

Abstract
Measurements of the orientational freedom with which a single molecule may rotate or ‘wobble’ about a fixed axis have provided researchers invaluable clues about the underlying behavior of a variety of biological systems. In this paper, we propose a measurement and data analysis procedure based on a widefield fluorescence microscope image for quantitatively distinguishing individual molecules that exhibit varying degrees of rotational mobility. Our proposed technique is especially applicable to cases in which the molecule undergoes rotational motions on a timescale much faster than the framerate of the camera used to record fluorescence images. Unlike currently available methods, sophisticated hardware for modulating the polarization of light illuminating the sample is not required. Additional polarization optics may be inserted in the microscope’s imaging pathway to achieve superior measurement precision, but are not essential. We present a theoretical analysis, and benchmark our technique with numerical simulations using typical experimental parameters for single-molecule imaging.
OCIS codes: (170.2520) Fluorescence microscopy, (100.0100) Image processing
1. Introduction
Ever since the first experimental measurements of single-molecule orientational dynamics [1], polarization optics have played a critical role in determining the rotational mobility of the fluorescent molecules under observation. A fluorescent molecule may be regarded to leading order as an oscillating electric dipole. Hence, the electric field emitted by a molecule has a characteristic polarized far field pattern according to the orientation of that molecule’s emission dipole moment. Furthermore, the efficiency with which a molecule may be excited by a light source will also depend upon the alignment of the polarization of the incident light relative to the molecule’s absorption dipole moment. Changes in fluorescence intensity, as a function of excitation/emission polarization, may thus be related to the orientational behavior of the molecule under observation. A simply measured quantity is the linear dichroism ( ) of a molecule, which may be computed as:
| (1) |
Where is the total measured intensity after fluorescence has passed through a polarizer, and is the emitted intensity measured when using a perpendicularly oriented polarizer. In practice, both and may be acquired simultaneously using a polarizing beamsplitter, and two separate photodetectors (or separate regions on a single image sensor). It is useful to know the linear dichroism of a molecule, since this quantity tends to approach zero as the molecule becomes more mobile. It may thus be used to establish a bound on the range of orientations visited by the fluorophore over the integration time of the photodetector. Furthermore, by recording multiple measurements of the same molecule [2–6] using different excitation polarizations, one may determine the underlying amount of rotational freedom with useful precision. For in-depth comparisons of various polarization configurations, and their relative precisions given limited signal, see [7,8].
In previous work, measurements have played a crucial role in helping researchers quantify the mechanical properties of DNA [2,9], and understand the complex mechanisms governing the movement of motor proteins [10,11]. However, there are some notable limitations to this technique. For example, consider the following (Fig. 1 ): measurements for three different molecules are acquired. The first molecule is rotationally immobile, and oriented parallel to the optical axis. The second molecule is oriented perpendicular to the optical axis, but is oriented at 45° with respect to the polarizing beamsplitter placed in the emission pathway (this molecule is also immobilized). Finally, the third molecule undergoes rotation about the optical axis on a timescale faster than the temporal resolution of the photodetectors. All three molecules will yield an measurement of zero, even though they each exhibit completely different orientational characteristics! In order to break such degeneracies, it is often necessary to introduce polarization modulation optics in the illumination pathway, and/or repeatedly measure the fluoresence emitted from the same molecule after a different excitation polarization has been applied. However, in widefield imaging studies, it may only be feasible to record a single measurement per molecule, using a single excitation polarization [12,13]. In this case, the potential to mis-interpret an data set is quite real, since a single measurement cannot completely characterize the rotational behavior of a molecule. Reliance on data alone may obscure relevant physical phenomena or, as we will demonstrate in a numerical experiment, may cause an experimenter to form patently incorrect conclusions about a specimen under observation.
Fig. 1.
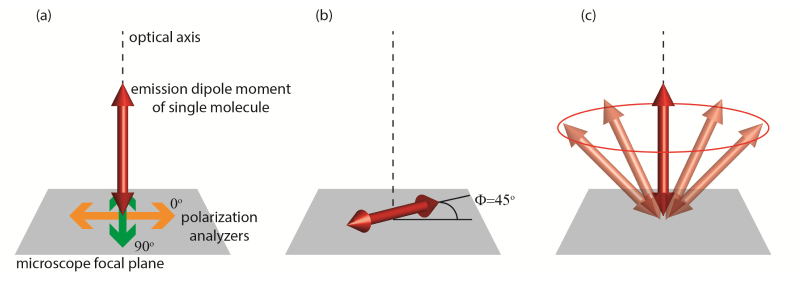
Examples of rotational behavior which yield identical linear dichroism measurements. (a) Immobile molecule aligned along the optical axis. Orientation of polarization analyzers indicated with respect to microscope focal plane. (b) Immobile molecule aligned in plane of coverslip, at 45° angle to each of the polarization analyzers. (c) Molecule rotating about the optical axis.
In order to avoid many of the ambiguities inherent in measurements, many researchers have turned to widefield image-based analysis in order to determine the orientation of single molecules [14–22]. The combination of image-based analysis with polarized detection configurations has been considered in [23]. Using slightly defocused images of single dye molecules in order to deduce orientation, researchers have studied the stepping behavior of the myosin V motor protein [24], and have gained insight into the optical biasing of Brownian rotations when molecules are attached to a thin polymer film [25]. Furthermore, defocused imaging has been recently proposed as a means of studying the photophysics of chiral molecules [26], and molecules containing multiple chromophores [27]. Applications of orientation imaging have assumed that a fluorophore is either fixed in orientation, or rotating at a rate far slower than the integration time of the camera. However, molecules commonly undergo rotational motions on timescales much faster than the ~ms temporal resolution of state-of-the-art image sensors. We address this apparent shortcoming by proposing a method to determine the amount of wobble that a molecule undergoes, in addition to that molecule’s mean orientation, using just one camera frame. Unlike alternative image-based approaches for determining rotational mobility [28], ours does not rely upon any specific rotational diffusion model, nor does it require a large library of ‘training’ single-molecule images of known orientations/mobilities.
This paper is organized as follows: In section 2, we describe our theoretical framework for simulating and characterizing single-molecule widefield fluorescence images arising from molecules of different orientations and rotational mobilities. In section 3, we perform a series of numerical experiments designed to showcase the method’s capacity to finely differentiate molecules exhibiting varying amounts of rotational freedom. We demonstrate that linear dichroism measurements would not provide sufficient information to distinguish such behavior. Furthermore, we examine how our method performs in moderate to low signal-to-noise imaging conditions, and suggest various means of optimizing our method, given a limited photon budget. Finally, in section 4 we discuss the practical hurdles that must be overcome in order to make our proposed method experimentally realizable.
2. Theoretical framework
In this section, we describe a simple, accurate and computationally efficient method for calculating the image of single-molecule fluorescence formed on a typical camera array detector such as an electron-multiplication charge coupled device (EMCCD) or a sCMOS detector. Our method can be applied to simulate images of both rotationally mobile and immobile molecules. In the first part of this section, we show how any single-molecule image can be fully characterized using a 3-by-3 symmetric, positive-semidefinite matrix, which we term . The relative magnitudes of the eigenvalues, , of this matrix may be used to quantify the mobility of any given molecule under observation. As an application of the matrix approach, we show how this formulation may be related to a more commonly employed, but less general, ‘constrained rotation within a cone’ model of molecular orientational dynamics. In the latter half of this section, we address to the problem of precisely determining the entries of the matrix (a necessary precursor to calculating the eigenvalues!). Importantly, may be inferred simply by solving a matrix pseudo-inversion problem of modest dimensions. This computational technique for determining will be indispensable to the later sections of this paper, in which we extract rotational mobility measurements from single noisy images of molecules.
2.1 The matrix formulation for characterizing images of rotating molecules
High numerical aperture ( ) imaging apparatuses based on oil-immerison or other immersion microscope objectives require special modeling considerations, in order to properly account for all of the optical effects that will influence the final intensity distribution recorded on a detector [29]. Furthermore, electric dipoles (and therefore fluorescent molecules) are highly anisotropic, emitting a far-field intensity distribution resembling that of an expanding torus [30] aligned perpendicular to the emission dipole moment of a given molecule. After propagation through a microscope composed of an objective/ tube lens pair, the electric field present at an image sensor can be calculated as follows:
| (2) |
In Eq. (2), are the x/y polarized electric fields present at a given point, , in the microscope’s image plane. are the x/y polarized electric fields associated with an immobile dipole oriented along the x, y or z axis of the optical system (see Fig. 2(a) ). Subscripts denote the polarization of the field, and superscripts denote the orientation of the dipole. This equation simply states that the field for an arbitrary dipole drection is a superposition of three effective dipoles corresponding to the three Carteisan projections of the dipole moment. Collectively, these fields may be assembled into a 2-by-3 matrix . The vector specifies the orientation of the emission dipole moment of a single molecule at a given instant in time as a point on a sphere. The amplitude, A, of the dipole moment can be calculated from as:
| (3) |
Alternatively, one may parameterize the instantaneous orientation of a molecule by the azimuthal and polar inclination, , of the dipole moment with respect to the imaging system’s coordinate frame (Fig. 2(a)):
| (4) |
In order to calculate the electric fields , one must, in general, rigorously model all components of the optical system, as well as any inhomogeneities within the sample itself (such as refractive index variations) [31–33]. Furthermore, the electric fields present at the image sensor will vary as a function of microscope defocus d, the distance between the objective lens focal plane and the single molecule being imaged (Fig. 2(b)). In this paper, we assume isotropic media, and calculate the electric fields using the procedure detailed in [22]. For completeness, we briefly summarize how the image plane electric fields are computed in the appendix of this paper. Finally, it ought to be noted that we have neglected to calculate the z-polarized portion of the electric field incident upon the microscope image plane. While a z-polarized field does exist, it is generally many orders of magnitude smaller than the x/y polarized electric fields, and need not be included in our analysis. Equipped with Eq. (2), the intensity, , present at the image plane (at a single instant in time) may be calculated as:
| (5) |
Rotation of the emission dipole moment is modeled as follows: If the molecule undergoes a total of absorption-emission cycles over the integration period of the image sensor, , we simply replace the outer-product enclosed in parentheses in Eq. (5) with the summation:
| (6) |
Where denotes the orientation of the emission dipole at the time of the ’th emission event. Note that up to this point we have made no assumptions regarding the rotational behavior of the molecule, or the polarization of excitation illumination. Such modeling considerations must eventually be taken into account in order to prescribe the relative frequencies of various . (In part b of this section, we will adopt a simplified ‘rotation within a cone’ model for this purpose.) However, key insights may be achieved before restricting our analysis. Substituting Eq. (6) into Eq. (5), the time-integrated image associated with a rotating molecule is:
| (7) |
Equation (7) is completely general. No matter how erratically a molecule behaves over the camera integration time, the final image visible on the detector can be calculated from the matrix . From Eq. (6), is the summation of the outer-products of 3-dimensional vectors . Therefore, must have an eigen-decomposition:
| (8) |
Where the are the eigenvalues of , indexed from largest to smallest in magnitude. The are the corresponding eigenvectors. One may interpret the as three, stationary dipoles arranged othogonally. The square roots of the are then understood to be the corresponding amplitudes of these three dipoles (see Figs. 2(d) and 2(f)). The relative sizes of the eigenvalues , yields a useful picture of the rotational mobility of the molecule in question.
Fig. 2.

Parameterizations of single-molecule orientation and rotational mobility. (a) A rotationally fixed single molecule may be modeled as a fixed dipole with polar orientation and azimuthal orientation . Alternatively, orientation may be described as a unit vector , with x, y and z components , and respectively. (b) Experimental schematic: A single molecule is placed a distance d from the focal plane of the objective, and a single widefield image is acquired. (c) Rotation within a cone model: A single molecule undergoes constrained rotation about some mean orientation . The molecule may deviate by an angle from the mean. (d) A molecule rotating in a cone may be alternatively parameterized by three orthogonal dipoles. One dipole will have amplitude equal to the square root of the largest eigenvalue of the M matrix, as defined in the main text. The other two dipoles will have amplitudes equal to the square root of the second largest eigenvalue. (e) In a more general case, a single molecule’s rotation may be confined to an elliptical region of the unit hemisphere, parameterized by two angles and . (f) For rotation within an elliptic region, the equivalent eigenvectors provide three different dipoles, each with a distinct amplitude determined from the square roots of the eigenvalues of the M matrix.
2.2 Example: Relationship of matrix to the ‘rotation within a cone’ model
We now discuss how one may interpret the magnitudes of the in order to deduce rotational mobility, by showing how a special case of the matrix approach is equivalent to the widely used ‘rotation within a cone’ model of rotational diffusion [34]. In many specimens of biological interest, fluorescent labels are neither entirely immobilized, nor entirely free to rotate. A molecule exhibiting some intermediate mobility may be approximated as follows: The molecule will have a mean orientation, specified by the angles: . Furthermore, we assume that the molecule may deviate from its mean orientation by an angle α, which specifies the half-aperture of a cone, within which the emission dipole is rotationally constrained (Fig. 2(c)). How is the matrix calculated for this case? We begin by assuming that the molecule visits each orientation within its constraint cone with uniform frequency. (We have chosen this elementary model in order to streamline the ensuing mathematical analysis, however our approach may be readily adopted to incorporate more sophisticated treatments of orientational dynamics involving rotational diffusion and fluorescence lifetime considerations. The reader is refered to [35].) This assumption permits us to convert the summation of Eq. (6) into an averaging over the solid angle circumscribed by α (the red region shown in Fig. 2(c)). In this case:
| (9) |
Where is the area of the solid angle, and:
| (10) |
In Eq. (9), we have chosen to work in a rotated coordinate system (Fig. 2(c)). That is, when the molecule assumes the orientation . This coordinate transformation is effected by the rotation matrix , and the relationship:
| (11) |
Explicitly, a suitable rotation matrix can be calculated using the axis/angle method [36]:
| (12) |
Where:
| (13) |
Since and are constant with respect to the variables of integration, they may be placed outside of the integral in Eq. (9). The remaining expression may be integrated analytically:
| (14) |
and do not affect the magnitudes of the eigenvalues, , because they are orthonormal. Hence,
| (15) |
Figure 3 shows single-molecule images simulated using the matrix approach. Five molecules are oriented at , each exhibiting a different α. As α increases, the images become increasingly symmetric. In Fig. 4(a) , the eigenvalues are plotted as a function of cone angle α (we assume that ). Note that two of the eigenvalues, and , are identical, and increase in magnitude with increasing α. The image of a molecule rotating within a cone will thus be identical to an image of three super-imposed dipoles—two of the dipoles will have equal amplitude, and a third dipole will have a distinct amplitude, greater than or equal to that of the other two. When , the rotating molecule effectively behaves as an isotropic emitter, as all three eigenvalues are identical, (corresponding to three dipoles of amplitude 1/3). When , We observe that , the other eigenvalues are zero, and the molecule behaves as a rotationally fixed dipole. In the intermediate regime, in which α is between and , the cone angle may be inferred simply by using a single measurement to read off α from the red curve in Fig. 4(a).
Fig. 3.

Images of single molecules simulated with mean orientation , and varying α. For these images, the defocus was set to d = 1.25 μm. All other simulation parameters are specified in section 3.
Fig. 4.
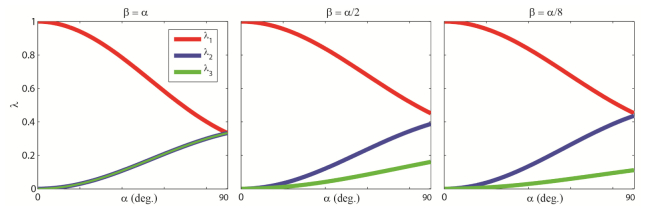
Analytical calculation of the eigenvalues of the M matrix for different cone angles α and β. Note that these parameters do not change as a function of mean orientation, . Furthermore, they are not affected by experimental variables such as defocus, emission wavelength, or microscope NA. (a)-(c) Eigenvalue calculations, β = α, β = α/2, and β = α/8 respectively.
How does the above analysis relate to more sophisticated single-molecule rotational diffusion models? Our assumption of a uniform emission probability distribution within the rotational constraint cone is only completely valid under select circumstances. For example, consider the case when the rotational correlation time [37], , of a molecule is much shorter that the molecule’s fluorescence lifetime, . That is:
| (16) |
In this case, the molecule will have completely explored the constraint cone between the time at which it absorbs a photon and when it emits a photon, and the emission probability within the constraint cone will therefore be uniform. On the other hand, if , then the polarization of the excitation illumination source relative to the mean orientation of the molecule will induce an asymmetric emission probablity distribution within the constraint cone [35]. In this situation, the cone angle α cannot be deduced from the eigenvalues of alone.
2.3 Beyond the rotation within a cone model with the matrix
The rotation within a cone model may be augmented to allow for the case in which there are three distinct eigenvalues of the matrix. Instead of assuming the molecule’s cone of rotational constraint is a circular region on a sphere, the dipole motion may instead demark an ellipse (Fig. 2(e)) parameterized by two angles, α and β. If we again assume that the molecule’s emission probability is uniform throughout this elliptical constraint region, the matrix may be calculated by changing the bounds of integration of Eq. (9):
| (17) |
is the area of the solid angle parameterized by α and β. The integral over may be evaluated straightforwardly, simplifying Eq. (17) to:
| (18) |
Where:
| (19) |
In this case, is computed as:
| (20) |
From inspection, the eigenvalues of are:
| (21) |
The one-dimensional integrals appearing in Eqs. (19) and (20) may be calculated numerically using a trapezoidal approximation scheme. In Figs. 4(b) and 4(c), we have calculated the three eigenvalues of for different α and β. Hence, the image formed from a molecule rotating within an elliptic constraint region will be identical to the image formed from three super-imposed dipoles, each of distinct amplitude (Fig. 2(f)).
2.4 The basis function formulation: determining the entries of the matrix
Given a single molecule’s corresponding matrix, it is straightforward to compute the eigenvalues of this matrix, and to deduce the images expected from the molecule. We now address the inverse problem of inferring the entries of the matrix from raw image data. Here we propose a simple approach based on matrix inversion. Carrying out the matrix multiplication in Eq. (7), and distributing terms yields the following expression for :
| (22) |
We define the following six ‘basis functions’:
| (23) |
Substituting Eqs. (23) into Eq. (22) allows us to express as an inner product:
| (24) |
Where is the vector containing the six unique elements of . Practically, the data set acquired by an image sensor is not a continuous function , but a set of N discrete pixel intensity readings , which we will denote by the vector . Such a data set may be interpreted as having been generated by the following matrix multiplication:
| (25) |
Where the columns of the matrix are composed of a discretized sampling of the six basis functions , evaluated at the N image sensor pixel locations. So long as the single-molecule image is sampled over more than six pixels, Eq. (25) defines an over-determined linear system, which may be solved by computing the pseudo-inverse of , which we denote :
| (26) |
We then recover the entries of by multiplying by the intensity data:
| (27) |
Evaluating Eq. (27) is equivalent to solving a linear least squares problem. Once the six unique entries of are found, analysis of the eigenvalues may proceed as detailed in section 2.b. Alternatively, the entries of may be inferred by solving a maximum likelihood estimation problem, subject to Poisson noise statistics, using an approach similar to [18, 20]. While maximum likelihood estimation theoretically achieves superior measurement precision, the strategy described here is computationally advantageous – maximum likelihood estimation requires iteratively optimizing an objective function, while determining from Eq. (27) requires only a single matrix multiplication of modest dimensions. (Strategies for handling the more complex MLE problem have been described in detail in [18], and GPUs can be leveraged to accelerate image analysis speed [38].)
We will also consider the effect of polarization optics, aligned along the x/y axes of our imaging system. The basis function approach to inferring may be trivially modified. Polarized images, , and their pixelated counterparts , may be simulated by ignoring the terms corresponding to y/x polarized light in Eq. (22) respectively. The polarized components of the six basis functions, , may be found in similar fashion. Assuming that both the x and y polarized images are recorded on different image sensors (or different regions of the same image sensor), the system of equations that is solved in order to determine may be augmented:
| (28) |
Where is found by computing the pseudo-inverse of . In Fig. 5 , representative basis functions have been generated using various amounts of objective lens defocus d (see the next section for the other simulation parameters, which were held constant throughout this paper). The six basis functions used to generate unpolarized images are shown alongside their polarized components. Note that as defocus increases, the basis functions become more diffuse, inhabiting a larger region of the image sensor. As will be shown in the next section, this effect will have different consequences, depending upon the signal and background levels.
Fig. 5.

The image of any single molecule, fixed or rotationally mobile, may be decomposed into a linear combination of six basis functions. These six basis functions have been calculated at three representative defocus depths, given the simulation parameters presented in the main text. The x/y polarized components of these basis functions are also shown with x/y superscripts. Units of intensity are scaled such that the brightest pixel for a dipole parallel to the focal plane at 0.55 μm defocus has a magnitude of 1.
3. Numerical experiments
In this section, we demonstate the efficacy of our proposed method using simulated data sets of single-molecule images. In the first trial, we simulate rotationally fixed molecules, while varying the microscope defocus distance, d. We determine the optimal defocus for various signal and background levels by measuring the orientation of each simulated molecule, and comparing our measurement to the molecule’s true orientation. Using a value of d that minimized mean angular measurement error for fixed molecules, we performed two additional numerical experiments using rotationally mobile molecules: First, we demonstrate that quantitative cone angle (α) measurements may be acquired even when signal is modest. Then, we show that two distinct populations of molecules may be differentiated by their rotational mobility. All simulations presented in this section were coded using the MATLAB programming language.
For all numerical trials, we assumed the experimental apparatus depicted in Fig. 6 , which simultaneously records two orthogonally polarized images. Alternatively, a single unpolarized image may be captured by removing the polarizing beamsplitter and one of the image sensors. We simulated an objective lens with 100 magnification and a numerical aperture of 1.40. Molecules were embedded in a medium identical to that of the objective’s immersion oil, of refractive index n = 1.518. The emission wavelength of the molecules was λ = 600 nm. We assumed an effective pixel size of 160 nm for our image sensor (which corresponds to the 16 μm pixel size of an Andor iXon Ultra 897 EMCCD detector, after 100 magnification). Images were simulated on a 33-by-33 pixel grid, assuming that the molecule was located at the midpoint of the central pixel. In order to model the effects of photon shot noise upon our measurements, we first normalized simulated (noiseless) images to a desired number of total signal photons, then added a specified amount of uniform background to each pixel. For each pixel, as our ‘measured’ number of photons detected, we drew a Poisson distributed random variable with mean equal to the calculated (noiseless) photon count. Specifically, the shot noise corrupted data at a given pixel is determined as:
| (29) |
Where denotes a random variable drawn from a Poisson distribution, with mean in brackets. In practice, the noise statistics of an EMCCD detector are a much more complicated function of signal photons, readout noise, and electron-multiplication gain [39]. However, the raw data from an EMCCD can be appropriately scaled such that a distribution is observed that can be closely approximated as Poisson [40]. Hence, we adopt this simplified model, and neglect other noise sources.
Fig. 6.
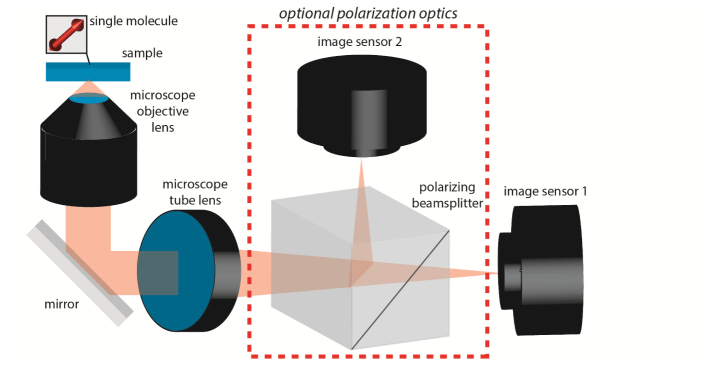
Experimental schematic assumed for all numerical experiments.
An additional modeling consideration arises due to the fact that the objective’s collection efficiency is a function of a molecule’s orientation. In isotropic media, far fewer photons will be collected for a molecule with an emission dipole moment parallel to the optical axis, than for a molecule oriented perpendicular to the optical axis (parallel to the plane of the coverslip), even if the two molecules emit the same total number of photons. This feature arises due to the fact that the objective collects only a cone of light, defined by the numerical aperture, emmanating from a given molecule [41]. In order to properly account for this effect, the basis components used to simulate (noiseless) single-molecule images must be properly normalized. For example, say a molecule oriented parallel to the coverslip emits a total of P photons. Then the normalized basis components will be:
| (30) |
The pixels of a noiseless image may thus be calculated from the normalized matrix as:
| (31) |
Normalizing the basis components for polarized images may also be performed simply by multiplying each component by a factor of . For the remainder of this paper, when signal photons are reported, we are referring to the mean signal for a molecule in the plane of the coverslip. Hence, the mean signal detected for molecules inclined towards the optical axis will be substantially less. For simplicity, we assume that molecules of all orientations receive equal excitation intensity. As a final data-processing step, the eigenvalue measurements are normalized such that they sum to 1. This normalization is equivalent to dividing by a factor of . This permits eigenvalues corresponding to different molecules emitting varying numbers of photons to be compared on the same scale.
3.1 Numerical experiment #1: Determining optimal microscope defocus
In this experiment, images of 1,000 rotationally immobilized molecules were simulated at orientations drawn uniformly at random from a unit hemisphere. That is, a given molecule’s orientation was selected by drawing and from the following distributions:
| (32) |
Where denotes a random variable drawn from a uniform distribution with support [0,1]. The orientation of each molecule was estimated from unpolarized and polarized image data as the eigenvector corresponding to the largest eigenvalue of . Since this simulation concerns immobilized molecules, we expect the second and third largest eigenvalues to be zero. However, due to Poisson noise, we found these eigenvalues to have magnitudes ~3-7% that of the largest eigenvalue. The angular error from the true orientation was calculated as:
| (33) |
Where and are unit vectors corresponding to the true and estimated orientations of the emission dipole moment respectively. For a molecule oriented in the plane of the coverslip the mean detected signal was 3,000 photons. The simulation was repeated for defocus values of d = 0.3-3.0 μm in 50 nm increments. Furthermore, tests were performed using a background of b = {0, 5, 10, 15, 20} photons per pixel. Results for polarized and unpolarized data are shown in Figs. 7(a) and 7(b) . Inspection of these plots reveals that polarized detection offers significant advantages, in terms of reducing measurement error for a given photon budget. Furthermore, these plots demonstrate the interesting feature that the optimal defocus (minimal mean error)is a function of signal and background. When there is no background, it is advantageous to defocus as much as possible. In this regime, increased defocus produces images that vary strikedly as a function of orientation, aiding image analysis and enhancing measurement precision. However, if a moderate to high amount of background is present, there is a ‘sweet-spot’ between d = 0.5 μm to d = 0.6 μm, at which the measurement error is minimized. When background is no longer negligible, the helpful effects of defocus must be balanced with the need to conentrate emitter intensity upon a smaller region of the image sensor, in order to maintain a reasonable signal-to-background ratio. From inspection of the results in Fig. 7, we choose a defocus of d = 0.55 μm for the remainder of our numerical experiments. Note that our chosen defocus parameter is much smaller in magnitude than what is generally employed in orientation-imaging applications (d ~1.0 μm in [16, 24, 25]). Before continuing, it ought to be stressed that the optimal defocus can be altered when one adjusts any of the imaging parameters such as emission wavelength, objective specifications or detector pixel size. Hence, when applying this technique to a different experimental system, it is necessary to re-calculate the plots in Fig. 7 accordingly. As an alternative to performing numerical simulations, as we have done here, the tradeoffs between defocus and background can be analyzed using Cramer-Rao lower bound calculations. The reader is referred to [7, 8, 42].
Fig. 7.
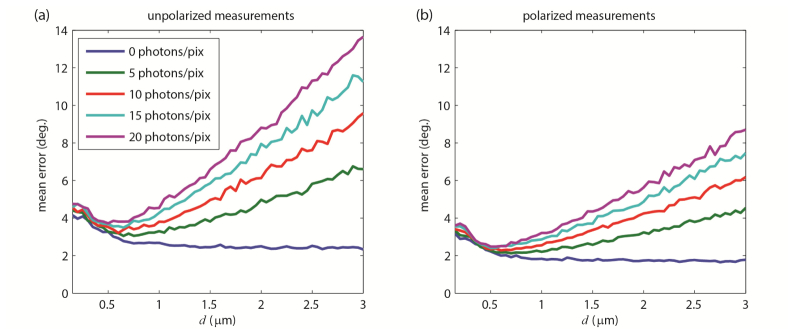
Results of numerical experiment 1. (a) Mean angular error as a function of defocus, d, for unpolarized and (b) polarized image data, with varying numbers of background photons per pixel.
3.2 Numerical experiment #2: Measuring the eigenvalues of in the presence of noise
To demonstrate that rotational mobility may be ascertained from single-molecule images, we simulate a corpus of molecules with cone angle, α, varied in 5° increments. The mean orientation, , of each molecule was drawn randomly. 100 molecules were simulated for each distinct α. The simulation was performed in two different signal regimes: First, we used a mean signal for a molecule in the plane of the coverslip of 10,000 photons, and a background of 0 photons. Next, we switched to a mean of 3,000 photons of signal, and 20 photons of background per pixel. This experiment was repeated for both polarized and unpolarized data. Figures 8(a) and 8(b) show the resulting eigenvalue measurements as a function of α. For comparison, we overlay the theoretically calculated eigenvalues as blue and red curves, as plotted in Fig. 4. We note that the eigenvalues obtained from simulated data cluster around the theoretical values, and that both high signal and polarized data contribute to increased precision of acquired eigenvalue measurements. To demonstrate the effects of noise on the raw data input into our algorithm for determining and its eigenvalues, in Fig. 8(c), we show representative simulated images of single molecules with different α. The cone angle α of a given molecule may be inferred from the largest eigenvalue, , alone (the red curve). However, measuring all three eigenvalues provides a much more robust means of ascertaining rotational mobility, especially if each eigenvalue has a significantly different magnitude—as this would imply that the rotation ‘within a cone’ model of Fig. 2(c) is invalid, and the more complex elliptic constraint region (Fig. 2(e)) is in force.
Fig. 8.

Results of numerical experiment 2. (a) and (b) Eigenvalue measurements from single-molecule images for unpolarized data (a) and polarized data (b). Overall standard deviations in eigenvalue measurements for each trial are noted on their respective plots. Error bars are . (c) Sample raw images of molecules with different α.
3.3 Numerical experiment #3: Differentiating sub-populations of molecules by their rotational mobility
Having demonstrated the ability to extract the eigenvalues of the matrix for a given single-molecule image, we now turn our attention to gauging whether our method can yield meaningful insight under realistic experimental conditions. We carried out the following trial: 3,000 single-molecule images were simulated, using 3,000 photons mean signal for a molecule parallel to the coverslip, and 20 photons of background per pixel. Of these molecules, 1,500 had a cone angle of α = 55°, and the other 1,500 had a cone angle of α = 65°. As before, the mean orientation of each molecule was drawn randomly. We computed the eigenvalues of the matrix using both polarized and unpolarized image data. Additonally, in order to benchmark our technique with an established method, we also computed the LD associated with each molecule. In our simulation of LD measurements, we incorporated the photon shot noise resulting from signal detected from a single molecule. However, in order to demonstrate that our image analysis technique outperforms a simple LD measurement, even under unfavorable signal-to-noise conditions, we did not include background when simulating noisy LD data. In Fig. 9(a) , we histogram the LD measurements associated with the 3,000 simulated molecules. From the LD data alone, it is difficult to infer that two populations of molecules are present: The histogram features a single peak at LD = 0, and the broadness of the distribution could potentially be a result of multiple populations of molecules with distinct rotational mobilities, a single population of molecules with a low rotational mobility, or noise associated with the LD measurements. In order to make any conclusions about this sample, more quantitative analysis and potentially a more sophisticated experimental setup is required. However, in Figs. 9(b) and 9(c), we histogram the largest eigenvalue, , of the matrix associated with each single-molecule image, computed using unpolarized and polarized data, respectively. When polarized data (Fig. 9(c)) is inspected, a bimodal distribution of measurements is clearly present, confirming the presence of two populations of molecules with different rotational mobilities. In comparison, two peaks in the eigenvalue histogram are not as readily evident when examining the unpolarized measurement results (Fig. 9(b)). This difference underscores the improved measurement capabilites of a polarized detection system. To better quantify the performance enhancement gained by acquiring polarized images, we fit Gaussian distributions (overlaid on Figs. 9(b) and 9(c)) to the 1,500 eigenvalue measurements taken for the set of molecules that had α = 55° (red curves) and α = 65° (green curves), using both the polarized and unpolarized results. While the means, η, of the Gaussians corresponding to different α are nearly identical when considering either polarized or unpolarized data, the standard deviations, σ, of the measurements differ. In the unpolarized case, the standard deviations in eigenvalue measurements are ± 0.061 while for polarized data, the standard deviations are ± 0.042. Practically, if one were to infer cone angle by consulting the red curve in Fig. 4(a), such precision in eigenvalue measurement would imply a precision of ± 6.3° (unpolarized data) or ± 4.3° (polarized data) in measurement of α. Thus when it is necessary to make fine distinctions in the rotational mobilities of different molecules, polarized detection is preferred.
Fig. 9.

Results of numerical experiment 3. (a) Linear dichroism histogram. From this data alone, the presence of two distinct populations of molecules is not clearly evident. (b) Histogram of largest eigenvalues measured for each single-molecule image using unpolarized data. (c) Histogram of largest eigenvalues measured for each single-molecule image using polarized data.
4. Discussion
We conclude by remarking on some of the challenges that, although out of the scope of this current work, will need to be addressed in order to realize the M matrix method in actual experiments.
-
Localization of molecules:
In our current simulation framework, we have assumed that our simulated data are precisely aligned with the basis functions used to infer the matrix. In experiment, however, we do not know the lateral (x-y) position of a given molecule with respect to the grid of image-sensor pixels, nor do we know the precise defocus of the molecule. In practice, each of these quantities will have to be estimated as a pre-processing step, then an appropriate set of basis functions generated accordingly. As has been shown in previous work [17–19,21], accurate methods for localizing molecules in three dimensions are feasible, even when orientational effects are prominent. Alternatively, one could envision employing maximum likelihood estimation or an expectation-maximization framework [43] to iteratively estimate both the position and the matrix from a single-molecule image.
-
Optical Aberrations:
The accuracy of the method hinges upon the ability to determine the true basis functions that are superimposed to form single-molecule images. In our current simulations, we have employed an idealized model for our objective lens, and assumed that there are no aberrations present in our system. In practice, the sample under investigation and the components in the imaging pathway of the microscope will introduce aberrations in the acquired single-molecule images. In order to avoid incurring systematic measurement errors as a result of these aberrations, the simulations used to generate accurate basis functions must be augmented to incorporate any aberrations that may have some impact on experimentally measured images [44–46]. A spatial-light modulator or deformable mirror could additionally be used to mitigate aberrations that may cause discrepancies between theoretical basis function calculations and experiment [47].
Notwithstanding the points noted above, the proposed method provides new insight into the orientational mobility of a molecule from a single image. It removes many of the ambiguities that arise when using more conventional linear dichroism (or bulk polarization anisotropy) measurements to ascertain the orientational dynamics of individual fluorescent molecules. As evidenced by our simulations, it is possible to acquire meaningful rotational mobility data when signal and background are at levels typical of single-molecule imaging experiments (3,000 photons signal, 20 photons per pixel of background). Our method may be applied to unpolarized data sets, however polarized detection enhances measurement precision. Future work will explore further augmentations to the optical system that will make our method feasible under circumstances when signal is severely limited, and when aberrations/localization uncertainty may be present.
Acknowledgments
The authors wish to thank Mikael Backlund, Matthew Lew, Steffen Sahl and Yoav Shechtman for helpful discussions. A. S. B. acknowledges support from a Simons Graduate Research Assistantship. This work was supported by National Institutes of Health, National Institute of General Medical Sciences Grant R01GM085437.
5. Appendix
In this section, we summarize how we determine the electric fields present at the image plane of a microscope for a rotationally fixed molecule. This calculation serves as a major building block for simulating the image of a molecule undergoing constrained rotation. The first step is to analytically evaluate the electric fields, , present at the microscope’s back focal plane [48,49]:
| (34) |
In Eqs. (34), the polar coordinates specify a point in space within the plane located one focal length behind the microscope’s objective lens. These formulas are used to calculate electric fields within the circular pupil specified by , Where is the numerical aperture of the objective, and is the refractive index of the objective’s immersion medium. Outside of this circle, the electric fields are zero. Furthermore, the fluorescence wavelength is , the wavenumber is , the defocus distance of the emitter from the (front) focal plane is d (where d < 0 specifies an emitter between the objective lens and its focal plane), and the refractive index of the medium surrounding the image sensor is (usually it is assumed that ). To calculate the image plane electric fields , the Fourier transforms of the back focal plane fields are calculated numerically, which can be performed efficiently using the fast Fourier transform algorithm:
| (35) |
In Eq. (35), denotes the Fourier transform operation, and specifies a point on the image plane. When numerically simulating single-molecule images using Eqs. (34) and (35), the units of length at the image plane will depend upon the density of sampling within the back focal plane, and the fluorescence wavelength, . For example, our simulations evaluate on a 512-by-512 grid, with a sample spacing of . After applying the fast Fourier transform, the resulting simulated images are sampled at Δr = 160 nm. If one were to also account for the magnification , of our optical system, this sampling would correspond to Δr = 160 μm, which matches the pixel size of the image sensor referred to in our numerical experiments.
References and links
- 1.Ha T., Enderle T., Chemla S., Selvin R., Weiss S., “Single molecule dynamics studied by polarization modulation,” Phys. Rev. Lett. 77(19), 3979–3982 (1996). 10.1103/PhysRevLett.77.3979 [DOI] [PubMed] [Google Scholar]
- 2.Ha T., Glass J., Enderle T., Chemla D. S., Weiss S., “Hindered rotational diffusion and rotational jumps of single molecules,” Phys. Rev. Lett. 80(10), 2093–2096 (1998). 10.1103/PhysRevLett.80.2093 [DOI] [Google Scholar]
- 3.Sosa H., Peterman E. J. G., Moerner W. E., Goldstein L. S. B., “ADP-induced rocking of the kinesin motor domain revealed by single-molecule fluorescence polarization microscopy,” Nat. Struct. Biol. 8(6), 540–544 (2001). 10.1038/88611 [DOI] [PubMed] [Google Scholar]
- 4.Peterman E. J. G., Sosa H., Goldstein L. S. B., Moerner W. E., “Polarized fluorescence microscopy of individual and many kinesin motors bound to axonemal microtubules,” Biophys. J. 81(5), 2851–2863 (2001). 10.1016/S0006-3495(01)75926-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forkey J. N., Quinlan M. E., Goldman Y. E., “Measurement of single macromolecule orientation by total internal reflection fluorescence polarization microscopy,” Biophys. J. 89(2), 1261–1271 (2005). 10.1529/biophysj.104.053470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg S. A., Quinlan M. E., Forkey J. N., Goldman Y. E., “Rotational motions of macro-molecules by single-molecule fluorescence microscopy,” Acc. Chem. Res. 38(7), 583–593 (2005). 10.1021/ar040137k [DOI] [PubMed] [Google Scholar]
- 7.Foreman M. R., Török P., “Fundamental limits in single-molecule orientation measurements,” New J. Phys. 13(9), 093013 (2011). 10.1088/1367-2630/13/9/093013 [DOI] [Google Scholar]
- 8.Agrawal A., Quirin S., Grover G., Piestun R., “Limits of 3D dipole localization and orientation estimation for single-molecule imaging: towards Green’s tensor engineering,” Opt. Express 20(24), 26667–26680 (2012). 10.1364/OE.20.026667 [DOI] [PubMed] [Google Scholar]
- 9.Phelps C., Lee W., Jose D., von Hippel P. H., Marcus A. H., “Single-molecule FRET and linear dichroism studies of DNA breathing and helicase binding at replication fork junctions,” Proc. Natl. Acad. Sci. U.S.A. 110(43), 17320–17325 (2013). 10.1073/pnas.1314862110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quinlan M. E., Forkey J. N., Goldman Y. E., “Orientation of the Myosin Light Chain Region by Single Molecule Total Internal Reflection Fluorescence Polarization Microscopy,” Biophys. J. 89(2), 1132–1142 (2005). 10.1529/biophysj.104.053496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beausang J. F., Shroder D. Y., Nelson P. C., Goldman Y. E., “Tilting and wobble of Myosin V by high-speed single-molecule polarized fluorescence microscopy,” Biophys. J. 104(6), 1263–1273 (2013). 10.1016/j.bpj.2013.01.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gould T. J., Gunewardene M. S., Gudheti M. V., Verkhusha V. V., Yin S. R., Gosse J. A., Hess S. T., “Nanoscale imaging of molecular positions and anisotropies,” Nat. Methods 5(12), 1027–1030 (2008). 10.1038/nmeth.1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Testa I., Schönle A., von Middendorff C., Geisler C., Medda R., Wurm C. A., Stiel A. C., Jakobs S., Bossi M., Eggeling C., Hell S. W., Egner A., “Nanoscale separation of molecular species based on their rotational mobility,” Opt. Express 16(25), 21093–21104 (2008). 10.1364/OE.16.021093 [DOI] [PubMed] [Google Scholar]
- 14.Dickson R. M., Norris D. J., Moerner W. E., “Simultaneous imaging of individual molecules aligned both parallel and perpendicular to the optic axis,” Phys. Rev. Lett. 81(24), 5322–5325 (1998). 10.1103/PhysRevLett.81.5322 [DOI] [Google Scholar]
- 15.Bartko A. P., Dickson R. M., “Imaging three-dimensional single molecule orientations,” J. Phys. Chem. B 103(51), 11237–11241 (1999). 10.1021/jp993364q [DOI] [Google Scholar]
- 16.Patra D., Gregor I., Enderlein J., “Image analysis of defocused single-molecule images for three-dimensional molecule orientation studies,” J. Phys. Chem. A 108(33), 6836–6841 (2004). 10.1021/jp048188m [DOI] [Google Scholar]
- 17.Aguet F., Geissbühler S., Märki I., Lasser T., Unser M., “Super-resolution orientation estimation and localization of fluorescent dipoles using 3-D steerable filters,” Opt. Express 17(8), 6829–6848 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Mortensen K. I., Churchman L. S., Spudich J. A., Flyvbjerg H., “Optimized localization analysis for single-molecule tracking and super-resolution microscopy,” Nat. Methods 7(5), 377–381 (2010). 10.1038/nmeth.1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Backlund M. P., Lew M. D., Backer A. S., Sahl S. J., Grover G., Agrawal A., Piestun R., Moerner W. E., “Simultaneous, accurate measurement of the 3D position and orientation of single molecules,” Proc. Natl. Acad. Sci. U.S.A. 109(47), 19087–19092 (2012). 10.1073/pnas.1216687109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Backer A. S., Backlund M. P., Lew M. D., Moerner W. E., “Single-molecule orientation measurements with a quadrated pupil,” Opt. Lett. 38(9), 1521–1523 (2013). 10.1364/OL.38.001521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Backer A. S., Backlund M. P., Diezmann A. R., Sahl S. J., Moerner W. E., “A bisected pupil for studying single-molecule orientational dynamics and its application to 3D super-resolution microscopy,” Appl. Phys. Lett. 104, 193701 (2014). 10.1063/1.4876440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Backer A. S., Moerner W. E., “Extending single-molecule microscopy using optical Fourier processing,” J. Phys. Chem. B 118(28), 8313–8329 (2014). 10.1021/jp501778z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stallinga S., Rieger B., “Position and orientation estimation of fixed dipole emitters using an effective Hermite point spread function model,” Opt. Express 20(6), 5896–5921 (2012). 10.1364/OE.20.005896 [DOI] [PubMed] [Google Scholar]
- 24.Toprak E., Enderlein J., Syed S., McKinney S. A., Petschek R. G., Ha T., Goldman Y. E., Selvin P. R., “Defocused orientation and position imaging (DOPI) of myosin V,” Proc. Natl. Acad. Sci. U.S.A. 103(17), 6495–6499 (2006). 10.1073/pnas.0507134103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hutchison J. A., Uji-i H., Deres A., Vosch T., Rocha S., Müller S., Bastian A. A., Enderlein J., Nourouzi H., Li C., Herrmann A., Müllen K., De Schryver F., Hofkens J., “A surface-bound molecule that undergoes optically biased Brownian rotation,” Nat. Nanotechnol. 9(2), 131–136 (2014). 10.1038/nnano.2013.285 [DOI] [PubMed] [Google Scholar]
- 26.Cyphersmith A., Maksov A., Hassey-Paradise R., McCarthy K. D., Barnes M. D., “Defocused emission patterns from chiral fluorophores: application to chiral axis orientation determination,” J. Phys. Chem. Lett. 2(6), 661–665 (2011). 10.1021/jz2001024 [DOI] [Google Scholar]
- 27.Ham S., Yang J., Schlosser F., Wurthner F., Kim D., “Reconstruction of the molecular structure of a multichromophoric system using single-molecule defocused wide-field imaging,” J. Phys. Chem. Lett. 5(16), 2830–2835 (2014). 10.1021/jz501233d [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y., Gu L., Chang H., Ji W., Chen Y., Zhang M., Yang L., Liu B., Chen L., Xu T., “Ultrafast, accurate, and robust localization of anisotropic dipoles,” Protein Cell 4(8), 598–606 (2013). 10.1007/s13238-013-3904-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richards B., Wolf E., “Electromagnetic diffraction in optical systems. II. Structure of the image field in an aplanatic system,” Proc. R. Soc. Lond. A Math. Phys. Sci. 253(1274), 358–379 (1959). 10.1098/rspa.1959.0200 [DOI] [Google Scholar]
- 30.Jackson J. D., Classical Electrodynamics (Wiley, 1962). [Google Scholar]
- 31.Hellen E. H., Axelrod D., “Fluorescence emission at dielectric and metal-film interfaces,” J. Opt. Soc. Am. B 4(3), 337–350 (1987). 10.1364/JOSAB.4.000337 [DOI] [Google Scholar]
- 32.Böhmer M., Enderlein J., “Orientation imaging of single molecules by wide-field epifluorescence microscopy,” J. Opt. Soc. Am. B 20(3), 554–559 (2003). 10.1364/JOSAB.20.000554 [DOI] [Google Scholar]
- 33.Novotny L., Hecht B., Principles of Nano-Optics (Cambridge University, 2007). [Google Scholar]
- 34.Kinosita K., Jr, Kawato S., Ikegami A., “A theory of fluorescence polarization decay in membranes,” Biophys. J. 20(3), 289–305 (1977). 10.1016/S0006-3495(77)85550-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lew M. D., Backlund M. P., Moerner W. E., “Rotational mobility of single molecules affects localization accuracy in super-resolution fluorescence microscopy,” Nano Lett. 13(9), 3967–3972 (2013). 10.1021/nl304359p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vince J., Geometric Algebra for Computer Graphics (Springer, 2008). [Google Scholar]
- 37.Lakowicz J. R., Principles of Fluorescence Spectroscopy (Kluwer Academic, 1999). [Google Scholar]
- 38.Smith C. S., Joseph N., Rieger B., Lidke K. A., “Fast, single-molecule localization that achieves theoretically minimum uncertainty,” Nat. Methods 7(5), 373–375 (2010). 10.1038/nmeth.1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirsch M., Wareham R. J., Martin-Fernandez M. L., Hobson M. P., Rolfe D. J., “A stochastic model for electron multiplication charge-coupled devices--from theory to practice,” PLoS ONE 8(1), e53671 (2013). 10.1371/journal.pone.0053671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang F., Hartwich T. M. P., Rivera-Molina F. E., Lin Y., Duim W. C., Long J. J., Uchil P. D., Myers J. R., Baird M. A., Mothes W., Davidson M. W., Toomre D., Bewersdorf J., “Video-rate nanoscopy using sCMOS camera-specific single-molecule localization algorithms,” Nat. Methods 10(7), 653–658 (2013). 10.1038/nmeth.2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Axelrod D., “Fluorescence polarization microscopy,” Methods Cell Biol. 30, 333–352 (1989). 10.1016/S0091-679X(08)60985-1 [DOI] [PubMed] [Google Scholar]
- 42.Ober R. J., Ram S., Ward E. S., “Localization accuracy in single-molecule microscopy,” Biophys. J. 86(2), 1185–1200 (2004). 10.1016/S0006-3495(04)74193-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bishop C., Pattern Recognition and Machine Learning (Springer, 2006). [Google Scholar]
- 44.Quirin S., Pavani S. R. P., Piestun R., “Optimal 3D single-molecule localization for superresolution microscopy with aberrations and engineered point spread functions,” Proc. Natl. Acad. Sci. U.S.A. 109(3), 675–679 (2012). 10.1073/pnas.1109011108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu S., Kromann E. B., Krueger W. D., Bewersdorf J., Lidke K. A., “Three dimensional single molecule localization using a phase retrieved pupil function,” Opt. Express 21(24), 29462–29487 (2013). 10.1364/OE.21.029462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang B., Perroud T. D., Zare R. N., “Photon counting histogram: one-photon excitation,” ChemPhysChem 5(10), 1523–1531 (2004). 10.1002/cphc.200400176 [DOI] [PubMed] [Google Scholar]
- 47.Izeddin I., El Beheiry M., Andilla J., Ciepielewski D., Darzacq X., Dahan M., “PSF shaping using adaptive optics for three-dimensional single-molecule super-resolution imaging and tracking,” Opt. Express 20(5), 4957–4967 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Lieb M. A., Zavislan J. M., Novotny L., “Single-molecule orientations determined by direct emission pattern imaging,” J. Opt. Soc. Am. B 21(6), 1210–1215 (2004). 10.1364/JOSAB.21.001210 [DOI] [Google Scholar]
- 49.Axelrod D., “Fluorescence excitation and imaging of single molecules near dielectric-coated and bare surfaces: a theoretical study,” J. Microsc. 247(2), 147–160 (2012). 10.1111/j.1365-2818.2012.03625.x [DOI] [PubMed] [Google Scholar]