induces activation of cGAS and results in its production of a cyclic dinucleotide 2′3′-cGAMP (cyclic [G(2′,5′)pA(3′,5′)p]) (Ablasser et al., 2013a; Diner et al., 2013; Wu et al., 2013b; Zhang et al., 2013), a second messenger that binds to a dimer of STING (Huang et al., 2012; Ouyang et al., 2012; Shang et al., 2012; Shu et al., 2012). cGAMP binding alters the conformation of STING in a manner that initiates downstream signaling and is coincident with the relocalization of STING to a currently unidentified membranous compartment (Ishikawa and Barber, 2008). cGAS has been shown to be essential for DNA-dependent IFN-I production in various murine and human cell types (Li et al., 2013b; Sun et al., 2013) and for the sensing of HSV-1 viruses in vivo (Li et al., 2013b) as well as retroviruses including HIV (Gao et al., 2013a; Lahaye et al., 2013). One intriguing aspect of this discovery is that other dinucleotides of bacterial rather than host cell origin, such as cyclic-di-GMP, can also activate STING, indicating a broader role for STING in sensing intracellular bacteria (McWhirter et al., 2009; Burdette et al., 2011; Jin et al., 2011; Diner et al., 2013; Gao et al., 2013b) and perhaps cGAS as evolutionary adaptation, co-opting the STING pathway (Wu et al., 2014). The structure of cGAS, solved independently by several groups, revealed a structure similar to the 2′-5′-oligoadenylate synthetase family of antiviral nucleotidyltransferases that belong to the ISG group and suggests this family has a conserved function in cell intrinsic immunity (Civril et al., 2013; Kranzusch et al., 2013; Li et al., 2013a; Hornung et al., 2014; Zhang et al., 2014b). cGAS itself is an ISG and its function in antiviral immunity was discovered independently in broad screen of the antiviral activities of ISGs (Schoggins et al., 2011). Interestingly, in a follow up study cGAS was shown to antagonize certain RNA viruses, indicating other functions for this enzyme in antiviral immunity (Schoggins et al., 2014). The discovery of a unique enzyme with antiviral properties that produces a second messenger that can act with defined mechanism to induce IFN-I production provides several points for potential pharmacological exploitation.
g. DEAD (Asp-Glu-Ala-Asp) box helicase 41.
A member of the DEXDc family of DExD/H-box helicases that functions in DCs, DDX41 binds both DNA and bacterial cyclic-dinucleotides to activate STING-dependent IFN-I production (Zhang et al., 2011c; Parvatiyar et al., 2012). The ability of DDX41 to sense foreign DNA is associated with HSV-1 sensing (Zhang et al., 2011c), whereas its direct binding to cyclic-di-AMP or cyclic-di-GMP is linked to detection of L. monocytogenes (Parvatiyar et al., 2012). DDX41 was shown to exist in the same subcellular compartment as STING, and the two proteins may interact directly (Zhang et al., 2011c). The identification of a second cyclic-dinucleotide binding protein in the STING-dependent pathway raises some interesting questions about the functions of these two proteins, particularly given that STING has also been shown to bind DNA directly (Abe et al., 2013). Further work is required to understand the interplay and physiologic functions of these two proteins that are able to bind DNA and nucleotide second messengers as well as each other to assist pathogen sensing by DCs.
A selection of other cytoplasmic proteins has been linked with DNA sensing in various systems. These include leucine-rich repeat (in FLII) interacting protein 1, a leucine-rich repeat-containing protein that can bind DNA and RNA in the cytoplasm and activate a β-catenin–dependent IFNβ production (Yang et al., 2010), and meiotic recombination 11 and RAD50, two proteins that function in the double strand DNA repair that may also function in innate sensing of DNA to activate IRF3 and NFκB, respectively (Kondo et al., 2013; Roth et al., 2014); high mobility group box proteins and LSM14A, SCD6 homolog A, may also assist in the sensing of intracellular NAs but in a manner that is less well understood (Yanai et al., 2009; Li et al., 2012b).
D. Cytoplasmic Nucleic Acid Sensor Mutations and Disease
There is a strong connection between polymorphism in genes associated with host defense and autoimmunity and the RLRs are no exception, particularly for Mda5. Several independent screens identified Mda5 polymorphisms associated with type I diabetes (Smyth et al., 2006; Concannon et al., 2008; Liu et al., 2009; Nejentsev et al., 2009). Mechanistically, it has been suggested that loss of functional MDA5 allows pancreatic islet damage by β-cell tropic viruses, although this remains to be proven (Loo and Gale, 2011; McCartney et al., 2011). On the other hand, gain of function MDA5 missense polymorphisms have also been found associated with a type I interferonopathy, Aicardi-Goutieres syndrome (AGS) (Rice et al., 2014). AGS is caused by type I interferon that can be produced as a direct result of a buildup of intracellular DNA (Behrendt and Roers, 2014), but the discovery of Mda5 alleles that can also initiate this disease indicates a general association with hyperactive intracellular nucleic acid sensing. Such a gain of function may confer enhanced protection against specific pathogens by driving a more rapid host innate immune response, the trade-off being enhanced susceptibility to autoinflammatory disease. Indeed, several of the patients in the study by Rice et al. (2014) show clinical signs of SLE, an observation confirmed by other studies of different Mda5 polymorphisms (Cen et al., 2013; Funabiki et al., 2014). Mda5 then has a clear and interesting association with autoimmunity and provides a useful example of how different selective pressures can affect both the provision of an appropriate inflammatory response to infection and the maintenance of tissue homeostasis. On the other hand, although numerous RigI polymorphisms in the human genome are present in SNP databases, only one common variant (R7C, >5% prevalence) is reported, and the functional consequence of this alteration is unclear, having different effects in different experimental systems. Loss-of-function missense polymorphisms appear to exist in the RigI gene, although these appear rare and have not to date been associated with disease susceptibility (Pothlichet et al., 2009; Shigemoto et al., 2009; Hu et al., 2010).
Innate immune DNA sensing is heavily associated with AGS and SLE. AGS is directly caused by mutations in proteins that control nucleic acid levels and quality. Disease-causing allelic variants in three prime repair exonuclease 1, ribonuclease H2 (subunits A, B, or C), SAM domain, and HD domain-containing protein 1, or RNA-specific adenosine deaminase are identified in approximately 90% of individuals with AGS (Behrendt and Roers, 2014). For example, one function of three prime repair exonuclease is to digest unwanted cytoplasmic DNA, acting as a negative regulator of innate immune DNA sensing and helping to set a threshold for DNA-triggered type 1 interferon production. Patients lacking the ability to successfully clear excess DNA trigger cytoplasmic nucleic acid sensing PRRs causing the devastating AGS pathology (Crow et al., 2006). In a similar vein, DNA-PK, and particularly Ku70/80, have a long-held association with SLE as one of the archetypical SLE autoantibodies present in SLE patients are against the Ku proteins (Reeves, 1992; Satoh et al., 1996). Several polymorphisms in the Ifi16 gene are associated with SLE and, in addition, IFI16 autoantibodies are present in the sera of some SLE patients (Seelig et al., 1994; Mondini et al., 2007; Kimkong et al., 2010). Although the molecular mechanisms that link DNA sensing with SLE are enigmatic, this is an active area of current research.
V. Pharmacological Manipulation of Pattern Recognition Receptors
The close association of PRRs with human disease, particularly chronic inflammatory disorders, makes them an ideal candidate for direct pharmacological targeting. Drugs with PRR specificity make an attractive proposition in terms of providing disease-specific treatments, potentially reducing treatment costs by replacing antibody based anti-inflammatory treatments, and reducing the repertoire of side effects that can result from broad spectrum anti-inflammatory therapies. For example, although glucocorticosteroids, IL-IR antagonists, and anti-TNF treatments can all produce clinical remission across a range of inflammatory disorders they can also induce widespread immunosuppression and an increased susceptibility to infection.
There are, however, challenges with developing novel pharmaceuticals that target PRRs. Classic quantitative pharmacology studies, such as binding data, are in their infancy in PRR biology. Many of the ligands that bind TLRs are molecules made by pathogens that are often contaminated with other pathogen molecules. Synthetic ligands are often complex molecules, for example lipid A, that can have high levels of nonspecific binding or make label attachment challenging. Successful protein expression of PRRs is difficult and often results in low yields that restrict the cost effectiveness of large-scale screening using techniques such as surface plasmon resonance. For many of the NLRs the ligands remain unknown. Much data have been generated from functional studies, but ligand dose response analysis is often not performed. TLR activation, for example, generally gives sharp concentration-dependent response curves as expected if oligomeric signaling complexes are formed, because dose relationships would be expected to be nonlinear with increasing ligand concentration. The lack of quantitative data also makes drug classification difficult especially for ligands with submaximal activation of PRRs. Does submaximal activation of a PRR by a ligand in a population of cells suggest the compound is a partial agonist or that not all the cells in a population have responded such that a reduced response will occur? Application of quantitative pharmacological and single cell analysis techniques to PRR biology should become more feasible as protein expression and image analysis techniques are optimized.
Nevertheless there are many drugs targeting PRRs under development to treat a range of human diseases. These include novel adjuvants, anticancer treatments, immunotherapeutic agents. and drugs targeting chronic respiratory diseases (O’Neill et al., 2009; Connolly and O’Neill, 2012). Although few are currently approved for use in patients, those that are in successful clinical use include Imiquimod (3M Pharmaceuticals), a TLR7 agonist used for the treatment of genital warts (Hemmi et al., 2002), and the vaccine adjuvants monophosphoryl lipid A (a TLR4 partial agonist) and alum (a proposed NLRP3 agonist) (Rappuoli et al., 2011).
To date, particular focus has been placed on the manipulation of TLR signaling with a number of therapeutic compounds targeting TLRs 4 and 2 entering clinical trials. The TLR4 antagonist (Eritoran; Eisai Co., Tokyo, Japan), however, failed to show efficacy in phase III clinical trials to treat sepsis, but another AV411 (Ibudilast; MediciNova Inc, La Jolla, CA) is being developed for neuropathic pain relief because opioids may induce proinflammatory effects through activation of TLR4 (Tanga et al., 2005; Connolly and O’Neill, 2012). It is controversial, however, as to whether opioids bind directly to MD2/TLR4 or whether modulation of TLR4 signaling pathways occurs through an indirect mechanism (Wang et al., 2012; Zwicker et al., 2014). Similarly, the classic opioid antagonists naloxone and naltrexone have been reported to antagonize TLR4 in some studies yet have no effect in others (Watkins et al., 2009; Skolnick et al., 2014).
TAK 242 (Resatorvid; Takeda Pharmaceutical Company Ltd, Deerfield, IL), a small molecule inhibitor of TLR4 that binds to Cys747 in the TLR4-TIR domain (Takashima et al., 2009), also failed in sepsis clinical trials, highlighting the complexity of targeting these signaling pathways for disease treatments. OPN305, developed by Opsona Therapeutics (Dublin, Ireland), a humanized anti-TLR2 monoclonal antibody that blocks TLR2, is in clinical trials for use in ischemia/reperfusion injuries (cardiac, renal, and organ transplantation) (Connolly and O’Neill, 2012; Navi et al., 2013). SMP105 (Dainippon Sumitomo Pharma, Tokyo, Japan), a TLR2 agonist, enhanced levels of cytotoxic T lymphocytes and IFN-producing cells to reduce tumor growth in mouse cancer models and is currently being investigated as an anticancer agent (Connolly and O’Neill, 2012).
The clinical success of Imiquimod has led to the production of other compounds targeting TLRs 7 and 8. Resiquimod (R-848; TLR7 and TLR8 agonist) showed a lack of efficacy in phase III trials, whereas 852A, a TLR7 agonist, is under study for use in cancer patients for its stimulatory effects on plasmacytoid dendritic cells. Another TLR7 agonist, ANA773, is being developed for the treatment of patients with chronic HCV infection and cancer (Connolly and O’Neill, 2012).
TLRs are also being investigated as treatment targets for allergy, partly due to the ability of receptors such as TLR7 to shift the balance of immune responses from Th2 to Th1 in the asthmatic airway (Drake et al., 2012). VTX1463, a TLR8 agonist, has similar immunomodulatory properties and is in development for the treatment of allergic rhinitis. Similarly, synthetic TLR9 agonists [AVE0675, SAR21609 (Sanofi-Aventis/Coley Pharmaceuticals, Bridgewater, NJ), QAX935 (Idera Pharmaceuticals, Cambridge, MA)] are also in development for treatment of allergic diseases. Meanwhile, Pollinex Quattro (Allergy Therapeutics, West Sussex, UK), a combination of the classic adjuvant monophosphoryl lipid A and the allergen ragweed pollen extract, is being trailed for the treatment of seasonal allergic rhinitis (Connolly and O’Neill, 2012).
TLR9 agonists (Dynavax; Idera Pharmaceuticals) are being developed as cancer therapeutics, adjuvants, and antiviral agents as well as for the treatment of steroid-resistant ulcerative colitis (InDex Pharmaceuticals, Stockholm, Sweden). Antagonists of TLR7, 8, and 9 (Dynavax; Idera Pharmaceuticals) are in trials for use in chronic inflammatory (psoriasis) and SLE diseases (Connolly and O’Neill, 2012). The similarity in ligand specificity between TLR7, TLR8, and to a lesser extent TLR9, means that care has to be taken in the evaluation of potential agonists and antagonists. Use of knockout cells is crucial to ensure specific, and not redundant, activation of signaling pathways, and the improved availability of recombinant receptors will assist in the more accurate determination of binding specificity, interaction affinity, and binding site location. Rintatolimod (Hemispherx Biopharma, Philadelphia, PA), a synthetic TLR3 agonist, is being developed for the treatment of a range of disorders including chronic fatigue syndrome, hepatitis, human papilloma virus, HIV, influenza, and cancer. TLR5 agonists (CBLB502, VAX102) are being developed for ischemia/reperfusion injury, cancer, and as flu vaccine adjuvants (Connolly and O’Neill, 2012).
The NLRs are also being looked at as possible targets for the development of new vaccine adjuvants. NOD2 responds to stimulation with the commonly used research vaccine complete Freunds adjuvant, and NLRP3 has been proposed to respond to alum, an adjuvant widely used in licensed human vaccines (Eisenbarth et al., 2008; Hornung et al., 2008; Li et al., 2008; Magalhaes et al., 2008; Sharp et al., 2009). Although MDP has long been known to be the active component of complete Freunds adjuvant, it was only recently shown that its functionality as an adjuvant is NOD2 dependent (Magalhaes et al., 2008). The molecular basis by which alum works as an adjuvant via NLRP3 is currently unknown, but it may be connected to the production and adjuvant-like activity of IL-1β (Staruch and Wood, 1983; Higgins and Mills, 2010). However, there is controversy as to whether alum does indeed specifically activate NLRP3 or whether this is simply an accessory effect (Oleszycka and Lavelle, 2014). Recently it was proposed that alum may actually function by stimulating DNA release from dying cells and thereby exert its adjuvant activity via indirect stimulation of nucleic acid sensing PRRs (Marichal et al., 2011). Deliberate and controlled activation of NOD2 and inflammasome-mediated IL-1β production is an attractive property for new adjuvants and has the potential to develop into a highly productive area of deliberative pharmacological manipulation of the immune system.
The endocytic function of CLRs provides potential for them to be used therapeutically to target antigens to antigen presenting cells. Several studies have examined this, and targeting an antigen to DCs via DEC-205 or the mannose receptor has been shown to result in cross-presentation of the antigen to CD8+ T cells in addition to the CD4+ T cell responses that are observed without specifically targeting the antigen via a CLR. NY-ESO-1, a cancer testis antigen that is widely used in cancer vaccine clinical trials, was fused to human antibodies to DEC-205 and the mannose receptor. Both of these products induced CD4 and CD8 T-cell responses (Tsuji et al., 2011). Phase I and phase II clinical trials are currently underway to examine the effect of CDX-1401, a DEC-205/NY-ESO-1 fusion protein (Celldex Therapeutics, Hampton, NJ) ,in patients with different cancers (myelodysplastic syndrome, acute myeloid leukemia, ovarian, fallopian tube, or primary peritoneal cancer in remission). In addition to cancer vaccines, an antibody to DEC-205 has also been fused to the HIV gag p24 protein to generate DCVax-001, which is a vaccine designed to prevent and potentially treat HIV infection (Bozzacco et al., 2007). Other adjuvants are used in each of these studies in addition to the targeted antigen.
β-Glucans (Dectin-1 ligands) are being used to prime/boost an immune response in combination with other cancer therapies. Various clinical trials are either completed or underway to examine their potency in this regard. Phase I clinical trials testing the safety of Imprime PGG, a β-1,3/1,6-glucan (Biothera, Eagan, MN) has been completed, and several Phase II/III clinical trials using this product are currently underway or completed in patients with various cancers (Non-Hodgkin lymphoma, chronic lymphocycitc leukemia, non-small cell lung cancer, colorectal cancer). Another β-1,3/1,6-glucan preparation, MacroForce plus IP6 (ImmuDyne, Florence, KY) has also been used in a Phase I/II clinical trial in patients with advanced malignancies.
Considering the more recent discovery of many of the intracellular sensors of nucleic acids, their application has yet to be fully investigated. However, the potential for therapeutic exploitation of these pathways is clear. First, RLR agonists such as short RNAs with 5′-triphosphates could make potential broad ranging antiviral therapies or vaccine adjuvants (Saito et al., 2008; Chakravarthy et al., 2010; Luke et al., 2011; Goulet et al., 2013; Martínez-Gil et al., 2013; Olagnier et al., 2014). Because DNA vaccination relies on the inflammation provided by intracellular DNA sensing pathways (Ishii et al., 2008; Ishikawa et al., 2009b), stimulation of intracellular DNA PRRs also provides a logical mechanism to improve DNA vaccines and to provide novel vaccine adjuvants in general. STING agonists such as cGAMP, 10-carboxymethyl-9-acridanone, and 5,6-dimethyl-9-oxo-9H-xanthen-4-yl)-acetic acid may be promising in this area (Roberts et al., 2007; Cavlar et al., 2013; Li et al., 2013b).
VI. In Conclusion
PRRs are without question critical to the development, maintenance, and resolution of a wide range of acute and chronic conditions. As our understanding of their molecular functionality continues to improve, we will not only come to understand how these receptors contribute to disease phenotypes but also develop ways to therapeutically target the signaling pathways to our own advantage. Whether this pharmacological manipulation will result in suppression or activation of a given pathway will entirely be dependent on the desired clinical outcome and we may well see the development of both agonist and antagonist molecules for the same target receptor. What we can be certain of is that PRRs will remain at the forefront of therapeutic research across a broad spectrum of diseases.
Acknowledgments
The authors thank members of the Bryant, Orr, Ferguson, and Monie groups for insight and discussion of the manuscript.
Abbreviations
- AGS
Aicardi-Goutieres syndrome
- AIM
absent in melanoma
- ALR
AIM2-like receptor
- ASC
apoptosis-associated speck-like protein containing a CARD
- CA-074-Me
methyl 1-[3-methyl-2-[[3-(propylcarbamoyl)oxirane-2-carbonyl]amino]pentanoyl]pyrrolidine-2-carboxylate
- CARD
caspase activation and recruitment domain
- cGAS
cGAMP-synthase
- CRD
carbohydrate recognition domain
- CLR
C-type lectin-like receptor
- CRX-527
(2S)-2-[[(3R)-3-decanoyloxytetradecanoyl]amino]-3-[(2R,3R,4R,5S,6R)-3-[[(3R)-3-decanoyloxytetradecanoyl]amino]-4-[(3R)-3-decanoyloxytetradecanoyl]oxy-6-(hydroxymethyl)-5-phosphonooxyoxan-2-yl]oxypropanoic acid
- CTD
C-terminal domain
- CTLD
C-type lectin domain (or C-type lectin-like domain)
- DAI
DNA-dependent activator of interferons
- DC
dendritic cell
- DCIR
dendritic cell immune receptor
- DC-SIGN
dendritic cell specific intracellular cell adhesion molecule-3 grabbing nonintegrin
- DD
death domain
- DDX
DEAD (Asp-Glu-Ala-Asp) box helicase
- Dectin
dendritic cell–associated C-type lectin
- DHX
DEAH (Asp-Glu-Ala-His) box polypeptide
- DNA-PK
DNA-dependent protein kinase
- FcRγ
Fc receptor γ
- HCV
hepatitis C virus
- HIN
hemopoietic expression, interferon-inducibility, nuclear localization
- HIV
human immunodeficiency virus
- HD
helical domain
- HSP
heat shock protein
- HSV
herpes simplex virus
- IBD
inflammatory bowel disease
- ie-DAP
γ-d-glutamyl-meso-diaminopimelic acid
- IFI16
γ-interferon–inducible protein 16
- IFN
interferon
- IL
interleukin
- IRAK
IL-1 receptor-associated kinase
- IRF
interferon response factor
- ISG
interferon-stimulated gene
- ITAM
immunoreceptor tyrosine-based activation motif
- ITIM
immunoreceptor tyrosine-based inhibitory motif
- JNK
c-Jun N-terminal kinase
- KSR
kinase suppressor of Ras
- LGP2
laboratory of genetics and physiology 2
- LPS
lipopolysaccharide
- LRR
leucine-rich repeat
- Mal
MyD88 adapter-like
- MALT1
mucosa-associated lymphoid tissue lymphoma translocation protein 1
- MAPK
mitogen-activated protein kinase
- MAVS
mitochondrial antiviral-signaling protein
- MCL
macrophage C-type lectin
- MDA5
melanoma differentiation associated antigen 5
- MD-2
myeloid differentiation protein 2
- MDP
muramyl dipeptide
- MyD88
myeloid differentiation primary response gene 88
- NA
nucleic acid
- NAIP
neuronal apoptosis inhibitory protein
- NFκB
nuclear factor κB
- NLR
nucleotide-binding domain and leucine-rich repeat-containing receptor
- NLRA
NLR containing an acidic domain
- NLRB
NLR containing baculovirus inhibitor of apoptosis repeats
- NLRC
NLR containing a CARD
- NLRP
NLR containing a pyrin
- NLRX1
NLR family member X1
- NBD
nucleotide-binding domain
- NOD
nucleotide oligomerization domain
- PDB
protein data bank
- PIDD
p53-induced death domain protein
- PKC
protein kinase C
- PRR
pattern recognition receptor
- PYD
pyrin domain
- RA
rheumatoid arthritis
- RIG-I
retinoic acid inducible gene I
- RIP2
receptor interacting protein 2
- RLR
RIG-I–like receptor
- ROS
reactive oxygen species
- RSV
respiratory syncitial virus
- SHP
Src homology region 2 domain-containing phosphatase
- siRNA
small interfering RNA
- SLE
systemic lupus erythromatosus
- SNP
single nucleotide polymorphism
- STING
stimulator of interferon genes
- Syk
spleen tyrosine kinase
- TAK
transforming growth factor-β–activated kinase
- TBK1
TANK-binding kinase 1
- TDB
trehalose-6,6′-dibehenate
- TDM
trehalose dimycolate
- TIR
Toll-IL1 receptor
- TLR
Toll-like receptor
- TNF
tumor necrosis factor
- TRAM
TRIF-related adapter molecule
- TRAF
TNF receptor–associated factor
- TRIF
TIR-domain protein inducing interferon
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Bryant, Orr, Ferguson, Symmons, Boyle, Monie.
Footnotes
This work was supported by the Wellcome Trust [Grants WT085090MA (to T.P.M.) and WT081744/Z/06/Z (to C.E.B.)]; the UK Medical Research Council [Grant G1000133 (to C.E.B.)]; and a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and Royal Society [Grant 099953/Z/12/Z (to S.O.)]. NC-IUPHAR is supported in part by Wellcome Trust Grant 099156/Z/12/Z.
References
- Abe T, Barber GN. (2014) Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol 88:5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe T, Harashima A, Xia T, Konno H, Konno K, Morales A, Ahn J, Gutman D, Barber GN. (2013) STING recognition of cytoplasmic DNA instigates cellular defense. Mol Cell 50:5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. (2009) RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol 10:1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner K-P, Ludwig J, Hornung V. (2013a) cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498:380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Hertrich C, Waßermann R, Hornung V. (2013b) Nucleic acid driven sterile inflammation. Clin Immunol 147:207–215. [DOI] [PubMed] [Google Scholar]
- Aglipay JA, Lee SW, Okada S, Fujiuchi N, Ohtsuka T, Kwak JC, Wang Y, Johnstone RW, Deng C, Qin J, et al. (2003) A member of the Pyrin family, IFI16, is a novel BRCA1-associated protein involved in the p53-mediated apoptosis pathway. Oncogene 22:8931–8938. [DOI] [PubMed] [Google Scholar]
- Ahn J, Gutman D, Saijo S, Barber GN. (2012) STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci USA 109:19386–19391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ, CGTP Collaborators (2013) The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol 170:1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. (2001) Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738. [DOI] [PubMed] [Google Scholar]
- Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP-Y. (2009) The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30:556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen-Nissen E, Hawn TR, Smith KD, Nachman A, Lampano AE, Uematsu S, Akira S, Aderem A. (2007a) Cutting edge: Tlr5-/- mice are more susceptible to Escherichia coli urinary tract infection. J Immunol 178:4717–4720. [DOI] [PubMed] [Google Scholar]
- Andersen-Nissen E, Smith KD, Bonneau R, Strong RK, Aderem A. (2007b) A conserved surface on Toll-like receptor 5 recognizes bacterial flagellin. J Exp Med 204:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA. (2000) TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet 25:187–191. [DOI] [PubMed] [Google Scholar]
- Bae JY, Park HH. (2011) Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J Biol Chem 286:39528–39536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamias G, Cominelli F. (2006) Novel strategies to attenuate immune activation in Crohn’s disease. Curr Opin Pharmacol 6:401–407. [DOI] [PubMed] [Google Scholar]
- Barnich N, Aguirre JE, Reinecker H-C, Xavier R, Podolsky DK. (2005) Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-kappaB activation in muramyl dipeptide recognition. J Cell Biol 170:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, Barberà-Cremades M, Yagüe J, Ruiz-Ortiz E, Antón J, et al. (2014) The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 15:738–748. [DOI] [PubMed] [Google Scholar]
- Barrett NA, Rahman OM, Fernandez JM, Parsons MW, Xing W, Austen KF, Kanaoka Y. (2011) Dectin-2 mediates Th2 immunity through the generation of cysteinyl leukotrienes. J Exp Med 208:593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez G, Hornung V. (2011) Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 187:613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behler F, Maus R, Bohling J, Knippenberg S, Kirchhof G, Nagata M, Jonigk D, Izykowski N, Mägel L, Welte T, et al. (2015) Macrophage-inducible C-type lectin Mincle-expressing dendritic cells contribute to control of splenic Mycobacterium bovis BCG infection in mice. Infect Immun 83:184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behler F, Steinwede K, Balboa L, Ueberberg B, Maus R, Kirchhof G, Yamasaki S, Welte T, Maus UA. (2012) Role of Mincle in alveolar macrophage-dependent innate immunity against mycobacterial infections in mice. J Immunol 189:3121–3129. [DOI] [PubMed] [Google Scholar]
- Behrendt R, Roers A. (2014) Mouse models for Aicardi-Goutières syndrome provide clues to the molecular pathogenesis of systemic autoimmunity. Clin Exp Immunol 175:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand MJM, Doiron K, Labbé K, Korneluk RG, Barker PA, Saleh M. (2009) Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 30:789–801. [DOI] [PubMed] [Google Scholar]
- Bhat N, Fitzgerald KA. (2014) Recognition of cytosolic DNA by cGAS and other STING-dependent sensors. Eur J Immunol 44:634–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielig H, Rompikuntal PK, Dongre M, Zurek B, Lindmark B, Ramstedt M, Wai SN, Kufer TA. (2011) NOD-like receptor activation by outer membrane vesicles from Vibrio cholerae non-O1 non-O139 strains is modulated by the quorum-sensing regulator HapR. Infect Immun 79:1418–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder M, Eberle F, Seitz S, Mücke N, Hüber CM, Kiani N, Kaderali L, Lohmann V, Dalpke A, Bartenschlager R. (2011) Molecular mechanism of signal perception and integration by the innate immune sensor retinoic acid-inducible gene-I (RIG-I). J Biol Chem 286:27278–27287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas A, Liu Y-J, Hao L, Mizoguchi A, Salzman NH, Bevins CL, Kobayashi KS. (2010) Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci USA 107:14739–14744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas A, Petnicki-Ocwieja T, Kobayashi KS. (2012) Nod2: a key regulator linking microbiota to intestinal mucosal immunity. J Mol Med (Berl) 90:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius AL, Beutler B. (2010) Intracellular toll-like receptors. Immunity 32:305–315. [DOI] [PubMed] [Google Scholar]
- Bloem K, Vuist IM, van den Berk M, Klaver EJ, van Die I, Knippels LMJ, Garssen J, García-Vallejo JJ, van Vliet SJ, van Kooyk Y. (2014) DCIR interacts with ligands from both endogenous and pathogenic origin. Immunol Lett 158:33–41. [DOI] [PubMed] [Google Scholar]
- Boneca IG, Dussurget O, Cabanes D, Nahori M-A, Sousa S, Lecuit M, Psylinakis E, Bouriotis V, Hugot J-P, Giovannini M, et al. (2007) A critical role for peptidoglycan N-deacetylation in Listeria evasion from the host innate immune system. Proc Natl Acad Sci USA 104:997–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonham KS, Orzalli MH, Hayashi K, Wolf AI, Glanemann C, Weninger W, Iwasaki A, Knipe DM, Kagan JC. (2014) A promiscuous lipid-binding protein diversifies the subcellular sites of toll-like receptor signal transduction. Cell 156:705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzacco L, Trumpfheller C, Siegal FP, Mehandru S, Markowitz M, Carrington M, Nussenzweig MC, Piperno AG, Steinman RM. (2007) DEC-205 receptor on dendritic cells mediates presentation of HIV gag protein to CD8+ T cells in a spectrum of human MHC I haplotypes. Proc Natl Acad Sci USA 104:1289–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. (2003) Dectin-1 mediates the biological effects of beta-glucans. J Exp Med 197:1119–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J, O’Callaghan CA, Marshall ASJ, Gilbert RJC, Siebold C, Gordon S, Brown GD, Jones EY. (2007) Structure of the fungal beta-glucan-binding immune receptor dectin-1: implications for function. Protein Sci 16:1042–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunette RL, Young JM, Whitley DG, Brodsky IE, Malik HS, Stetson DB. (2012) Extensive evolutionary and functional diversity among mammalian AIM2-like receptors. J Exp Med 209:1969–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, et al. (2009) An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol 10:266–272. [DOI] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. (2011) STING is a direct innate immune sensor of cyclic di-GMP. Nature 478:515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buwitt-Beckmann U, Heine H, Wiesmüller KH, Jung G, Brock R, Akira S, Ulmer AJ. (2006) TLR1- and TLR6-independent recognition of bacterial lipopeptides. J Biol Chem 281:9049–9057. [DOI] [PubMed] [Google Scholar]
- Buwitt-Beckmann U, Heine H, Wiesmüller KH, Jung G, Brock R, Akira S, Ulmer AJ. (2005a) Toll-like receptor 6-independent signaling by diacylated lipopeptides. Eur J Immunol 35:282–289. [DOI] [PubMed] [Google Scholar]
- Buwitt-Beckmann U, Heine H, Wiesmüller KH, Jung G, Brock R, Ulmer AJ. (2005b) Lipopeptide structure determines TLR2 dependent cell activation level. FEBS J 272:6354–6364. [DOI] [PubMed] [Google Scholar]
- Cáliz R, Canet LM, Lupiañez CB, Canhão H, Escudero A, Filipescu I, Segura-Catena J, Soto-Pino MJ, Expósito-Ruiz M, Ferrer MÁ, et al. (2013) Gender-specific effects of genetic variants within Th1 and Th17 cell-mediated immune response genes on the risk of developing rheumatoid arthritis. PLoS ONE 8:e72732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caminschi I, Lahoud MH, Shortman K. (2009) Enhancing immune responses by targeting antigen to DC. Eur J Immunol 39:931–938. [DOI] [PubMed] [Google Scholar]
- Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, Carter AB, Rothman PB, Flavell RA, Sutterwala FS. (2008) The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci USA 105:9035–9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavlar T, Deimling T, Ablasser A, Hopfner K-P, Hornung V. (2013) Species-specific detection of the antiviral small-molecule compound CMA by STING. EMBO J 32:1440–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen H, Wang W, Leng R-X, Wang T-Y, Pan H-F, Fan Y-G, Wang B, Ye D-Q. (2013) Association of IFIH1 rs1990760 polymorphism with susceptibility to autoimmune diseases: a meta-analysis. Autoimmunity 46:455–462. [DOI] [PubMed] [Google Scholar]
- Chakravarthy KV, Bonoiu AC, Davis WG, Ranjan P, Ding H, Hu R, Bowzard JB, Bergey EJ, Katz JM, Knight PR, et al. (2010) Gold nanorod delivery of an ssRNA immune activator inhibits pandemic H1N1 influenza viral replication. Proc Natl Acad Sci USA 107:10172–10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, Ogura Y, Kawasaki A, Fukase K, Kusumoto S, et al. (2003a) An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol 4:702–707. [DOI] [PubMed] [Google Scholar]
- Chamaillard M, Philpott D, Girardin SE, Zouali H, Lesage S, Chareyre F, Bui TH, Giovannini M, Zaehringer U, Penard-Lacronique V, et al. (2003b) Gene-environment interaction modulated by allelic heterogeneity in inflammatory diseases. Proc Natl Acad Sci USA 100:3455–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavarría-Smith J, Vance RE. (2013) Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog 9:e1003452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs KS, Randall RE, Goodbourn S. (2013) LGP2 plays a critical role in sensitizing mda-5 to activation by double-stranded RNA. PLoS ONE 8:e64202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu Y-H, Macmillan JB, Chen ZJ. (2009) RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138:576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F, Bennett M, Moldt M, Deimling T, Witte G, Schiesser S, Carell T, Hopfner K-P. (2011) The RIG-I ATPase domain structure reveals insights into ATP-dependent antiviral signalling. EMBO Rep 12:1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, Hopfner K-P. (2013) Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498:332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke DL, Davis NHE, Campion CL, Foster ML, Heasman SC, Lewis AR, Anderson IK, Corkill DJ, Sleeman MA, May RD, et al. (2014) Dectin-2 sensing of house dust mite is critical for the initiation of airway inflammation. Mucosal Immunol 7:558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole LE, Shirey KA, Barry E, Santiago A, Rallabhandi P, Elkins KL, Puche AC, Michalek SM, Vogel SN. (2007) Toll-like receptor 2-mediated signaling requirements for Francisella tularensis live vaccine strain infection of murine macrophages. Infect Immun 75:4127–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concannon P, Onengut-Gumuscu S, Todd JA, Smyth DJ, Pociot F, Bergholdt R, Akolkar B, Erlich HA, Hilner JE, Julier C, et al. Type 1 Diabetes Genetics Consortium (2008) A human type 1 diabetes susceptibility locus maps to chromosome 21q22.3. Diabetes 57:2858–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly DJ, O’Neill LA. (2012) New developments in Toll-like receptor targeted therapeutics. Curr Opin Pharmacol 12:510–518. [DOI] [PubMed] [Google Scholar]
- Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJP, Campbell BJ, Jewell D, Simmons A. (2010) NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 16:90–97. [DOI] [PubMed] [Google Scholar]
- Coulombe F, Divangahi M, Veyrier F, de Léséleuc L, Gleason JL, Yang Y, Kelliher MA, Pandey AK, Sassetti CM, Reed MB, et al. (2009) Increased NOD2-mediated recognition of N-glycolyl muramyl dipeptide. J Exp Med 206:1709–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens NP, Mowers JC, McDonald C, Nuñez G, Ramaswamy S. (2007) Crystal structure of the Nod1 caspase activation and recruitment domain. Biochem Biophys Res Commun 353:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cridland JA, Curley EZ, Wykes MN, Schroder K, Sweet MJ, Roberts TL, Ragan MA, Kassahn KS, Stacey KJ. (2012) The mammalian PYHIN gene family: phylogeny, evolution and expression. BMC Evol Biol 12:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, et al. (2006) Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat Genet 38:917–920. [DOI] [PubMed] [Google Scholar]
- Cunha C, Di Ianni M, Bozza S, Giovannini G, Zagarella S, Zelante T, D’Angelo C, Pierini A, Pitzurra L, Falzetti F, et al. (2010) Dectin-1 Y238X polymorphism associates with susceptibility to invasive aspergillosis in hematopoietic transplantation through impairment of both recipient- and donor-dependent mechanisms of antifungal immunity. Blood 116:5394–5402. [DOI] [PubMed] [Google Scholar]
- Dalmasso G, Nguyen HTT, Charrier-Hisamuddin L, Yan Y, Laroui H, Demoulin B, Sitaraman SV, Merlin D. (2010) PepT1 mediates transport of the proinflammatory bacterial tripeptide L-Ala-gamma-D-Glu-meso-DAP in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 299:G687–G696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danot O, Marquenet E, Vidal-Ingigliardi D, Richet E. (2009) Wheel of Life, Wheel of Death: A Mechanistic Insight into Signaling by STAND Proteins. Structure 17:172–182. [DOI] [PubMed] [Google Scholar]
- da Silva Correia J, Miranda Y, Leonard N, Ulevitch R. (2007) SGT1 is essential for Nod1 activation. Proc Natl Acad Sci USA 104:6764–6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BK, Wen H, Ting JP-Y. (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29:707–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alba E. (2009) Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC). J Biol Chem 284:32932–32941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Fresno C, Soulat D, Roth S, Blazek K, Udalova I, Sancho D, Ruland J, Ardavín C. (2013) Interferon-β production via Dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity 38:1176–1186. [DOI] [PubMed] [Google Scholar]
- Delaloye J, Roger T, Steiner-Tardivel Q-G, Le Roy D, Knaup Reymond M, Akira S, Petrilli V, Gomez CE, Perdiguero B, Tschopp J, et al. (2009) Innate immune sensing of modified vaccinia virus Ankara (MVA) is mediated by TLR2-TLR6, MDA-5 and the NALP3 inflammasome. PLoS Pathog 5:e1000480. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- de Sousa M da GT, Belda W, Jr, Spina R, Lota PR, Valente NS, Brown GD, Criado PR, Benard G. (2014) Topical application of imiquimod as a treatment for chromoblastomycosis. Clin Infect Dis 58:1734–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennehy KM, Ferwerda G, Faro-Trindade I, Pyz E, Willment JA, Taylor PR, Kerrigan A, Tsoni SV, Gordon S, Meyer-Wentrup F, et al. (2008) Syk kinase is required for collaborative cytokine production induced through Dectin-1 and Toll-like receptors. Eur J Immunol 38:500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA, van der Meer JWM. (2013) Treating inflammation by blocking interleukin-1 in humans. Semin Immunol 25:469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. (2013) The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Reports 3:1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, Suva M-L, Stehle J-C, Kopf M, Stamenkovic I, et al. (2009) Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS ONE 4:e6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Osualdo A, Weichenberger CX, Wagner RN, Godzik A, Wooley J, Reed JC. (2011) CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PLoS ONE 6:e27396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake MG, Kaufman EH, Fryer AD, Jacoby DB. (2012) The therapeutic potential of Toll-like receptor 7 stimulation in asthma. Inflamm Allergy Drug Targets 11:484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drickamer K, Fadden AJ. (2002) Genomic analysis of C-type lectins. Biochem Soc Symp 69:59–72. [DOI] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M, et al. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464:1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duluc D, Joo H, Ni L, Yin W, Upchurch K, Li D, Xue Y, Klucar P, Zurawski S, Zurawski G, et al. (2014) Induction and activation of human Th17 by targeting antigens to dendritic cells via dectin-1. J Immunol 192:5776–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle ME, Dalpke AH. (2012) Dectin-1 stimulation induces suppressor of cytokine signaling 1, thereby modulating TLR signaling and T cell responses. J Immunol 188:5644–5654. [DOI] [PubMed] [Google Scholar]
- Eibl C, Grigoriu S, Hessenberger M, Wenger J, Puehringer S, Pinheiro AS, Wagner RN, Proell M, Reed JC, Page R, et al. (2012) Structural and functional analysis of the NLRP4 pyrin domain. Biochemistry 51:7330–7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eibl C, Hessenberger M, Wenger J, Brandstetter H. (2014) Structures of the NLRP14 pyrin domain reveal a conformational switch mechanism regulating its molecular interactions. Acta Crystallogr D Biol Crystallogr 70:2007–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. (2008) Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453:1122–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklöw C, Makrygiannakis D, Bäckdahl L, Padyukov L, Ulfgren A-K, Lorentzen JC, Malmström V. (2008) Cellular distribution of the C-type II lectin dendritic cell immunoreceptor (DCIR) and its expression in the rheumatic joint: identification of a subpopulation of DCIR+ T cells. Ann Rheum Dis 67:1742–1749. [DOI] [PubMed] [Google Scholar]
- Engering A, Geijtenbeek TBH, van Vliet SJ, Wijers M, van Liempt E, Demaurex N, Lanzavecchia A, Fransen J, Figdor CG, Piguet V, et al. (2002) The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J Immunol 168:2118–2126. [DOI] [PubMed] [Google Scholar]
- Esteban A, Popp MW, Vyas VK, Strijbis K, Ploegh HL, Fink GR. (2011) Fungal recognition is mediated by the association of dectin-1 and galectin-3 in macrophages. Proc Natl Acad Sci USA 108:14270–14275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman-Williams ME, Guenther U-P, Jankowsky E. (2010) SF1 and SF2 helicases: family matters. Curr Opin Struct Biol 20:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhat K, Riekenberg S, Heine H, Debarry J, Lang R, Mages J, Buwitt-Beckmann U, Röschmann K, Jung G, Wiesmüller KH, et al. (2008) Heterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. J Leukoc Biol 83:692–701. [DOI] [PubMed] [Google Scholar]
- Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, Reed JC. (2007) Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell 25:713–724. [DOI] [PubMed] [Google Scholar]
- Feinberg H, Mitchell DA, Drickamer K, Weis WI. (2001) Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science 294:2163–2166. [DOI] [PubMed] [Google Scholar]
- Feng Q, Hato SV, Langereis MA, Zoll J, Virgen-Slane R, Peisley A, Hur S, Semler BL, van Rij RP, van Kuppeveld FJM. (2012) MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Reports 2:1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL. (2012) DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife 1:e00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458:509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, et al. (2010) The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol 11:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferwerda B, Ferwerda G, Plantinga TS, Willment JA, van Spriel AB, Venselaar H, Elbers CC, Johnson MD, Cambi A, Huysamen C, et al. (2009) Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med 361:1760–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferwerda B, McCall MB, Verheijen K, Kullberg BJ, van der Ven AJ, Van der Meer JW, Netea MG. (2008) Functional consequences of toll-like receptor 4 polymorphisms. Mol Med 14:346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger JN, Lich JD, Dare LC, Cook MN, Brown KK, Duraiswami C, Bertin J, Gough PJ. (2012) Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J Biol Chem 287:25030–25037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Muñoz-Planillo R, Núñez G. (2012) Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13:325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke A, McGovern DPB, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. (2010) Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet 42:1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR, Al-Amoudi A, et al. (2014) The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 15:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridh V, Rittinger K. (2012) The tandem CARDs of NOD2: intramolecular interactions and recognition of RIP2. PLoS ONE 7:e34375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikado N, Saijo S, Yonezawa T, Shimamori K, Ishii A, Sugai S, Kotaki H, Sudo K, Nose M, Iwakura Y. (2008) Dcir deficiency causes development of autoimmune diseases in mice due to excess expansion of dendritic cells. Nat Med 14:176–180. [DOI] [PubMed] [Google Scholar]
- Fujita T, Onoguchi K, Onomoto K, Hirai R, Yoneyama M. (2007) Triggering antiviral response by RIG-I-related RNA helicases. Biochimie 89:754–760. [DOI] [PubMed] [Google Scholar]
- Funabiki M, Kato H, Miyachi Y, Toki H, Motegi H, Inoue M, Minowa O, Yoshida A, Deguchi K, Sato H, et al. (2014) Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 40:199–212. [DOI] [PubMed] [Google Scholar]
- Furukawa A, Kamishikiryo J, Mori D, Toyonaga K, Okabe Y, Toji A, Kanda R, Miyake Y, Ose T, Yamasaki S, et al. (2013) Structural analysis for glycolipid recognition by the C-type lectins Mincle and MCL. Proc Natl Acad Sci USA 110:17438–17443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. (2003) Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med 197:1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Wu J, Wu Y-T, Du F, Aroh C, Yan N, Sun L, Chen ZJ. (2013a) Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341:903–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, et al. (2013b) Structure-function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell 154:748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Vallejo JJ, van Kooyk Y. (2013) The physiological role of DC-SIGN: a tale of mice and men. Trends Immunol 34:482–486. [DOI] [PubMed] [Google Scholar]
- Gay NJ, Gangloff M. (2007) Structure and function of Toll receptors and their ligands. Annu Rev Biochem 76:141–165. [DOI] [PubMed] [Google Scholar]
- Gay NJ, Symmons MF, Gangloff M, Bryant CE. (2014) Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol 14:546–558. [DOI] [PubMed] [Google Scholar]
- Gazzinelli RT, Mendonça-Neto R, Lilue J, Howard J, Sher A. (2014) Innate resistance against Toxoplasma gondii: an evolutionary tale of mice, cats, and men. Cell Host Microbe 15:132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee P, Chua PK, Gevorkyan J, Klumpp K, Najera I, Swinney DC, Deval J. (2008) Essential role of the N-terminal domain in the regulation of RIG-I ATPase activity. J Biol Chem 283:9488–9496. [DOI] [PubMed] [Google Scholar]
- Geijtenbeek TBH, Gringhuis SI. (2009) Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol 9:465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessner MA, Werner JL, Lilly LM, Nelson MP, Metz AE, Dunaway CW, Chan YR, Ouyang W, Brown GD, Weaver CT, et al. (2012) Dectin-1-dependent interleukin-22 contributes to early innate lung defense against Aspergillus fumigatus. Infect Immun 80:410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbard RJ, Morley PJ, Gay NJ. (2006) Conserved features in the extracellular domain of human toll-like receptor 8 are essential for pH-dependent signaling. J Biol Chem 281:27503–27511. [DOI] [PubMed] [Google Scholar]
- Gioannini TL, Teghanemt A, Zhang D, Coussens NP, Dockstader W, Ramaswamy S, Weiss JP. (2004) Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc Natl Acad Sci USA 101:4186–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard R, Pedron T, Uematsu S, Balloy V, Chignard M, Akira S, Chaby R. (2003) Lipopolysaccharides from Legionella and Rhizobium stimulate mouse bone marrow granulocytes via Toll-like receptor 2. J Cell Sci 116:293–302. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jéhanno M, Viala J, Tedin K, Taha MK, Labigne A, Zähringer U, et al. (2003a) Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300:1584–1587. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. (2003b) Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 278:8869–8872. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Jéhanno M, Mengin-Lecreulx D, Sansonetti PJ, Alzari PM, Philpott DJ. (2005) Identification of the critical residues involved in peptidoglycan detection by Nod1. J Biol Chem 280:38648–38656. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Travassos LH, Hervé M, Blanot D, Boneca IG, Philpott DJ, Sansonetti PJ, Mengin-Lecreulx D. (2003c) Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem 278:41702–41708. [DOI] [PubMed] [Google Scholar]
- Glocker E-O, Hennigs A, Nabavi M, Schäffer AA, Woellner C, Salzer U, Pfeifer D, Veelken H, Warnatz K, Tahami F, et al. (2009) A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med 361:1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorjestani S, Yu M, Tang B, Zhang D, Wang D, Lin X. (2011) Phospholipase Cγ2 (PLCγ2) is key component in Dectin-2 signaling pathway, mediating anti-fungal innate immune responses. J Biol Chem 286:43651–43659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubau D, Deddouche S, Reis e Sousa C. (2013) Cytosolic sensing of viruses. Immunity 38:855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, et al. (2014) Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 514:372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulet M-L, Olagnier D, Xu Z, Paz S, Belgnaoui SM, Lafferty EI, Janelle V, Arguello M, Paquet M, Ghneim K, et al. (2013) Systems analysis of a RIG-I agonist inducing broad spectrum inhibition of virus infectivity. PLoS Pathog 9:e1003298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham LM, Brown GD. (2009) The Dectin-2 family of C-type lectins in immunity and homeostasis. Cytokine 48:148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham LM, Gupta V, Schafer G, Reid DM, Kimberg M, Dennehy KM, Hornsell WG, Guler R, Campanero-Rhodes MA, Palma AS, et al. (2012) The C-type lectin receptor CLECSF8 (CLEC4D) is expressed by myeloid cells and triggers cellular activation through Syk kinase. J Biol Chem 287:25964–25974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes CL, Ariyananda LdeZ, Melnyk JE, O’Shea EK. (2012) The innate immune protein Nod2 binds directly to MDP, a bacterial cell wall fragment. J Am Chem Soc 134:13535–13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Geijtenbeek TBH. (2009) Carbohydrate-specific signaling through the DC-SIGN signalosome tailors immunity to Mycobacterium tuberculosis, HIV-1 and Helicobacter pylori. Nat Immunol 10:1081–1088. [DOI] [PubMed] [Google Scholar]
- Gringhuis SI, den Dunnen J, Litjens M, van Het Hof B, van Kooyk Y, Geijtenbeek TBH. (2007) C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity 26:605–616. [DOI] [PubMed] [Google Scholar]
- Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T, Geijtenbeek TBH. (2012) Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat Immunol 13:246–254. [DOI] [PubMed] [Google Scholar]
- Gringhuis SI, van der Vlist M, van den Berg LM, den Dunnen J, Litjens M, Geijtenbeek TBH. (2010) HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat Immunol 11:419–426. [DOI] [PubMed] [Google Scholar]
- Gringhuis SI, Wevers BA, Kaptein TM, van Capel TMM, Theelen B, Boekhout T, de Jong EC, Geijtenbeek TBH. (2011) Selective C-Rel activation via Malt1 controls anti-fungal T(H)-17 immunity by dectin-1 and dectin-2. PLoS Pathog 7:e1001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Shan YA, Zhou J, Jiang DP, Zhang L, Du DY, Wang ZG, Jiang JX. (2007) Functional significance of gene polymorphisms in the promoter of myeloid differentiation-2. Ann Surg 246:151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Ranoa DRE, Jiang S, Mutha SK, Li X, Baudry J, Tapping RI. (2010) Human TLRs 10 and 1 share common mechanisms of innate immune sensing but not signaling. J Immunol 184:5094–5103. [DOI] [PubMed] [Google Scholar]
- Guo J, Wu X, Too CL, Yin F, Lu X, He J, Li R, Liu X, Murad S, Padyukov L, et al. (2012) A replication study confirms the association of dendritic cell immunoreceptor (DCIR) polymorphisms with ACPA - negative RA in a large Asian cohort. PLoS ONE 7:e41228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutte PGM, Jurt S, Grütter MG, Zerbe O. (2014) Unusual structural features revealed by the solution NMR structure of the NLRC5 caspase recruitment domain. Biochemistry 53:3106–3117. [DOI] [PubMed] [Google Scholar]
- Haas T, Metzger J, Schmitz F, Heit A, Müller T, Latz E, Wagner H. (2008) The DNA sugar backbone 2′ deoxyribose determines toll-like receptor 9 activation. Immunity 28:315–323. [DOI] [PubMed] [Google Scholar]
- Halff EF, Diebolder CA, Versteeg M, Schouten A, Brondijk THC, Huizinga EG. (2012) Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N- and C-terminal regions of flagellin. J Biol Chem 287:38460–38472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9:857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann L, Kumpf O, Müller M, Visintin A, Eckert J, Schlag PM, Schumann RR. (2004) A coding mutation within the first exon of the human MD-2 gene results in decreased lipopolysaccharide-induced signaling. Genes Immun 5:283–288. [DOI] [PubMed] [Google Scholar]
- Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y, Mizuguchi J, Schweighoffer E, Tybulewicz V, Mitsuyama M. (2013) Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol 14:1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, Guiet C, Brière F, Vlach J, Lebecque S, et al. (2005) Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol 174:2942–2950. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Asai Y, Ogawa T. (2004) Separation and structural analysis of lipoprotein in a lipopolysaccharide preparation from Porphyromonas gingivalis. Int Immunol 16:1431–1437. [DOI] [PubMed] [Google Scholar]
- Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. (2001) The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099–1103. [DOI] [PubMed] [Google Scholar]
- Haziot A, Ferrero E, Köntgen F, Hijiya N, Yamamoto S, Silver J, Stewart CL, Goyert SM. (1996) Resistance to endotoxin shock and reduced dissemination of gram-negative bacteria in CD14-deficient mice. Immunity 4:407–414. [DOI] [PubMed] [Google Scholar]
- Hedl M, Li J, Cho JH, Abraham C. (2007) Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proc Natl Acad Sci USA 104:19440–19445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitmann L, Schoenen H, Ehlers S, Lang R, Hölscher C. (2013) Mincle is not essential for controlling Mycobacterium tuberculosis infection. Immunobiology 218:506–516. [DOI] [PubMed] [Google Scholar]
- Hellman J, Tehan MM, Warren HS. (2003) Murein lipoprotein, peptidoglycan-associated lipoprotein, and outer membrane protein A are present in purified rough and smooth lipopolysaccharides. J Infect Dis 188:286–289. [DOI] [PubMed] [Google Scholar]
- Hellmich KA, Levinsohn JL, Fattah R, Newman ZL, Maier N, Sastalla I, Liu S, Leppla SH, Moayeri M. (2012) Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS ONE 7:e49741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. (2002) Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol 3:196–200. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, et al. (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408:740–745. [DOI] [PubMed] [Google Scholar]
- Higgins SC, Mills KHG. (2010) TLR, NLR Agonists, and Other Immune Modulators as Infectious Disease Vaccine Adjuvants. Curr Infect Dis Rep 12:4–12. [DOI] [PubMed] [Google Scholar]
- Hiller S, Kohl A, Fiorito F, Herrmann T, Wider G, Tschopp J, Grütter MG, Wüthrich K. (2003) NMR structure of the apoptosis- and inflammation-related NALP1 pyrin domain. Structure 11:1199–1205. [DOI] [PubMed] [Google Scholar]
- Hirschfeld M, Weis JJ, Toshchakov V, Salkowski CA, Cody MJ, Ward DC, Qureshi N, Michalek SM, Vogel SN. (2001) Signaling by toll-like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect Immun 69:1477–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, Sovath S, Shamel L, Hartung T, Zähringer U, et al. (2005) CD36 is a sensor of diacylglycerides. Nature 433:523–527. [DOI] [PubMed] [Google Scholar]
- Homer CR, Richmond AL, Rebert NA, Achkar J-P, McDonald C. (2010) ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 139:1630–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M, Yoon S-I, Wilson IA. (2012) Structure and functional characterization of the RNA-binding element of the NLRX1 innate immune modulator. Immunity 36:337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann K-K, Schlee M, et al. (2006) 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994–997. [DOI] [PubMed] [Google Scholar]
- Hornung V, Hartmann R, Ablasser A, Hopfner K-P. (2014) OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat Rev Immunol 14:521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. (1999) Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162:3749–3752. [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang Q-X, Chen ZJ. (2011) MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146:448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y-MS, Zhang Y, You Y, Wang D, Li H, Duramad O, Qin X-F, Dong C, Lin X. (2007) The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol 8:198–205. [DOI] [PubMed] [Google Scholar]
- Hu J, Nistal-Villán E, Voho A, Ganee A, Kumar M, Ding Y, García-Sastre A, Wetmur JG. (2010) A common polymorphism in the caspase recruitment domain of RIG-I modifies the innate immune response of human dendritic cells. J Immunol 185:424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Yan C, Liu P, Huang Z, Ma R, Zhang C, Wang R, Zhang Y, Martinon F, Miao D, et al. (2013) Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 341:172–175. [DOI] [PubMed] [Google Scholar]
- Huang Y-H, Liu X-Y, Du X-X, Jiang Z-F, Su X-D. (2012) The structural basis for the sensing and binding of cyclic di-GMP by STING. Nat Struct Mol Biol 19:728–730. [DOI] [PubMed] [Google Scholar]
- Hugot J-P, Laurent-Puig P, Gower-Rousseau C, Olson JM, Lee JC, Beaugerie L, Naom I, Dupas J-L, Van Gossum A, Orholm M, et al. (1996) Mapping of a susceptibility locus for Crohn’s disease on chromosome 16. Nature 379:821–823. [DOI] [PubMed] [Google Scholar]
- Ichinohe T, Pang IK, Iwasaki A. (2010) Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol 11:404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifrim DC, Bain JM, Reid DM, Oosting M, Verschueren I, Gow NAR, van Krieken JH, Brown GD, Kullberg B-J, Joosten LAB, et al. (2014) Role of Dectin-2 for host defense against systemic infection with Candida glabrata. Infect Immun 82:1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifrim DC, Joosten LAB, Kullberg B-J, Jacobs L, Jansen T, Williams DL, Gow NAR, van der Meer JWM, Netea MG, Quintin J. (2013) Candida albicans primes TLR cytokine responses through a Dectin-1/Raf-1-mediated pathway. J Immunol 190:4129–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, Brown J, Becker CA, Fleshner PR, Dubinsky M, et al. (2012) Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336:1314–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal M, Philbin VJ, Withanage GS, Wigley P, Beal RK, Goodchild MJ, Barrow P, McConnell I, Maskell DJ, Young J, et al. (2005) Identification and functional characterization of chicken toll-like receptor 5 reveals a fundamental role in the biology of infection with Salmonella enterica serovar typhimurium. Infect Immun 73:2344–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving AT, Mimuro H, Kufer TA, Lo C, Wheeler R, Turner LJ, Thomas BJ, Malosse C, Gantier MP, Casillas LN, et al. (2014) The immune receptor NOD1 and kinase RIP2 interact with bacterial peptidoglycan on early endosomes to promote autophagy and inflammatory signaling. Cell Host Microbe 15:623–635. [DOI] [PubMed] [Google Scholar]
- Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, et al. (2006) A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol 7:40–48. [DOI] [PubMed] [Google Scholar]
- Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, Uematsu S, Takeuchi O, Takeshita F, Coban C, et al. (2008) TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451:725–729. [DOI] [PubMed] [Google Scholar]
- Ishikawa E, Ishikawa T, Morita YS, Toyonaga K, Yamada H, Takeuchi O, Kinoshita T, Akira S, Yoshikai Y, Yamasaki S. (2009a) Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med 206:2879–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. (2009b) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, et al. (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39:311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Kaczanowska S, Davila E. (2014) IL-1 Receptor-Associated Kinase Signaling and Its Role in Inflammation, Cancer Progression, and Therapy Resistance. Front Immunol 5:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen MR, Bak RO, Andersen A, Berg RK, Jensen SB, Tengchuan J, Laustsen A, Hansen K, Ostergaard L, Fitzgerald KA, et al. (2013) IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc Natl Acad Sci USA 110:E4571–E4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA., Jr (1989) Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 54:1–13. [DOI] [PubMed] [Google Scholar]
- Jiang F, Ramanathan A, Miller MT, Tang G-Q, Gale M, Jr, Patel SS, Marcotrigiano J. (2011) Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 479:423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, Grishin NV, Chen ZJ. (2012) Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 36:959–973 Elsevier Inc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, et al. (2005) CD14 is required for MyD88-independent LPS signaling. Nat Immunol 6:565–570. [DOI] [PubMed] [Google Scholar]
- Jin L, Hill KK, Filak H, Mogan J, Knowles H, Zhang B, Perraud A-L, Cambier JC, Lenz LL. (2011) MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. J Immunol 187:2595–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik S-G, Lee H, Lee J-O. (2007) Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130:1071–1082. [DOI] [PubMed] [Google Scholar]
- Jin T, Curry J, Smith P, Jiang J, Xiao TS. (2013) Structure of the NLRP1 caspase recruitment domain suggests potential mechanisms for its association with procaspase-1. Proteins 81:1266–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, Jiang Z, Horvath G, Rathinam VA, Johnstone RW, et al. (2012) Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 36:561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CM, Lyle EA, Omueti KO, Stepensky VA, Yegin O, Alpsoy E, Hamann L, Schumann RR, Tapping RI. (2007) Cutting edge: A common polymorphism impairs cell surface trafficking and functional responses of TLR1 but protects against leprosy. J Immunol 178:7520–7524. [DOI] [PubMed] [Google Scholar]
- Johnstone RW, Wei W, Greenway A, Trapani JA. (2000) Functional interaction between p53 and the interferon-inducible nucleoprotein IFI 16. Oncogene 19:6033–6042. [DOI] [PubMed] [Google Scholar]
- Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, et al. (2010) Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci USA 107:9771–9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. (2012) Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 287:36617–36622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, Lipford G, Bauer S. (2002) Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol 3:499. [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Mocarski ES. (2008) Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol 181:6427–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa N, Okazaki T, Nishimura H, Tashiro K, Inaba K, Miyachi Y. (2002) DCIR acts as an inhibitory receptor depending on its immunoreceptor tyrosine-based inhibitory motif. J Invest Dermatol 118:261–266. [DOI] [PubMed] [Google Scholar]
- Kang JY, Nan X, Jin MS, Youn S-J, Ryu YH, Mah S, Han SH, Lee H, Paik S-G, Lee J-O. (2009) Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31:873–884 [DOI] [PubMed] [Google Scholar]
- Kanneganti T-D, Lamkanfi M, Kim Y-G, Chen G, Park J-H, Franchi L, Vandenabeele P, Núñez G. (2007) Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 26:433–443. [DOI] [PubMed] [Google Scholar]
- Kaparakis M, Turnbull L, Carneiro L, Firth S, Coleman HA, Parkington HC, Le Bourhis L, Karrar A, Viala J, Mak J, et al. (2010) Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol 12:372–385. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Forman M, Arav-Boger R. (2014) Activation of nucleotide oligomerization domain 2 (NOD2) by human cytomegalovirus initiates innate immune responses and restricts virus replication. PLoS ONE 9:e92704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. [DOI] [PubMed] [Google Scholar]
- Kato K, Ishii R, Goto E, Ishitani R, Tokunaga F, Nureki O. (2013) Structural and functional analyses of DNA-sensing and immune activation by human cGAS. PLoS ONE 8:e76983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–650. [DOI] [PubMed] [Google Scholar]
- Kawane K, Motani K, Nagata S. (2014) DNA degradation and its defects. Cold Spring Harb Perspect Biol 6:a016394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, Yoshikawa H, Nagata S. (2006) Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443:998–1002. [DOI] [PubMed] [Google Scholar]
- Kawata K, Illarionov P, Yang G-X, Kenny TP, Zhang W, Tsuda M, Ando Y, Leung PSC, Ansari AA, Eric Gershwin M. (2012) Mincle and human B cell function. J Autoimmun 39:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerscher B, Willment JA, Brown GD. (2013) The Dectin-2 family of C-type lectin-like receptors: an update. Int Immunol 25:271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersse K, Verspurten J, Vanden Berghe T, Vandenabeele P. (2011) The death-fold superfamily of homotypic interaction motifs. Trends Biochem Sci 36:541–552. [DOI] [PubMed] [Google Scholar]
- Kimkong I, Avihingsanon Y, Hirankarn N. (2010) Association of IFI200 gene polymorphisms with susceptibility to systemic lupus erythematosus. J Rheumatol 37:1544–1547. [DOI] [PubMed] [Google Scholar]
- Kirschning CJ, Wesche H, Merrill Ayres T, Rothe M. (1998) Human toll-like receptor 2 confers responsiveness to bacterial lipopolysaccharide. J Exp Med 188:2091–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Yuliwulandari R, Yanai H, Lien LT, Hang NT, Hijikata M, Keicho N, Tokunaga K. (2011) Association of CD209 polymorphisms with tuberculosis in an Indonesian population. Hum Immunol 72:741–745. [DOI] [PubMed] [Google Scholar]
- Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nuñez G, Flavell RA. (2005) Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307:731–734. [DOI] [PubMed] [Google Scholar]
- Kofoed EM, Vance RE. (2011) Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477:592–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro A, Horvath CM. (2006) RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J Virol 80:12332–12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, Barber GN, Komatsu K, Akira S, Kawai T. (2013) DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci USA 110:2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kormann MSD, Depner M, Hartl D, Klopp N, Illig T, Adamski J, Vogelberg C, Weiland SK, von Mutius E, Kabesch M. (2008) Toll-like receptor heterodimer variants protect from childhood asthma. J Allergy Clin Immunol 122:86–92, e1–e8. [DOI] [PubMed] [Google Scholar]
- Kovarova M, Hesker PR, Jania L, Nguyen M, Snouwaert JN, Xiang Z, Lommatzsch SE, Huang MT, Ting JP-Y, Koller BH. (2012) NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J Immunol 189:2006–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, Gerlier D, Cusack S. (2011) Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147:423–435. [DOI] [PubMed] [Google Scholar]
- Kranzusch PJ, Lee AS-Y, Berger JM, Doudna JA. (2013) Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Reports 3:1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg A, Correa RG, Garrison JB, Le Negrate G, Welsh K, Huang Z, Knoefel WT, Reed JC. (2009) XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA 106:14524–14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufer TA, Kremmer E, Adam AC, Philpott DJ, Sansonetti PJ. (2008) The pattern-recognition molecule Nod1 is localized at the plasma membrane at sites of bacterial interaction. Cell Microbiol 10:477–486. [DOI] [PubMed] [Google Scholar]
- Lahaye X, Satoh T, Gentili M, Cerboni S, Conrad C, Hurbain I, El Marjou A, Lacabaratz C, Lelièvre J-D, Manel N. (2013) The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity 39:1132–1142. [DOI] [PubMed] [Google Scholar]
- Lambert AA, Azzi A, Lin S-X, Allaire G, St-Gelais KP, Tremblay MJ, Gilbert C. (2013) Dendritic cell immunoreceptor is a new target for anti-AIDS drug development: identification of DCIR/HIV-1 inhibitors. PLoS ONE 8:e67873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AA, Barabé F, Gilbert C, Tremblay MJ. (2011) DCIR-mediated enhancement of HIV-1 infection requires the ITIM-associated signal transduction pathway. Blood 117:6589–6599. [DOI] [PubMed] [Google Scholar]
- Lambert AA, Gilbert C, Richard M, Beaulieu AD, Tremblay MJ. (2008) The C-type lectin surface receptor DCIR acts as a new attachment factor for HIV-1 in dendritic cells and contributes to trans- and cis-infection pathways. Blood 112:1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. (2014) Mechanisms and functions of inflammasomes. Cell 157:1013–1022. [DOI] [PubMed] [Google Scholar]
- Lang R. (2013) Recognition of the mycobacterial cord factor by Mincle: relevance for granuloma formation and resistance to tuberculosis. Front Immunol 4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laroui H, Yan Y, Narui Y, Ingersoll SA, Ayyadurai S, Charania MA, Zhou F, Wang B, Salaita K, Sitaraman SV, et al. (2011) L-Ala-γ-D-Glu-meso-diaminopimelic acid (DAP) interacts directly with leucine-rich region domain of nucleotide-binding oligomerization domain 1, increasing phosphorylation activity of receptor-interacting serine/threonine-protein kinase 2 and its interaction with nucleotide-binding oligomerization domain 1. J Biol Chem 286:31003–31013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein DCG, Monks BG, McKnight CJ, Lamphier MS, Duprex WP, et al. (2007) Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat Immunol 8:772–779. [DOI] [PubMed] [Google Scholar]
- Latz E, Xiao TS, Stutz A. (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor KE, Vince JE. (2014) Ambiguities in NLRP3 inflammasome regulation: is there a role for mitochondria? Biochim Biophys Acta 1840:1433–1440. [DOI] [PubMed] [Google Scholar]
- Lécine P, Esmiol S, Métais J-Y, Nicoletti C, Nourry C, McDonald C, Nunez G, Hugot J-P, Borg J-P, Ollendorff V. (2007) The NOD2-RICK complex signals from the plasma membrane. J Biol Chem 282:15197–15207. [DOI] [PubMed] [Google Scholar]
- Lee CC, Avalos AM, Ploegh HL. (2012a) Accessory molecules for Toll-like receptors and their function. Nat Rev Immunol 12:168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G-S, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, Chae JJ. (2012b) The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492:123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Lee J, Tobias PS. (2002) Two lipoproteins extracted from Escherichia coli K-12 LCD25 lipopolysaccharide are the major components responsible for Toll-like receptor 2-mediated signaling. J Immunol 168:4012–4017. [DOI] [PubMed] [Google Scholar]
- Lee J, Tattoli I, Wojtal KA, Vavricka SR, Philpott DJ, Girardin SE. (2009) pH-dependent internalization of muramyl peptides from early endosomes enables Nod1 and Nod2 signaling. J Biol Chem 284:23818–23829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K-H, Biswas A, Liu Y-J, Kobayashi KS. (2012c) Proteasomal degradation of Nod2 protein mediates tolerance to bacterial cell wall components. J Biol Chem 287:39800–39811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W-B, Kang J-S, Yan J-J, Lee MS, Jeon B-Y, Cho S-N, Kim Y-J. (2012d) Neutrophils Promote Mycobacterial Trehalose Dimycolate-Induced Lung Inflammation via the Mincle Pathway. PLoS Pathog 8:e1002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E, Tybulewicz V, Brown GD, Ruland J, et al. (2007) Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol 8:630–638. [DOI] [PubMed] [Google Scholar]
- Lenz LL, Mohammadi S, Geissler A, Portnoy DA. (2003) SecA2-dependent secretion of autolytic enzymes promotes Listeria monocytogenes pathogenesis. Proc Natl Acad Sci USA 100:12432–12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Zouali H, Cézard J-P, Colombel J-F, Belaiche J, Almer S, Tysk C, O’Morain C, Gassull M, Binder V, et al. EPWG-IBD Group. EPIMAD Group. GETAID Group (2002) CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet 70:845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, Moayeri M. (2012) Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 8:e1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Willingham SB, Ting JP-Y, Re F. (2008) Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol 181:17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Diner BA, Chen J, Cristea IM. (2012a) Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci USA 109:10558–10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, Zuo X, Kao CC, Herr AB, Li P. (2013a) Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39:1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Utomo A, Cullere X, Choi MM, Milner DA, Jr, Venkatesh D, Yun S-H, Mayadas TN. (2011) The β-glucan receptor Dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe 10:603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-D, Wu J, Gao D, Wang H, Sun L, Chen ZJ. (2013b) Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341:1390–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chen R, Zhou Q, Xu Z, Li C, Wang S, Mao A, Zhang X, He W, Shu HB. (2012b) LSm14A is a processing body-associated sensor of viral nucleic acids that initiates cellular antiviral response in the early phase of viral infection. Proc Natl Acad Sci USA 109:11770–11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR, Ma Y, Pannicke U, Schwarz K. (2003) Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 4:712–720. [DOI] [PubMed] [Google Scholar]
- Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, Henry T, Sun YH, Cado D, Dietrich WF, et al. (2008) Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol 9:1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilic D. (2012) Unravelling fungal immunity through primary immune deficiencies. Curr Opin Microbiol 15:420–426. [DOI] [PubMed] [Google Scholar]
- Lin K-M, Hu W, Troutman TD, Jennings M, Brewer T, Li X, Nanda S, Cohen P, Thomas JA, Pasare C. (2014) IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci USA 111:775–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S-C, Lo Y-C, Wu H. (2010) Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 465:885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YT, Verma A, Hodgkinson CP. (2012) Toll-like receptors and human disease: lessons from single nucleotide polymorphisms. Curr Genomics 13:633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski T, Fitieh A, St Pierre J, Ostergaard HL, Bundle DR, Touret N. (2013) Enhanced immunogenicity of a tricomponent mannan tetanus toxoid conjugate vaccine targeted to dendritic cells via Dectin-1 by incorporating β-glucan. J Immunol 190:4116–4128. [DOI] [PubMed] [Google Scholar]
- Lippmann J, Rothenburg S, Deigendesch N, Eitel J, Meixenberger K, van Laak V, Slevogt H, N’guessan PD, Hippenstiel S, Chakraborty T, et al. (2008) IFNbeta responses induced by intracellular bacteria or cytosolic DNA in different human cells do not require ZBP1 (DLM-1/DAI). Cell Microbiol 10:2579–2588. [DOI] [PubMed] [Google Scholar]
- Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, Davies DR. (2008) Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science 320:379–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Wang H, Jin Y, Podolsky R, Reddy MVPL, Pedersen J, Bode B, Reed J, Steed D, Anderson S, et al. (2009) IFIH1 polymorphisms are significantly associated with type 1 diabetes and IFIH1 gene expression in peripheral blood mononuclear cells. Hum Mol Genet 18:358–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobato-Pascual A, Saether PC, Fossum S, Dissen E, Daws MR. (2013) Mincle, the receptor for mycobacterial cord factor, forms a functional receptor complex with MCL and FcεRI-γ. Eur J Immunol 43:3167–3174. [DOI] [PubMed] [Google Scholar]
- Long KM, Whitmore AC, Ferris MT, Sempowski GD, McGee C, Trollinger B, Gunn B, Heise MT. (2013) Dendritic cell immunoreceptor regulates Chikungunya virus pathogenesis in mice. J Virol 87:5697–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo Y-M, Gale M., Jr (2011) Immune signaling by RIG-I-like receptors. Immunity 34:680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Castejon G, Luheshi NM, Compan V, High S, Whitehead RC, Flitsch S, Kirov A, Prudovsky I, Swanton E, Brough D. (2013) Deubiquitinases regulate the activity of caspase-1 and interleukin-1β secretion via assembly of the inflammasome. J Biol Chem 288:2721–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz E, Mira JP, Cornish KL, Arbour NC, Schwartz DA. (2000) A novel polymorphism in the toll-like receptor 2 gene and its potential association with staphylococcal infection. Infect Immun 68:6398–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schröder GF, Fitzgerald KA, Wu H, Egelman EH. (2014) Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156:1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Xu H, Ranjith-Kumar CT, Brooks MT, Hou TY, Hu F, Herr AB, Strong RK, Kao CC, Li P. (2010) The structural basis of 5′ triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure 18:1032–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke JM, Simon GG, Söderholm J, Errett JS, August JT, Gale M, Jr, Hodgson CP, Williams JA. (2011) Coexpressed RIG-I agonist enhances humoral immune response to influenza virus DNA vaccine. J Virol 85:1370–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D, Ding SC, Vela A, Kohlway A, Lindenbach BD, Pyle AM. (2011) Structural insights into RNA recognition by RIG-I. Cell 147:409–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupfer C, Kanneganti T-D. (2013) The expanding role of NLRs in antiviral immunity. Immunol Rev 255:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H, et al. (2013) Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol 14:480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Becker C, Reyes C, Underhill DM. (2014) Cutting edge: FYCO1 recruitment to dectin-1 phagosomes is accelerated by light chain 3 protein and regulates phagosome maturation and reactive oxygen production. J Immunol 192:1356–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhaes JG, Fritz JH, Le Bourhis L, Sellge G, Travassos LH, Selvanantham T, Girardin SE, Gommerman JL, Philpott DJ. (2008) Nod2-dependent Th2 polarization of antigen-specific immunity. J Immunol 181:7925–7935. [DOI] [PubMed] [Google Scholar]
- Maglinao M, Klopfleisch R, Seeberger PH, Lepenies B. (2013) The C-type lectin receptor DCIR is crucial for the development of experimental cerebral malaria. J Immunol 191:2551–2559. [DOI] [PubMed] [Google Scholar]
- Mailaparambil B, Krueger M, Heinze J, Forster J, Heinzmann A. (2008) Polymorphisms of toll like receptors in the genetics of severe RSV associated diseases. Dis Markers 25:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhas AN, Abuknesha RA, Price RG. (2002) Interaction of the leucine-rich repeats of polycystin-1 with extracellular matrix proteins: possible role in cell proliferation. J Am Soc Nephrol 13:19–26. [DOI] [PubMed] [Google Scholar]
- Man SM, Hopkins LJ, Nugent E, Cox S, Glück IM, Tourlomousis P, Wright JA, Cicuta P, Monie TP, Bryant CE. (2014) Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci USA 111:7403–7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manon F, Favier A, Núñez G, Simorre J-P, Cusack S. (2007) Solution structure of NOD1 CARD and mutational analysis of its interaction with the CARD of downstream kinase RICK. J Mol Biol 365:160–174. [DOI] [PubMed] [Google Scholar]
- Mansour MK, Tam JM, Khan NS, Seward M, Davids PJ, Puranam S, Sokolovska A, Sykes DB, Dagher Z, Becker C, et al. (2013) Dectin-1 activation controls maturation of β-1,3-glucan-containing phagosomes. J Biol Chem 288:16043–16054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza AR, Barrio MM, Moutel S, Bover L, Weck M, Brossart P, Teillaud JL, Mordoh J. (2004) CD63 tetraspanin slows down cell migration and translocates to the endosomal-lysosomal-MIICs route after extracellular stimuli in human immature dendritic cells. Blood 104:1183–1190. [DOI] [PubMed] [Google Scholar]
- Marakalala MJ, Guler R, Matika L, Murray G, Jacobs M, Brombacher F, Rothfuchs AG, Sher A, Brown GD. (2011) The Syk/CARD9-coupled receptor Dectin-1 is not required for host resistance to Mycobacterium tuberculosis in mice. Microbes Infect 13:198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marichal T, Ohata K, Bedoret D, Mesnil C, Sabatel C, Kobiyama K, Lekeux P, Coban C, Akira S, Ishii KJ, et al. (2011) DNA released from dying host cells mediates aluminum adjuvant activity. Nat Med 17:996–1002. [DOI] [PubMed] [Google Scholar]
- Martin MP, Lederman MM, Hutcheson HB, Goedert JJ, Nelson GW, van Kooyk Y, Detels R, Buchbinder S, Hoots K, Vlahov D, et al. (2004) Association of DC-SIGN promoter polymorphism with increased risk for parenteral, but not mucosal, acquisition of human immunodeficiency virus type 1 infection. J Virol 78:14053–14056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Gil L, Goff PH, Hai R, García-Sastre A, Shaw ML, Palese P. (2013) A Sendai virus-derived RNA agonist of RIG-I as a virus vaccine adjuvant. J Virol 87:1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10:417–426. [DOI] [PubMed] [Google Scholar]
- Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, et al. (2010) Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol 11:897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur R, Oh H, Zhang D, Park SG, Seo J, Koblansky A, Hayden MS, Ghosh S. (2012) A mouse model of Salmonella typhi infection. Cell 151:590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayle S, Boyle JP, Sekine E, Zurek B, Kufer TA, Monie TP. (2014) Engagement of nucleotide-binding oligomerization domain-containing protein 1 (NOD1) by receptor-interacting protein 2 (RIP2) is insufficient for signal transduction. J Biol Chem 289:22900–22914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor A, Martinon F, De Smedt T, Pétrilli V, Tschopp J. (2007) A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat Immunol 8:497–503. [DOI] [PubMed] [Google Scholar]
- McCartney SA, Vermi W, Lonardi S, Rossini C, Otero K, Calderon B, Gilfillan S, Diamond MS, Unanue ER, Colonna M. (2011) RNA sensor-induced type I IFN prevents diabetes caused by a β cell-tropic virus in mice. J Clin Invest 121:1497–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald C, Chen FF, Ollendorff V, Ogura Y, Marchetto S, Lécine P, Borg J-P, Nuñez G. (2005) A role for Erbin in the regulation of Nod2-dependent NF-kappaB signaling. J Biol Chem 280:40301–40309. [DOI] [PubMed] [Google Scholar]
- McWhirter SM, Barbalat R, Monroe KM, Fontana MF, Hyodo M, Joncker NT, Ishii KJ, Akira S, Colonna M, Chen ZJ, et al. (2009) A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J Exp Med 206:1899–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–397. [DOI] [PubMed] [Google Scholar]
- Merx S, Neumaier M, Wagner H, Kirschning CJ, Ahmad-Nejad P. (2007) Characterization and investigation of single nucleotide polymorphisms and a novel TLR2 mutation in the human TLR2 gene. Hum Mol Genet 16:1225–1232. [DOI] [PubMed] [Google Scholar]
- Meyer-Wentrup F, Benitez-Ribas D, Tacken PJ, Punt CJA, Figdor CG, de Vries IJM, Adema GJ. (2008) Targeting DCIR on human plasmacytoid dendritic cells results in antigen presentation and inhibits IFN-alpha production. Blood 111:4245–4253. [DOI] [PubMed] [Google Scholar]
- Meyer-Wentrup F, Cambi A, Joosten B, Looman MW, de Vries IJM, Figdor CG, Adema GJ. (2009) DCIR is endocytosed into human dendritic cells and inhibits TLR8-mediated cytokine production. J Leukoc Biol 85:518–525. [DOI] [PubMed] [Google Scholar]
- Meyer-Wentrup F, Figdor CG, Ansems M, Brossart P, Wright MD, Adema GJ, van Spriel AB. (2007) Dectin-1 interaction with tetraspanin CD37 inhibits IL-6 production. J Immunol 178:154–162. [DOI] [PubMed] [Google Scholar]
- Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A. (2010) Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA 107:3076–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitoma H, Hanabuchi S, Kim T, Bao M, Zhang Z, Sugimoto N, Liu Y-J. (2013) The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity 39:123–135 Elsevier Inc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Y, Toyonaga K, Mori D, Kakuta S, Hoshino Y, Oyamada A, Yamada H, Ono K, Suyama M, Iwakura Y, et al. (2013) C-type lectin MCL is an FcRγ-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity 38:1050–1062. [DOI] [PubMed] [Google Scholar]
- Mo J, Boyle JP, Howard CB, Monie TP, Davis BK, Duncan JA. (2012) Pathogen sensing by nucleotide-binding oligomerization domain-containing protein 2 (NOD2) is mediated by direct binding to muramyl dipeptide and ATP. J Biol Chem 287:23057–23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanan V, Grimes CL. (2014) The molecular chaperone HSP70 binds to and stabilizes NOD2, an important protein involved in Crohn disease. J Biol Chem 289:18987–18998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondini M, Vidali M, Airò P, De Andrea M, Riboldi P, Meroni PL, Gariglio M, Landolfo S. (2007) Role of the interferon-inducible gene IFI16 in the etiopathogenesis of systemic autoimmune disorders. Ann N Y Acad Sci 1110:47–56. [DOI] [PubMed] [Google Scholar]
- Monie TP. (2013) NLR activation takes a direct route. Trends Biochem Sci 38:131–139. [DOI] [PubMed] [Google Scholar]
- Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC. (2014) IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343:428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morath S, Stadelmaier A, Geyer A, Schmidt RR, Hartung T. (2002) Synthetic lipoteichoic acid from Staphylococcus aureus is a potent stimulus of cytokine release. J Exp Med 195:1635–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrone SR, Wang T, Constantoulakis LM, Hooy RM, Delannoy MJ, Sohn J. (2014) Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc Natl Acad Sci USA 111:E62–E71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Planillo R, Franchi L, Miller LS, Núñez G. (2009) A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol 183:3942–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. (2013) K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38:1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T. (2012) Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci USA 109:11282–11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, Kitamura T, Kosugi A, Kimoto M, Miyake K. (2002) Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol 3:667–672. [DOI] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12:222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Lill JR, Phung Q, Jiang Z, Bakalarski C, de Mazière A, Klumperman J, Schlatter M, Delamarre L, Mellman I. (2014) Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature 509:240–244. [DOI] [PubMed] [Google Scholar]
- Navi A, Patel H, Shaw S, Baker D, Tsui J. (2013) Therapeutic role of toll-like receptor modification in cardiovascular dysfunction. Vascul Pharmacol 58:231–239. [DOI] [PubMed] [Google Scholar]
- Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. (2009) Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324:387–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Wijmenga C, O’Neill LA. (2012) Genetic variation in Toll-like receptors and disease susceptibility. Nat Immunol 13:535–542. [DOI] [PubMed] [Google Scholar]
- Nigro G, Fazio LL, Martino MC, Rossi G, Tattoli I, Liparoti V, De Castro C, Molinaro A, Philpott DJ, Bernardini ML. (2008) Muramylpeptide shedding modulates cell sensing of Shigella flexneri. Cell Microbiol 10:682–695. [DOI] [PubMed] [Google Scholar]
- Norimoto A, Hirose K, Iwata A, Tamachi T, Yokota M, Takahashi K, Saijo S, Iwakura Y, Nakajima H. (2014) Dectin-2 promotes house dust mite-induced T helper type 2 and type 17 cell differentiation and allergic airway inflammation in mice. Am J Respir Cell Mol Biol 51:201–209. [DOI] [PubMed] [Google Scholar]
- Núñez C, Oliver J, Mendoza JL, Gómez-García M, Taxonera C, Gómez LM, López-Nevot MA, de la Concha EG, Urcelay E, Martínez A, et al. (2007) CD209 in inflammatory bowel disease: a case-control study in the Spanish population. BMC Med Genet 8:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LAJ, Bowie AG. (2007) The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 7:353–364. [DOI] [PubMed] [Google Scholar]
- O’Neill LAJ, Bryant CE, Doyle SL. (2009) Therapeutic targeting of Toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol Rev 61:177–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohto U, Tanji H, Shimizu T. (2014) Structure and function of toll-like receptor 8. Microbes Infect 16:273–282. [DOI] [PubMed] [Google Scholar]
- Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF., 3rd (2001) The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem 276:10229–10233. [DOI] [PubMed] [Google Scholar]
- Olagnier D, Scholte FEM, Chiang C, Albulescu IC, Nichols C, He Z, Lin R, Snijder EJ, van Hemert MJ, Hiscott J. (2014) Inhibition of dengue and chikungunya virus infections by RIG-I-mediated type I interferon-independent stimulation of the innate antiviral response. J Virol 88:4180–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleszycka E, Lavelle EC. (2014) Immunomodulatory properties of the vaccine adjuvant alum. Curr Opin Immunol 28:1–5. [DOI] [PubMed] [Google Scholar]
- De Oliveira LF, De Lima CPS, Azevedo RDSS, De Mendonça DSF, Rodrigues SG, Carvalho VL, Pinto EV, Maia AL, Maia MHT, Vasconcelos JM, et al. (2014) Polymorphism of DC-SIGN (CD209) Promoter in Association with Clinical Symptoms of Dengue Fever. Viral Immunol 27:1–5. [DOI] [PubMed] [Google Scholar]
- Orr SJ, Burg AR, Chan T, Quigley L, Jones GW, Ford JW, Hodge D, Razzook C, Sarhan J, Jones YL, et al. (2013) LAB/NTAL facilitates fungal/PAMP-induced IL-12 and IFN-γ production by repressing β-catenin activation in dendritic cells. PLoS Pathog 9:e1003357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, DeLuca NA, Knipe DM. (2012) Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci USA 109:E3008–E3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio F, Reis e Sousa C. (2011) Myeloid C-type lectin receptors in pathogen recognition and host defense. Immunity 34:651–664. [DOI] [PubMed] [Google Scholar]
- Ouyang S, Song X, Wang Y, Ru H, Shaw N, Jiang Y, Niu F, Zhu Y, Qiu W, Parvatiyar K, et al. (2012) Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity 36:1073–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. (2000) The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci USA 97:13766–13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludan SR, Bowie AG. (2013) Immune sensing of DNA. Immunity 38:870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. (2011) Recognition of herpesviruses by the innate immune system. Nat Rev Immunol 11:143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi B-S, Lee H, Lee J-O. (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458:1191–1195. [DOI] [PubMed] [Google Scholar]
- Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J, Wu H. (2007) Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 128:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Hahn WH, Suh JS, Kim MJ, Kang SW, Lee JS, Kim JW, Chung JH, Cho BS. (2011) Association between toll-like receptor 10 (TLR10) gene polymorphisms and childhood IgA nephropathy. Eur J Pediatr 170:503–509. [DOI] [PubMed] [Google Scholar]
- Parkhouse R, Boyle JP, Mayle S, Sawmynaden K, Rittinger K, Monie TP. (2014a) Interaction between NOD2 and CARD9 involves the NOD2 NACHT and the linker region between the NOD2 CARDs and NACHT domain. FEBS Lett 588:2830–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhouse R, Boyle JP, Monie TP. (2014b) Blau syndrome polymorphisms in NOD2 identify nucleotide hydrolysis and helical domain 1 as signalling regulators. FEBS Lett 588:3382–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MW, Li L, Wallace AM, Lee MJ, Katz HR, Fernandez JM, Saijo S, Iwakura Y, Austen KF, Kanaoka Y, et al. (2014) Dectin-2 regulates the effector phase of house dust mite-elicited pulmonary inflammation independently from its role in sensitization. J Immunol 192:1361–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvatiyar K, Zhang Z, Teles RM, Ouyang S, Jiang Y, Iyer SS, Zaver SA, Schenk M, Zeng S, Zhong W, et al. (2012) The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat Immunol 13:1155–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JR, Jain A, Chou YY, Baum A, Ha T, García-Sastre A. (2013) ATPase-driven oligomerization of RIG-I on RNA allows optimal activation of type-I interferon. EMBO Rep 14:780–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisley A, Lin C, Wu B, Orme-Johnson M, Liu M, Walz T, Hur S. (2011) Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci USA 108:21010–21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisley A, Wu B, Xu H, Chen ZJ, Hur S. (2014) Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 509:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisley A, Wu B, Yao H, Walz T, Hur S. (2013) RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol Cell 51:573–583. [DOI] [PubMed] [Google Scholar]
- Petnicki-Ocwieja T, Hrncir T, Liu Y-J, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, Kobayashi KS. (2009) Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci USA 106:15813–15818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. (2014) NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol 14:9–23. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, Reis e Sousa C. (2006) RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997–1001. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan C-P, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, Reis e Sousa C. (2009) Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol 83:10761–10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro AS, Eibl C, Ekman-Vural Z, Schwarzenbacher R, Peti W. (2011) The NLRP12 pyrin domain: structure, dynamics, and functional insights. J Mol Biol 413:790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro AS, Proell M, Eibl C, Page R, Schwarzenbacher R, Peti W. (2010) Three-dimensional structure of the NLRP7 pyrin domain: insight into pyrin-pyrin-mediated effector domain signaling in innate immunity. J Biol Chem 285:27402–27410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantinga TS, Fransen J, Takahashi N, Stienstra R, van Riel PL, van den Berg WB, Netea MG, Joosten LA. (2010) Functional consequences of DECTIN-1 early stop codon polymorphism Y238X in rheumatoid arthritis. Arthritis Res Ther 12:R26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantinga TS, van der Velden WJFM, Ferwerda B, van Spriel AB, Adema G, Feuth T, Donnelly JP, Brown GD, Kullberg B-J, Blijlevens NM, et al. (2009) Early stop polymorphism in human DECTIN-1 is associated with increased candida colonization in hematopoietic stem cell transplant recipients. Clin Infect Dis 49:724–732. [DOI] [PubMed] [Google Scholar]
- Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, Hannesschläger N, Schlee M, Rothenfusser S, Barchet W, et al. (2010) Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol 11:63–69. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088. [DOI] [PubMed] [Google Scholar]
- Pothlichet J, Burtey A, Kubarenko AV, Caignard G, Solhonne B, Tangy F, Ben-Ali M, Quintana-Murci L, Heinzmann A, Chiche J-D, et al. (2009) Study of human RIG-I polymorphisms identifies two variants with an opposite impact on the antiviral immune response. PLoS ONE 4:e7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, et al. (2011) Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332:65–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Py BF, Kim M-S, Vakifahmetoglu-Norberg H, Yuan J. (2013) Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 49:331–338. [DOI] [PubMed] [Google Scholar]
- Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y. (1999) Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 399:549–557. [DOI] [PubMed] [Google Scholar]
- Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, Lamkanfi M, Louie S, Kayagaki N, Liu J, Kömüves L, et al. (2012) Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490:539–542. [DOI] [PubMed] [Google Scholar]
- Qureshi ST, Larivière L, Leveque G, Clermont S, Moore KJ, Gros P, Malo D. (1999) Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J Exp Med 189:615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz CR, Whitfield C. (2002) Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan JV, Rodriguez D, Miao EA, Aderem A. (2011) The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J Virol 85:4167–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rallabhandi P, Awomoyi A, Thomas KE, Phalipon A, Fujimoto Y, Fukase K, Kusumoto S, Qureshi N, Sztein MB, Vogel SN. (2008) Differential activation of human TLR4 by Escherichia coli and Shigella flexneri 2a lipopolysaccharide: combined effects of lipid A acylation state and TLR4 polymorphisms on signaling. J Immunol 180:1139–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rallabhandi P, Phillips RL, Boukhvalova MS, Pletneva LM, Shirey KA, Gioannini TL, Weiss JP, Chow JC, Hawkins LD, Vogel SN, et al. (2012) Respiratory syncytial virus fusion protein-induced toll-like receptor 4 (TLR4) signaling is inhibited by the TLR4 antagonists Rhodobacter sphaeroides lipopolysaccharide and eritoran (E5564) and requires direct interaction with MD-2. MBio 3:e00218–e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan D, Tang MS, Bowcutt R, Loke P, Cadwell K. (2014) Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity 41:311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappuoli R, Mandl CW, Black S, De Gregorio E. (2011) Vaccines for the twenty-first century society. Nat Rev Immunol 11:865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VAK, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, et al. (2010) The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayamajhi M, Zak DE, Chavarria-Smith J, Vance RE, Miao EA. (2013) Cutting edge: Mouse NAIP1 detects the type III secretion system needle protein. J Immunol 191:3986–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsamen M, Heinz LX, Meylan E, Michallet M-C, Schroder K, Hofmann K, Vazquez J, Benedict CA, Tschopp J. (2009) DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep 10:916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves WH. (1992) Antibodies to the p70/p80 (Ku) antigens in systemic lupus erythematosus. Rheum Dis Clin North Am 18:391–414. [PubMed] [Google Scholar]
- Reubold TF, Hahne G, Wohlgemuth S, Eschenburg S. (2014) Crystal structure of the leucine-rich repeat domain of the NOD-like receptor NLRP1: implications for binding of muramyl dipeptide. FEBS Lett 588:3327–3332. [DOI] [PubMed] [Google Scholar]
- Rice GI, del Toro Duany Y, Jenkinson EM, Forte GMA, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, et al. (2014) Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet 46:503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard M, Thibault N, Veilleux P, Breton R, Beaulieu AD. (2003) The ITIM-bearing CLECSF6 (DCIR) is down-modulated in neutrophils by neutrophil activating agents. Biochem Biophys Res Commun 310:767–773. [DOI] [PubMed] [Google Scholar]
- Ritter M, Gross O, Kays S, Ruland J, Nimmerjahn F, Saijo S, Tschopp J, Layland LE, Prazeres da Costa C. (2010) Schistosoma mansoni triggers Dectin-2, which activates the Nlrp3 inflammasome and alters adaptive immune responses. Proc Natl Acad Sci USA 107:20459–20464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera A, Hohl TM, Collins N, Leiner I, Gallegos A, Saijo S, Coward JW, Iwakura Y, Pamer EG. (2011) Dectin-1 diversifies Aspergillus fumigatus-specific T cell responses by inhibiting T helper type 1 CD4 T cell differentiation. J Exp Med 208:369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, Hood LE, Aderem A. (2005) The evolution of vertebrate Toll-like receptors. Proc Natl Acad Sci USA 102:9577–9582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, et al. (2009) HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323:1057–1060. [DOI] [PubMed] [Google Scholar]
- Roberts ZJ, Goutagny N, Perera P-Y, Kato H, Kumar H, Kawai T, Akira S, Savan R, van Echo D, Fitzgerald KA, et al. (2007) The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J Exp Med 204:1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MJ, Osorio F, Rosas M, Freitas RP, Schweighoffer E, Gross O, Verbeek JS, Ruland J, Tybulewicz V, Brown GD, et al. (2009) Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J Exp Med 206:2037–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers MA, Bowman JW, Fujita H, Orazio N, Shi M, Liang Q, Amatya R, Kelly TJ, Iwai K, Ting J, et al. (2014) The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med 211:1333–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez KR, Bruns AM, Horvath CM. (2014) MDA5 and LGP2: accomplices and antagonists of antiviral signal transduction. J Virol 88:8194–8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosentul DC, Plantinga TS, Papadopoulos A, Joosten LAB, Antoniadou A, Venselaar H, Kullberg B-J, van der Meer JWM, Giamarellos-Bourboulis EJ, Netea MG. (2011) Variation in genes of β-glucan recognition pathway and susceptibility to opportunistic infections in HIV-positive patients. Immunol Invest 40:735–750. [DOI] [PubMed] [Google Scholar]
- Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, Schubert K, Schöneberg T, Schaefer M, Krügel U, et al. (2012) Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun 3:1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S, Rottach A, Lotz-Havla AS, Laux V, Muschaweckh A, Gersting SW, Muntau AC, Hopfner K-P, Jin L, Vanness K, et al. (2014) Rad50-CARD9 interactions link cytosolic DNA sensing to IL-1β production. Nat Immunol 15:538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA. (2005) The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol 175:5260–5268. [DOI] [PubMed] [Google Scholar]
- Rothfuchs AG, Bafica A, Feng CG, Egen JG, Williams DL, Brown GD, Sher A. (2007) Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J Immunol 179:3463–3471. [DOI] [PubMed] [Google Scholar]
- Ru H, Ni X, Zhao L, Crowley C, Ding W, Hung L-W, Shaw N, Cheng G, Liu Z-J. (2013) Structural basis for termination of AIM2-mediated signaling by p202. Cell Res 23:855–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino SJ, Magalhaes JG, Philpott D, Bahr GM, Blanot D, Girardin SE. (2013) Identification of a synthetic muramyl peptide derivative with enhanced Nod2 stimulatory capacity. Innate Immun 19:493–503. [DOI] [PubMed] [Google Scholar]
- Ryan EJ, Dring M, Ryan CM, McNulty C, Stevenson NJ, Lawless MW, Crowe J, Nolan N, Hegarty JE, O’Farrelly C. (2010) Variant in CD209 promoter is associated with severity of liver disease in chronic hepatitis C virus infection. Hum Immunol 71:829–832. [DOI] [PubMed] [Google Scholar]
- Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, Xiang Y, Bose S. (2009) Activation of innate immune antiviral responses by Nod2. Nat Immunol 10:1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo S, Fujikado N, Furuta T, Chung SH, Kotaki H, Seki K, Sudo K, Akira S, Adachi Y, Ohno N, et al. (2007) Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol 8:39–46. [DOI] [PubMed] [Google Scholar]
- Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, Fujikado N, Kusaka T, Kubo S, Chung SH, et al. (2010) Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 32:681–691. [DOI] [PubMed] [Google Scholar]
- Sainz J, Lupiáñez CB, Segura-Catena J, Vazquez L, Ríos R, Oyonarte S, Hemminki K, Försti A, Jurado M. (2012) Dectin-1 and DC-SIGN polymorphisms associated with invasive pulmonary Aspergillosis infection. PLoS ONE 7:e32273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M., Jr (2008) Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho D, Reis e Sousa C. (2012) Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu Rev Immunol 30:491–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho-Shimizu V, Pérez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, Fabrega S, Cardon A, Maluenda J, Tatematsu M, et al. (2011) Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest 121:4889–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Ajmani AK, Stojanov L, Langdon JJ, Ogasawara T, Wang J, Dooley MA, Richards HB, Winfield JB, Carter TH, et al. (1996) Autoantibodies that stabilize the molecular interaction of Ku antigen with DNA-dependent protein kinase catalytic subunit. Clin Exp Immunol 105:460–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, Tsujimura T, Fujita T, Akira S, Takeuchi O. (2010) LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci USA 107:1512–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer J-D, Pereyre S, Archer KA, Burke TP, Hanson B, Lauer P, Portnoy DA. (2011a) Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proc Natl Acad Sci USA 108:12419–12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer J-D, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, et al. (2011b) The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun 79:688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenen H, Bodendorfer B, Hitchens K, Manzanero S, Werninghaus K, Nimmerjahn F, Agger EM, Stenger S, Andersen P, Ruland J, et al. (2010) Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J Immunol 184:2756–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, et al. (2014) Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505:691–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Muruve DA, Tschopp J. (2009) Innate immunity: cytoplasmic DNA sensing by the AIM2 inflammasome. Curr Biol 19:R262–R265. [DOI] [PubMed] [Google Scholar]
- Schroder K, Sagulenko V, Zamoshnikova A, Richards AA, Cridland JA, Irvine KM, Stacey KJ, Sweet MJ. (2012) Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 217:1325–1329. [DOI] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. (2010) The inflammasomes. Cell 140:821–832. [DOI] [PubMed] [Google Scholar]
- Schroder K, Zhou R, Tschopp J. (2010) The NLRP3 inflammasome: a sensor for metabolic danger? Science 327:296–300. [DOI] [PubMed] [Google Scholar]
- Schröder NW, Hermann C, Hamann L, Göbel UB, Hartung T, Schumann RR. (2003) High frequency of polymorphism Arg753Gln of the Toll-like receptor-2 gene detected by a novel allele-specific PCR. J Mol Med (Berl) 81:368–372. [DOI] [PubMed] [Google Scholar]
- Schröder NW, Schumann RR. (2005) Single nucleotide polymorphisms of Toll-like receptors and susceptibility to infectious disease. Lancet Infect Dis 5:156–164. [DOI] [PubMed] [Google Scholar]
- Schumann RR, Leong SR, Flaggs GW, Gray PW, Wright SD, Mathison JC, Tobias PS, Ulevitch RJ. (1990) Structure and function of lipopolysaccharide binding protein. Science 249:1429–1431. [DOI] [PubMed] [Google Scholar]
- Schweneker K, Gorka O, Schweneker M, Poeck H, Tschopp J, Peschel C, Ruland J, Gross O. (2013) The mycobacterial cord factor adjuvant analogue trehalose-6,6′-dibehenate (TDB) activates the Nlrp3 inflammasome. Immunobiology 218:664–673. [DOI] [PubMed] [Google Scholar]
- Seelig HP, Ehrfeld H, Renz M. (1994) Interferon-gamma-inducible protein p16. A new target of antinuclear antibodies in patients with systemic lupus erythematosus. Arthritis Rheum 37:1672–1683. [DOI] [PubMed] [Google Scholar]
- Shang G, Zhu D, Li N, Zhang J, Zhu C, Lu D, Liu C, Yu Q, Zhao Y, Xu S, et al. (2012) Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat Struct Mol Biol 19:725–727. [DOI] [PubMed] [Google Scholar]
- Sharma A, Steichen AL, Jondle CN, Mishra BB, Sharma J. (2014) Protective role of Mincle in bacterial pneumonia by regulation of neutrophil mediated phagocytosis and extracellular trap formation. J Infect Dis 209:1837–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, DeOliveira RB, Kalantari P, Parroche P, Goutagny N, Jiang Z, Chan J, Bartholomeu DC, Lauw F, Hall JP, et al. (2011) Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity 35:194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, Singh M, O’Hagan DT, Pétrilli V, Tschopp J, et al. (2009) Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci USA 106:870–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw G, Morse S, Ararat M, Graham FL. (2002) Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J 16:869–871. [DOI] [PubMed] [Google Scholar]
- Shenderov K, Barber DL, Mayer-Barber KD, Gurcha SS, Jankovic D, Feng CG, Oland S, Hieny S, Caspar P, Yamasaki S, et al. (2013) Cord factor and peptidoglycan recapitulate the Th17-promoting adjuvant activity of mycobacteria through mincle/CARD9 signaling and the inflammasome. J Immunol 190:5722–5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C-S, Shenderov K, Huang N-N, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. (2012) Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto T, Kageyama M, Hirai R, Zheng J, Yoneyama M, Fujita T. (2009) Identification of loss of function mutations in human genes encoding RIG-I and MDA5: implications for resistance to type I diabetes. J Biol Chem 284:13348–13354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et al. (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36:401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. (1999) MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med 189:1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin DM, Yang CS, Yuk JM, Lee JY, Kim KH, Shin SJ, Takahara K, Lee SJ, Jo EK. (2008) Mycobacterium abscessus activates the macrophage innate immune response via a physical and functional interaction between TLR2 and dectin-1. Cell Microbiol 10:1608–1621. [DOI] [PubMed] [Google Scholar]
- Shu C, Yi G, Watts T, Kao CC, Li P. (2012) Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat Struct Mol Biol 19:722–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skolnick P, Davis H, Arnelle D, Deaver D. (2014) Translational potential of naloxone and naltrexone as TLR4 antagonists. Trends Pharmacol Sci 35:431–432. [DOI] [PubMed] [Google Scholar]
- Smirnova I, Mann N, Dols A, Derkx HH, Hibberd ML, Levin M, Beutler B. (2003) Assay of locus-specific genetic load implicates rare Toll-like receptor 4 mutations in meningococcal susceptibility. Proc Natl Acad Sci USA 100:6075–6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, Guja C, Ionescu-Tirgoviste C, Widmer B, Dunger DB, et al. (2006) A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet 38:617–619. [DOI] [PubMed] [Google Scholar]
- Song DH, Lee JO. (2012) Sensing of microbial molecular patterns by Toll-like receptors. Immunol Rev 250:216–229. [DOI] [PubMed] [Google Scholar]
- Sousa MdaG, Reid DM, Schweighoffer E, Tybulewicz V, Ruland J, Langhorne J, Yamasaki S, Taylor PR, Almeida SR, Brown GD. (2011) Restoration of pattern recognition receptor costimulation to treat chromoblastomycosis, a chronic fungal infection of the skin. Cell Host Microbe 9:436–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srimathi T, Robbins SL, Dubas RL, Hasegawa M, Inohara N, Park YC. (2008) Monomer/dimer transition of the caspase-recruitment domain of human Nod1. Biochemistry 47:1319–1325. [DOI] [PubMed] [Google Scholar]
- Staruch MJ, Wood DD. (1983) The adjuvanticity of interleukin 1 in vivo. J Immunol 130:2191–2194. [PubMed] [Google Scholar]
- Stetson DB, Medzhitov R. (2006) Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24:93–103. [DOI] [PubMed] [Google Scholar]
- Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, et al. (2010) CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 11:155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockhammer OW, Zakrzewska A, Hegedûs Z, Spaink HP, Meijer AH. (2009) Transcriptome profiling and functional analyses of the zebrafish embryonic innate immune response to Salmonella infection. J Immunol 182:5641–5653. [DOI] [PubMed] [Google Scholar]
- Stöver AG, Da Silva Correia J, Evans JT, Cluff CW, Elliott MW, Jeffery EW, Johnson DA, Lacy MJ, Baldridge JR, Probst P, et al. (2004) Structure-activity relationship of synthetic toll-like receptor 4 agonists. J Biol Chem 279:4440–4449. [DOI] [PubMed] [Google Scholar]
- Strasser D, Neumann K, Bergmann H, Marakalala MJ, Guler R, Rojowska A, Hopfner KP, Brombacher F, Urlaub H, Baier G, et al. (2012) Syk kinase-coupled C-type lectin receptors engage protein kinase C-σ to elicit Card9 adaptor-mediated innate immunity. Immunity 36:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strijbis K, Tafesse FG, Fairn GD, Witte MD, Dougan SK, Watson N, Spooner E, Esteban A, Vyas VK, Fink GR, et al. (2013) Bruton’s Tyrosine Kinase (BTK) and Vav1 contribute to Dectin1-dependent phagocytosis of Candida albicans in macrophages. PLoS Pathog 9:e1003446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M-Y, Kuo C-I, Chang C-F, Chang C-I. (2013) Three-dimensional structure of human NLRP10/PYNOD pyrin domain reveals a homotypic interaction site distinct from its mouse homologue. PLoS ONE 8:e67843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Franchi L, He Y, Muñoz-Planillo R, Mimuro H, Suzuki T, Sasakawa C, Núñez G. (2014) Shigella type III secretion protein MxiI is recognized by Naip2 to induce Nlrc4 inflammasome activation independently of Pkcδ. PLoS Pathog 10:e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaan PW, Bensman T, Bahadduri PM, Hall MW, Sarkar A, Bao S, Khantwal CM, Ekins S, Knoell DL. (2008) Bacterial peptide recognition and immune activation facilitated by human peptide transporter PEPT2. Am J Respir Cell Mol Biol 39:536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, et al. (2007) DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448:501–505. [DOI] [PubMed] [Google Scholar]
- Takashima K, Matsunaga N, Yoshimatsu M, Hazeki K, Kaisho T, Uekata M, Hazeki O, Akira S, Iizawa Y, Ii M. (2009) Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br J Pharmacol 157:1250–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. (1999) Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11:443–451. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Kawai T, Mühlradt PF, Morr M, Radolf JD, Zychlinsky A, Takeda K, Akira S. (2001) Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol 13:933–940. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Chamaillard M, Ogura Y, Zhu L, Qiu S, Masumoto J, Ghosh P, Moran A, Predergast MM, Tromp G, et al. (2004) Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J 23:1587–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Chen ZJ. (2012) STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5:ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. (2005) The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci USA 102:5856–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanji H, Ohto U, Shibata T, Miyake K, Shimizu T. (2013) Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 339:1426–1429. [DOI] [PubMed] [Google Scholar]
- Tao M, Scacheri PC, Marinis JM, Harhaj EW, Matesic LE, Abbott DW. (2009) ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influence inflammatory signaling pathways. Curr Biol 19:1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KM, Irving PM. (2011) Optimization of conventional therapy in patients with IBD. Nat Rev Gastroenterol Hepatol 8:646–656. [DOI] [PubMed] [Google Scholar]
- Taylor PR, Roy S, Leal SM, Jr, Sun Y, Howell SJ, Cobb BA, Li X, Pearlman E. (2014) Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORγt and dectin-2. Nat Immunol 15:143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, Haynes K, Steele C, Botto M, Gordon S, et al. (2007) Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol 8:31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenthorey JL, Kofoed EM, Daugherty MD, Malik HS, Vance RE. (2014) Molecular basis for specific recognition of bacterial ligands by NAIP/NLRC4 inflammasomes. Mol Cell 54:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PG, Dash P, Aldridge JR, Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, et al. (2009) The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30:566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MR, Sharma S, Atianand M, Jensen SB, Carpenter S, Knipe DM, Fitzgerald KA, Kurt-Jones EA. (2014) Interferon γ-inducible protein (IFI) 16 transcriptionally regulates type i interferons and other interferon-stimulated genes and controls the interferon response to both DNA and RNA viruses. J Biol Chem 289:23568–23581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigno-Aranjuez JT, Asara JM, Abbott DW. (2010) Inhibition of RIP2’s tyrosine kinase activity limits NOD2-driven cytokine responses. Genes Dev 24:2666–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigno-Aranjuez JT, Bai X, Abbott DW. (2013) A discrete ubiquitin-mediated network regulates the strength of NOD2 signaling. Mol Cell Biol 33:146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JP-Y, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, et al. (2008) The NLR gene family: a standard nomenclature. Immunity 28:285–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travassos LH, Carneiro LAM, Ramjeet M, Hussey S, Kim Y-G, Magalhães JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al. (2010) Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 11:55–62. [DOI] [PubMed] [Google Scholar]
- Tsuji T, Matsuzaki J, Kelly MP, Ramakrishna V, Vitale L, He L-Z, Keler T, Odunsi K, Old LJ, Ritter G, et al. (2011) Antibody-targeted NY-ESO-1 to mannose receptor or DEC-205 in vitro elicits dual human CD8+ and CD4+ T cell responses with broad antigen specificity. J Immunol 186:1218–1227. [DOI] [PubMed] [Google Scholar]
- Uematsu S, Jang MH, Chevrier N, Guo Z, Kumagai Y, Yamamoto M, Kato H, Sougawa N, Matsui H, Kuwata H, et al. (2006) Detection of pathogenic intestinal bacteria by Toll-like receptor 5 on intestinal CD11c+ lamina propria cells. Nat Immunol 7:868–874. [DOI] [PubMed] [Google Scholar]
- Unterholzner L. (2013) The interferon response to intracellular DNA: why so many receptors? Immunobiology 218:1312–1321. [DOI] [PubMed] [Google Scholar]
- Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, et al. (2010) IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11:997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES. (2012) DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11:290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg LM, Gringhuis SI, Geijtenbeek TBH. (2012) An evolutionary perspective on C-type lectins in infection and immunity. Ann N Y Acad Sci 1253:149–158. [DOI] [PubMed] [Google Scholar]
- Van Limbergen J, Wilson DC, Satsangi J. (2009) The genetics of Crohn’s disease. Annu Rev Genomics Hum Genet 10:89–116. [DOI] [PubMed] [Google Scholar]
- Vannberg FO, Chapman SJ, Khor CC, Tosh K, Floyd S, Jackson-Sillah D, Crampin A, Sichali L, Bah B, Gustafson P, et al. (2008) CD209 genetic polymorphism and tuberculosis disease. PLoS ONE 3:e1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga N, Sutkeviciute I, Ribeiro-Viana R, Berzi A, Ramdasi R, Daghetti A, Vettoretti G, Amara A, Clerici M, Rojo J, et al. (2014) A multivalent inhibitor of the DC-SIGN dependent uptake of HIV-1 and Dengue virus. Biomaterials 35:4175–4184. [DOI] [PubMed] [Google Scholar]
- Vasl J, Prohinar P, Gioannini TL, Weiss JP, Jerala R. (2008) Functional activity of MD-2 polymorphic variant is significantly different in soluble and TLR4-bound forms: decreased endotoxin binding by G56R MD-2 and its rescue by TLR4 ectodomain. J Immunol 180:6107–6115. [DOI] [PubMed] [Google Scholar]
- Vavricka SR, Musch MW, Chang JE, Nakagawa Y, Phanvijhitsiri K, Waypa TS, Merlin D, Schneewind O, Chang EB. (2004) hPepT1 transports muramyl dipeptide, activating NF-kappaB and stimulating IL-8 secretion in human colonic Caco2/bbe cells. Gastroenterology 127:1401–1409. [DOI] [PubMed] [Google Scholar]
- Ver Heul AM, Fowler CA, Ramaswamy S, Piper RC. (2013) Ubiquitin regulates caspase recruitment domain-mediated signaling by nucleotide-binding oligomerization domain-containing proteins NOD1 and NOD2. J Biol Chem 288:6890–6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Mémet S, Huerre MR, Coyle AJ, et al. (2004) Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 5:1166–1174. [DOI] [PubMed] [Google Scholar]
- Vijayan D, Radford KJ, Beckhouse AG, Ashman RB, Wells CA. (2012) Mincle polarizes human monocyte and neutrophil responses to Candida albicans. Immunol Cell Biol 90:889–895. [DOI] [PubMed] [Google Scholar]
- Vogel SN, Johnson D, Perera PY, Medvedev A, Larivière L, Qureshi ST, Malo D. (1999) Cutting edge: functional characterization of the effect of the C3H/HeJ defect in mice that lack an Lpsn gene: in vivo evidence for a dominant negative mutation. J Immunol 162:5666–5670. [PubMed] [Google Scholar]
- Walsh C, Gangloff M, Monie T, Smyth T, Wei B, McKinley TJ, Maskell D, Gay N, Bryant C. (2008) Elucidation of the MD-2/TLR4 interface required for signaling by lipid IVa. J Immunol 181:1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, LeBert V, Hung CY, Galles K, Saijo S, Lin X, Cole GT, Klein BS, Wüthrich M. (2014) C-type lectin receptors differentially induce th17 cells and vaccine immunity to the endemic mycosis of North America. J Immunol 192:1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Huang C-Y, Hasegawa M, Inohara N, Fujimoto Y, Fukase K. (2013) Glycan sequence-dependent Nod2 activation investigated by using a chemically synthesized bacterial peptidoglycan fragment library. ChemBioChem 14:482–488. [DOI] [PubMed] [Google Scholar]
- Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, Reddy A, Somogyi AA, Hutchinson MR, Watkins LR, et al. (2012) Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc Natl Acad Sci USA 109:6325–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ludwig J, Schuberth C, Goldeck M, Schlee M, Li H, Juranek S, Sheng G, Micura R, Tuschl T, et al. (2010) Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat Struct Mol Biol 17:781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Choi MK, Ban T, Yanai H, Negishi H, Lu Y, Tamura T, Takaoka A, Nishikura K, Taniguchi T. (2008) Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc Natl Acad Sci USA 105:5477–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Rice KC, Maier SF. (2009) The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci 30:581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis WI, Drickamer K. (1996) Structural basis of lectin-carbohydrate recognition. Annu Rev Biochem 65:441–473. [DOI] [PubMed] [Google Scholar]
- Wells CA, Salvage-Jones JA, Li X, Hitchens K, Butcher S, Murray RZ, Beckhouse AG, Lo Y-L-S, Manzanero S, Cobbold C, et al. (2008) The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J Immunol 180:7404–7413. [DOI] [PubMed] [Google Scholar]
- Wen H, Miao EA, Ting JP-Y. (2013) Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity 39:432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werts C, Tapping RI, Mathison JC, Chuang TH, Kravchenko V, Saint Girons I, Haake DA, Godowski PJ, Hayashi F, Ozinsky A, et al. (2001) Leptospiral lipopolysaccharide activates cells through a TLR2-dependent mechanism. Nat Immunol 2:346–352. [DOI] [PubMed] [Google Scholar]
- Wevers BA, Kaptein TM, Zijlstra-willems EM, Theelen B, Boekhout T, Geijtenbeek TBH, Gringhuis SI. (2014) Fungal engagement of the C-type lectin mincle suppresses dectin-1-induced antifungal immunity. Cell Host Microbe 15:494–505. [DOI] [PubMed] [Google Scholar]
- Wojtal KA, Eloranta JJ, Hruz P, Gutmann H, Drewe J, Staumann A, Beglinger C, Fried M, Kullak-Ublick GA, Vavricka SR. (2009) Changes in mRNA expression levels of solute carrier transporters in inflammatory bowel disease patients. Drug Metab Dispos 37:1871–1877. [DOI] [PubMed] [Google Scholar]
- Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. (1990) CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249:1431–1433. [DOI] [PubMed] [Google Scholar]
- Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C, Chu F, Walz T, Hur S. (2013a) Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 152:276–289. [DOI] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. (2013b) Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339:826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Wu F-H, Wang X, Wang L, Siedow JN, Zhang W, Pei Z-M. (2014) Molecular evolutionary and structural analysis of the cytosolic DNA sensor cGAS and STING. Nucleic Acids Res 42:8243–8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki S, Matsumoto M, Takeuchi O, Matsuzawa T, Ishikawa E, Sakuma M, Tateno H, Uno J, Hirabayashi J, Mikami Y, et al. (2009) C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc Natl Acad Sci USA 106:1897–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, Nakasato M, Lu Y, Hangai S, Koshiba R, et al. (2009) HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 462:99–103. [DOI] [PubMed] [Google Scholar]
- Yang J, Zhao Y, Shi J, Shao F. (2013a) Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci USA 110:14408–14413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, An H, Liu X, Wen M, Zheng Y, Rui Y, Cao X. (2010) The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a beta-catenin-dependent pathway. Nat Immunol 11:487–494. [DOI] [PubMed] [Google Scholar]
- Yang RB, Mark MR, Gray A, Huang A, Xie MH, Zhang M, Goddard A, Wood WI, Gurney AL, Godowski PJ. (1998) Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature 395:284–288. [DOI] [PubMed] [Google Scholar]
- Yang S, Wang B, Humphries F, Jackson R, Healy ME, Bergin R, Aviello G, Hall B, McNamara D, Darby T, et al. (2013b) Pellino3 ubiquitinates RIP2 and mediates Nod2-induced signaling and protective effects in colitis. Nat Immunol 14:927–936. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo Y-M, Gale M, Jr, Akira S, et al. (2005) Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 175:2851–2858. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737. [DOI] [PubMed] [Google Scholar]
- Yoon SI, Kurnasov O, Natarajan V, Hong M, Gudkov AV, Osterman AL, Wilson IA. (2012) Structural basis of TLR5-flagellin recognition and signaling. Science 335:859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. (2005) Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol 6:49–56. [DOI] [PubMed] [Google Scholar]
- Zähringer U, Lindner B, Inamura S, Heine H, Alexander C. (2008) TLR2 - promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 213:205–224. [DOI] [PubMed] [Google Scholar]
- Zelensky AN, Gready JE. (2005) The C-type lectin-like domain superfamily. FEBS J 272:6179–6217. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhao J, Song Y, Wang X, Wang L, Xu J, Song C, Liu F. (2014a) The E3 ligase RNF34 is a novel negative regulator of the NOD1 pathway. Cell Physiol Biochem 33:1954–1962. [DOI] [PubMed] [Google Scholar]
- Zhang X, Brann TW, Zhou M, Yang J, Oguariri RM, Lidie KB, Imamichi H, Huang D-W, Lempicki RA, Baseler MW, et al. (2011a) Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J Immunol 186:4541–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ. (2013) Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell 51:226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, Brautigam CA, Zhang X, Chen ZJ. (2014b) The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Reports 6:421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Kim T, Bao M, Facchinetti V, Jung SY, Ghaffari AA, Qin J, Cheng G, Liu Y-J. (2011b) DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity 34:866–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu Y-J. (2011c) The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 12:959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X-Q, Zhu L-L, Chang Q, Jiang C, You Y, Luo T, Jia X-M, Lin X. (2014) C-type lectin receptor dectin-3 mediates trehalose 6,6′-dimycolate (TDM)-induced Mincle expression through CARD9/Bcl10/MALT1-dependent nuclear factor (NF)-κB activation. J Biol Chem 289:30052–30062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. (2011) The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600. [DOI] [PubMed] [Google Scholar]
- Zhernakova A, Festen EME, Franke L, Trynka G, van Diemen CC, Monsuur AJ, Bevova M, Nijmeijer RM, van ’t Slot R, Heijmans R, et al. (2008) Genetic analysis of innate immunity in Crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am J Hum Genet 82:1202–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang Y-Y, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, et al. (2008) The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29:538–550. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Kinio A, Saleh M. (2013) Functions of NOD-Like Receptors in Human Diseases. Front Immunol 4:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. (2010) Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11:136–140. [DOI] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–225. [DOI] [PubMed] [Google Scholar]
- Zhu L-L, Zhao X-Q, Jiang C, You Y, Chen X-P, Jiang Y-Y, Jia X-M, Lin X. (2013) C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity 39:324–334. [DOI] [PubMed] [Google Scholar]
- Zurek B, Proell M, Wagner RN, Schwarzenbacher R, Kufer TA. (2012) Mutational analysis of human NOD1 and NOD2 NACHT domains reveals different modes of activation. Innate Immun 18:100–111. [DOI] [PubMed] [Google Scholar]
- Züst R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, et al. (2011) Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol 12:137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwicker JD, Zhang Y, Ren J, Hutchinson MR, Rice KC, Watkins LR, Greer JJ, Funk GD. (2014) Glial TLR4 signaling does not contribute to opioid-induced depression of respiration. J Appl Physiol (1985) 117:857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]